
Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer
 ,
, 
Simple Summary
Abstract
1. Introduction
2. Tumor Microenvironment
2.1. Exosome Biogenesis and Secretion in the TME
2.2. Mechanisms of Exosome-Mediated Intercellular Communication in TME
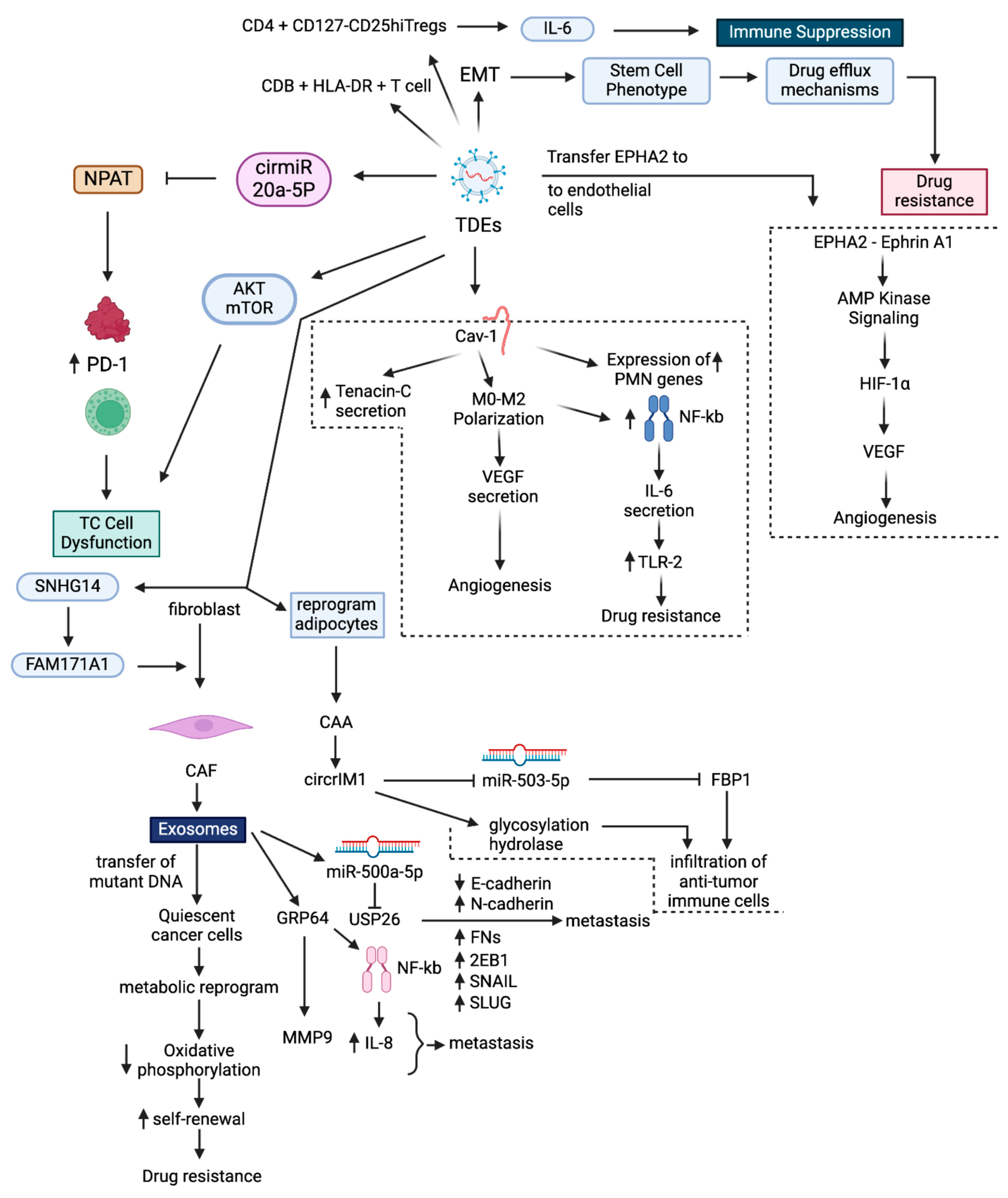
3. Exosomes Impart Drug Resistance in BC
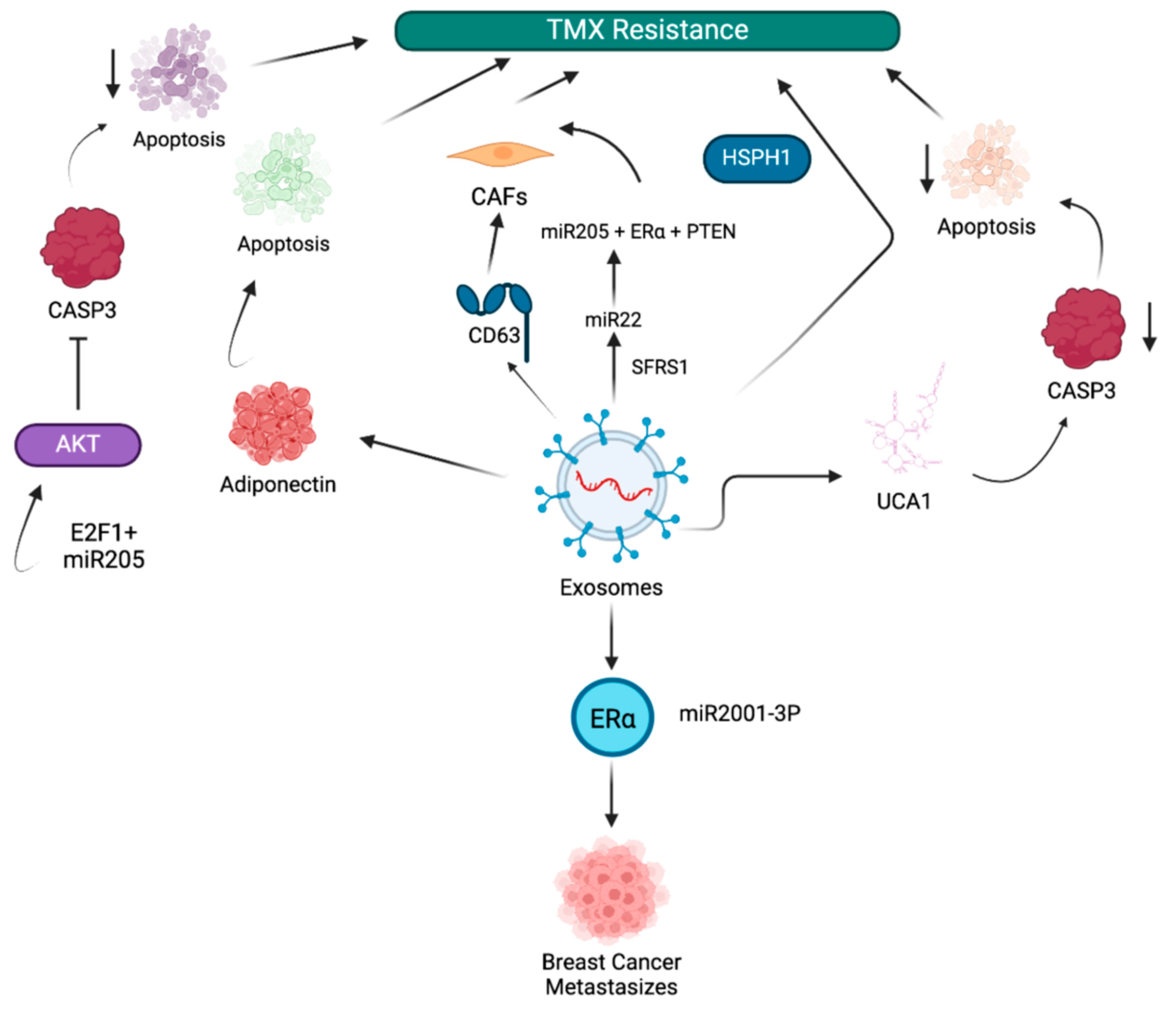
3.1. Exosome-Mediated Tamoxifen Resistance in BC
3.2. Exosome-Mediated Doxorubicin Resistance in BC
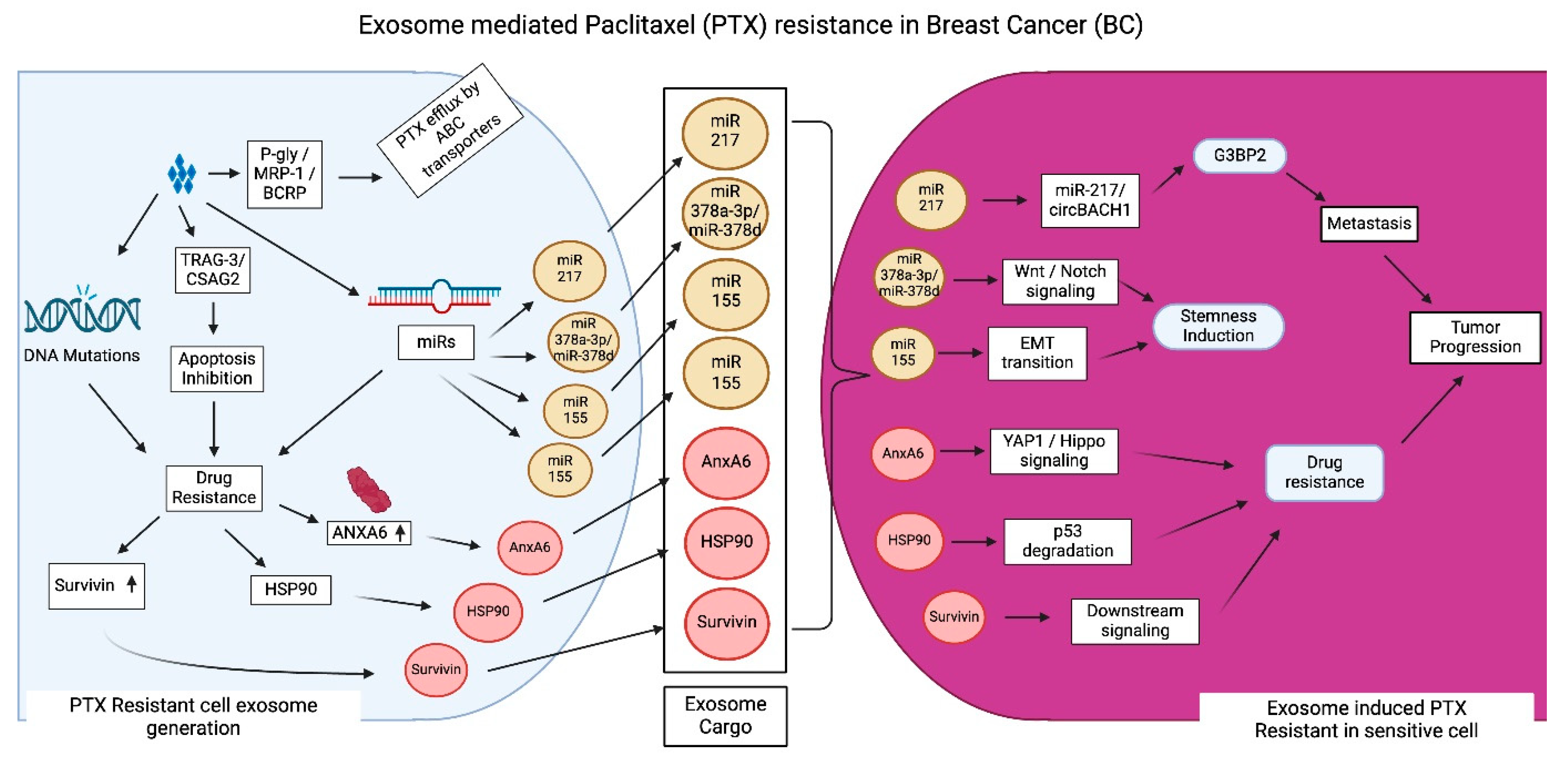
3.3. Exosome-Mediated PTX Resistance in BC
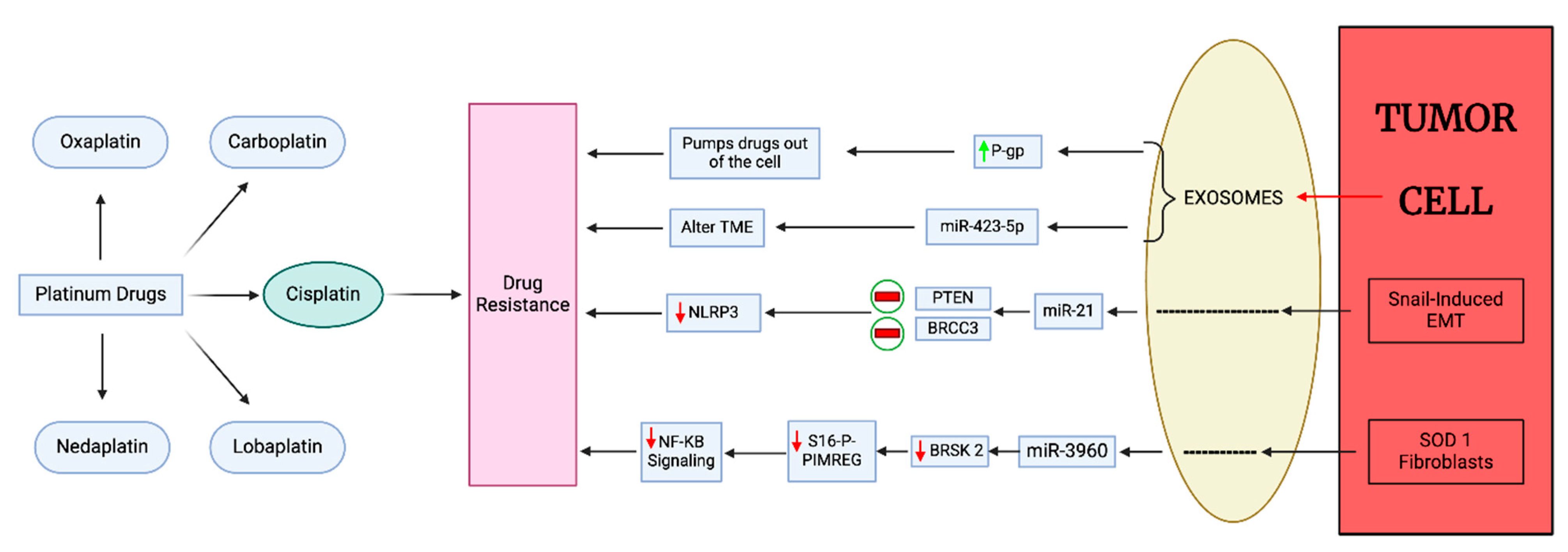
3.4. Exosome-Mediated Platinum Drug Resistance in BC
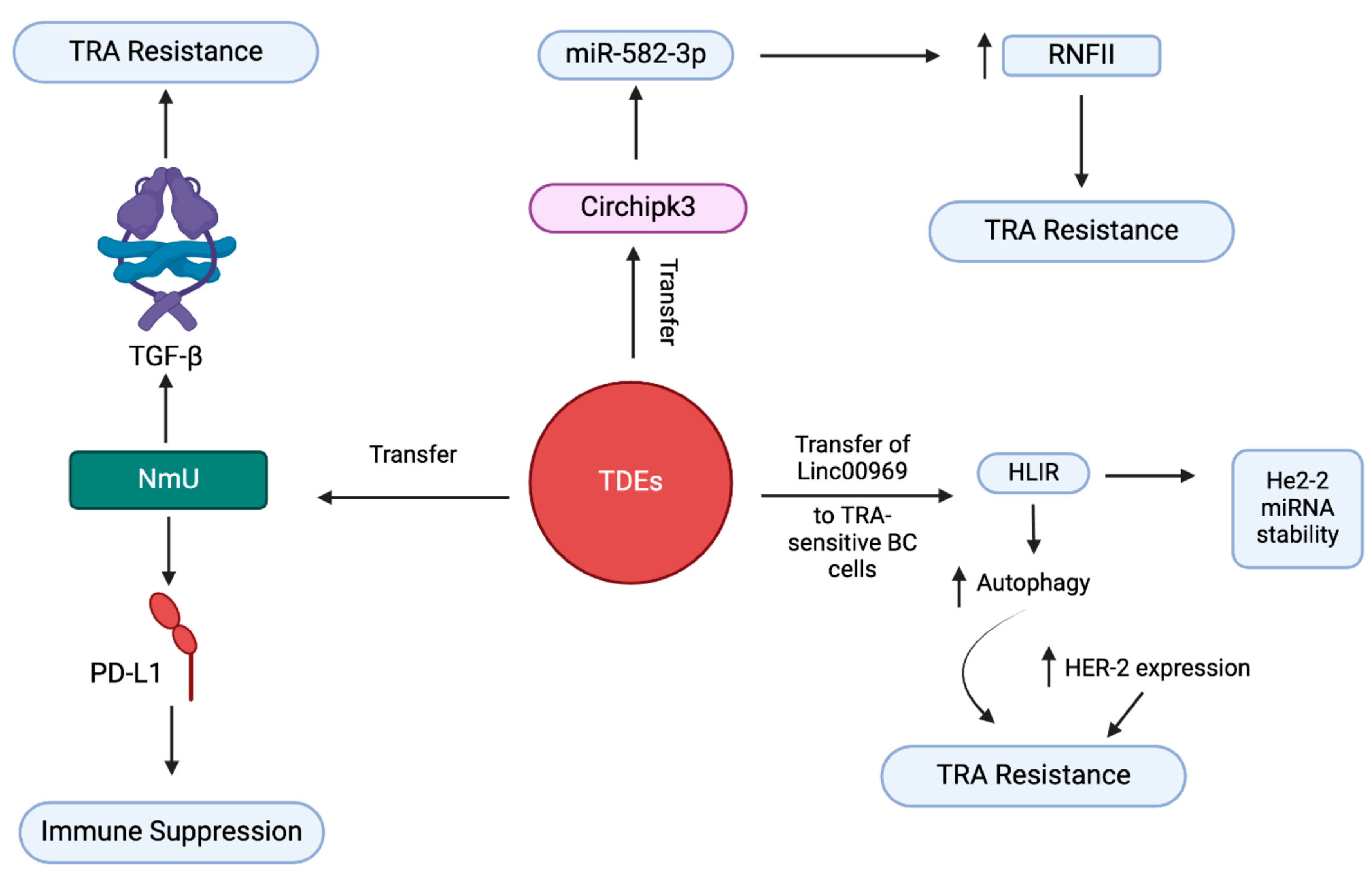
3.5. Exosome-Mediated Trastuzumab Resistance in BC
3.6. Exosome-Mediated ICI Resistance in BC
4. Targeting Exosome Formation and Secretion in BC
4.1. Inhibition of Exosome Secretion
4.2. Sensitization of Exosome-Mediated DR in BC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Antagonist Name | Target | Mechanism/Mode of Action | Ref. | |
|---|---|---|---|---|
| Inhibition of exosome secretion | Macitentan (MAC) | Type 1 ETA PD-L1 |
| [116] |
| Sulfisoxazole (SFX) | Biogenesis of exosomes type 1 ETA |
| [117] | |
| Sensitization of exosome mediated drug resistance in BC | Guggulsterone + Bexarotene (GS + BXT) | FXR and RXR |
| [118] |
| d-Rhamnose-β-hederin (DRβ-H) | miR-16 containing exosomes |
| [119] | |
| GW4869 | nSMase |
| [120] | |
| Nexinhib 20 | Rab27 |
| [120] |
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ugai, T.; Sasamoto, N.; Lee, H.Y.; Ando, M.; Song, M.; Tamimi, R.M.; Kawachi, I.; Campbell, P.T.; Giovannucci, E.L.; Weiderpass, E.; et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat. Rev. Clin. Oncol. 2022, 19, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Lee, J.; Park, Y.H. Trastuzumab deruxtecan for HER2+ advanced breast cancer. Future Oncol. 2022, 18, 7–19. [Google Scholar] [CrossRef]
- Lev, S. Targeted therapy and drug resistance in triple-negative breast cancer: The EGFR axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J.; Leyh, B. The impact of tumor stroma on drug response in breast cancer. Semin. Cancer Biol. 2015, 31, 3–15. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist. Updat. 2020, 50, 100682. [Google Scholar] [CrossRef]
- Battista, T.; Fiorillo, A.; Chiarini, V.; Genovese, I.; Ilari, A.; Colotti, G. Roles of Sorcin in Drug Resistance in Cancer: One Protein, Many Mechanisms, for a Novel Potential Anticancer Drug Target. Cancers 2020, 12, 887. [Google Scholar] [CrossRef]
- Navas, T.; Kinders, R.J.; Lawrence, S.M.; Ferry-Galow, K.V.; Borgel, S.; Hollingshead, M.G.; Srivastava, A.K.; Alcoser, S.Y.; Makhlouf, H.R.; Chuaqui, R.; et al. Clinical Evolution of Epithelial-Mesenchymal Transition in Human Carcinomas. Cancer Res. 2020, 80, 304–318. [Google Scholar] [PubMed]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [PubMed]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar]
- Chung, S.W.; Xie, Y.; Suk, J.S. Overcoming physical stromal barriers to cancer immunotherapy. Drug Deliv. Transl. Res. 2021, 11, 2430–2447. [Google Scholar]
- Bogdanov, A.; Bogdanov, A.; Chubenko, V.; Volkov, N.; Moiseenko, F.; Moiseyenko, V. Tumor acidity: From hallmark of cancer to target of treatment. Front. Oncol. 2022, 12, 979154. [Google Scholar]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar]
- Logozzi, M.; Mizzoni, D.; Angelini, D.F.; Di Raimo, R.; Falchi, M.; Battistini, L.; Fais, S. Microenvironmental pH and Exosome Levels Interplay in Human Cancer Cell Lines of Different Histotypes. Cancers 2018, 10, 370. [Google Scholar] [CrossRef]
- Kumar, A.; Deep, G. Exosomes in hypoxia-induced remodeling of the tumor microenvironment. Cancer Lett. 2020, 488, 1–8. [Google Scholar]
- Kumar, A.; Deep, G. Hypoxia in tumor microenvironment regulates exosome biogenesis: Molecular mechanisms and translational opportunities. Cancer Lett. 2020, 479, 23–30. [Google Scholar]
- Khaledian, B.; Thibes, L.; Shimono, Y. Adipocyte regulation of cancer stem cells. Cancer Sci. 2023, 114, 4134–4144. [Google Scholar] [PubMed]
- Melwani, P.K.; Checker, R.; Balla, M.M.S.; Pandey, B.N. Crosstalk Between Macrophages and Breast Cancer Cells: Networking Within Tumors. Results Probl. Cell Differ. 2024, 74, 213–238. [Google Scholar] [PubMed]
- Zhou, M.; He, X.; Mei, C.; Ou, C. Exosome derived from tumor-associated macrophages: Biogenesis, functions, and therapeutic implications in human cancers. Biomark. Res. 2023, 11, 100. [Google Scholar]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef]
- Srinivas, L.; Deepthi, S.; Deepak KG, K.; Malla, R.R. Current Perspectives of Exosomes as Therapeutic Targets and Drug Delivery Vehicles for Pancreatic Cancer. Crit. Rev. Oncog. 2019, 24, 179–190. [Google Scholar]
- Lee, Y.J.; Shin, K.J.; Chae, Y.C. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp. Mol. Med. 2024, 56, 877–889. [Google Scholar]
- Kenific, C.M.; Zhang, H.; Lyden, D. An exosome pathway without an ESCRT. Cell Res. 2021, 31, 105–106. [Google Scholar]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [PubMed]
- Lobert, V.H.; Stenmark, H. The ESCRT machinery mediates polarization of fibroblasts through regulation of myosin light chain. J. Cell Sci. 2012, 125, 29–36. [Google Scholar]
- Tamai, K.; Tanaka, N.; Nakano, T.; Kakazu, E.; Kondo, Y.; Inoue, J.; Shiina, M.; Fukushima, K.; Hoshino, T.; Sano, K. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem. Biophys. Res. Commun. 2010, 399, 384–390. [Google Scholar]
- Ludwig, N.; Rubenich, D.S.; Zaręba, Ł.; Siewiera, J.; Pieper, J.; Braganhol, E.; Reichert, T.E.; Szczepański, M.J. Potential roles of tumor cell-and stroma cell-derived small extracellular vesicles in promoting a pro-angiogenic tumor microenvironment. Cancers 2020, 12, 3599. [Google Scholar] [CrossRef] [PubMed]
- La Torre, M.; Burla, R.; Saggio, I. Preserving Genome Integrity: Unveiling the Roles of ESCRT Machinery. Cells 2024, 13, 1307. [Google Scholar] [CrossRef]
- Malla, R.R.; Pandrangi, S.; Kumari, S.; Gavara, M.M.; Badana, A.K. Exosomal tetraspanins as regulators of cancer progression and metastasis and novel diagnostic markers. Asia Pac. J. Clin. Oncol. 2018, 14, 383–391. [Google Scholar] [PubMed]
- Zhao, K.; Wang, Z.; Hackert, T.; Pitzer, C.; Zöller, M. Tspan8 and Tspan8/CD151 knockout mice unravel the contribution of tumor and host exosomes to tumor progression. J. Exp. Clin. Cancer Res. 2018, 37, 312. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arroyo, O.; Selma-Soriano, E.; Ortega, A.; Cortes, R.; Redon, J. Small Rab GTPases in Intracellular Vesicle Trafficking: The Case of Rab3A/Raphillin-3A Complex in the Kidney. Int. J. Mol. Sci. 2021, 22, 7679. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, Y.; Wang, Q.; Jayasinghe, U.; Luo, X.; Wei, Q.; Wang, J.; Xiong, H.; Chen, C.; Xu, B.; et al. Exosome: Emerging biomarker in breast cancer. Oncotarget 2017, 8, 41717–41733. [Google Scholar]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar]
- Malla, R.R.; Shailender, G.; Kamal, M.A. Exosomes: Critical Mediators of Tumour Microenvironment Reprogramming. Curr. Med. Chem. 2021, 28, 8182–8202. [Google Scholar]
- Jena, B.C.; Mandal, M. The emerging roles of exosomes in anti-cancer drug resistance and tumor progression: An insight towards tumor-microenvironment interaction. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188488. [Google Scholar]
- Wang, Y.; Li, Y.; Zhong, J.; Li, M.; Zhou, Y.; Lin, Q.; Zong, S.; Luo, W.; Wang, J.; Wang, K.; et al. Tumor-derived Cav-1 promotes pre-metastatic niche formation and lung metastasis in breast cancer. Theranostics 2023, 13, 1684–1697. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O'Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Sánchez, P.; Catalán, E.; Uranga-Murillo, I.; Aguiló, N.; Santiago, L.; Lanuza, P.M.; de Miguel, D.; A Arias, M.; Pardo, J. Antigen-specific primed cytotoxic T cells eliminate tumour cells in vivo and prevent tumour development, regardless of the presence of anti-apoptotic mutations conferring drug resistance. Cell Death Differ. 2018, 25, 1536–1548. [Google Scholar] [CrossRef]
- Li, W.; Han, G.; Li, F.; Bu, P.; Hao, Y.; Huang, L.; Bai, X. Cancer cell-derived exosomal miR-20a-5p inhibits CD8(+) T-cell function and confers anti-programmed cell death 1 therapy resistance in triple-negative breast cancer. Cancer Sci. 2024, 115, 347–356. [Google Scholar] [CrossRef]
- Choudhury, A.; Chatterjee, S.; Dalui, S.; Ghosh, P.; Daptary, A.H.; Mollah, G.K.; Bhattacharyya, A. Breast cancer cell derived exosomes reduces glycolysis of activated CD8 + T cells in a AKT-mTOR dependent manner. Cell Biol. Int. 2025, 49, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.; Gazinska, P.; Zhang, B.; Khiabany, A.; Sinha, S.; Alaguthurai, T.; Flores-Borja, F.; Vicencio, J.; Beuron, F.; Roxanis, I.; et al. Serum-derived extracellular vesicles from breast cancer patients contribute to differential regulation of T-cell-mediated immune-escape mechanisms in breast cancer subtypes. Front. Immunol. 2023, 14, 1204224. [Google Scholar] [CrossRef]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and breast cancer drug resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef]
- Dong, H.; Yang, C.; Chen, X.; Sun, H.; He, X.; Wang, W. Breast cancer-derived exosomal lncRNA SNHG14 induces normal fibroblast activation to cancer-associated fibroblasts via the EBF1/FAM171A1 axis. Breast Cancer 2023, 30, 1028–1040. [Google Scholar] [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-associated fibroblasts in breast cancer: Challenges and opportunities. Cancer Commun. 2022, 42, 401–434. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [PubMed]
- Xi, L.; Peng, M.; Liu, S.; Liu, Y.; Wan, X.; Hou, Y.; Qin, Y.; Yang, L.; Chen, S.; Zeng, H.; et al. Hypoxia-stimulated ATM activation regulates autophagy-associated exosome release from cancer-associated fibroblasts to promote cancer cell invasion. J. Extracell. Vesicles 2021, 10, e12146. [Google Scholar] [PubMed]
- Han, B.; Zhang, H.; Tian, R.; Liu, H.; Wang, Z.; Wang, Z.; Tian, J.; Cui, Y.; Ren, S.; Zuo, X.; et al. Exosomal EPHA2 derived from highly metastatic breast cancer cells promotes angiogenesis by activating the AMPK signaling pathway through Ephrin A1-EPHA2 forward signaling. Theranostics 2022, 12, 4127–4146. [Google Scholar]
- Li, Y.; Jiang, B.; Zeng, L.; Tang, Y.; Qi, X.; Wan, Z.; Feng, W.; Xie, L.; He, R.; Zhu, H.; et al. Adipocyte-derived exosomes promote the progression of triple-negative breast cancer through circCRIM1-dependent OGA activation. Environ. Res. 2023, 239 Pt 1, 117266. [Google Scholar]
- Liu, L.; Jiang, D.; Bai, S.; Zhang, X.; Kang, Y. Research progress of exosomes in drug resistance of breast cancer. Front. Bioeng. Biotechnol. 2023, 11, 1214648. [Google Scholar]
- Salimifard, S.; Masjedi, A.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Irandoust, M.; Azizi, G.; Mohammadi, H.; Keramati, M.R.; Jadidi-Niaragh, F. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathol. Res. Pract. 2020, 216, 152915. [Google Scholar]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar]
- Xu, Z.; Chen, Y.; Ma, L.; Chen, Y.; Liu, J.; Guo, Y.; Yu, T.; Zhang, L.; Zhu, L.; Shu, Y. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol. Ther. 2022, 30, 3133–3154. [Google Scholar]
- Abdul-Rahman, T.; Roy, P.; Herrera-Calderón, R.E.; Khidri, F.F.; Omotesho, Q.A.; Rumide, T.S.; Fatima, M.; Roy, S.; Wireko, A.A.; Atallah, O.; et al. Extracellular vesicle-mediated drug delivery in breast cancer theranostics. Discov. Oncol. 2024, 15, 181. [Google Scholar]
- Hekmatirad, S.; Moloudizargari, M.; Moghadamnia, A.A.; Kazemi, S.; Mohammadnia-Afrouzi, M.; Baeeri, M.; Moradkhani, F.; Asghari, M.H. Inhibition of Exosome Release Sensitizes U937 Cells to PEGylated Liposomal Doxorubicin. Front. Immunol. 2021, 12, 692654. [Google Scholar]
- Jordan, V.C. Tamoxifen: Catalyst for the change to targeted therapy. Eur. J. Cancer 2008, 44, 30–38. [Google Scholar] [PubMed]
- Zamanian, M.Y.; Golmohammadi, M.; Nili-Ahmadabadi, A.; Alameri, A.A.; Al-Hassan, M.; Alshahrani, S.H.; Hasan, M.S.; Ramírez-Coronel, A.A.; Qasim, Q.A.; Heidari, M.; et al. Targeting autophagy with tamoxifen in breast cancer: From molecular mechanisms to targeted therapy. Fundam. Clin. Pharmacol. 2023, 37, 1092–1108. [Google Scholar]
- Hu, K.; Liu, X.; Li, Y.; Li, Q.; Xu, Y.; Zeng, W.; Zhong, G.; Yu, C. Exosomes Mediated Transfer of Circ_UBE2D2 Enhances the Resistance of Breast Cancer to Tamoxifen by Binding to MiR-200a-3p. Med. Sci. Monit. 2020, 26, e922253. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, S.; Tang, W.; Huang, Q.; Mei, Y.; Yang, H. Exosomes from tamoxifen-resistant breast cancer cells transmit drug resistance partly by delivering miR-9-5p. Cancer Cell Int. 2021, 21, 55. [Google Scholar]
- Xu, C.G.; Yang, M.F.; Ren, Y.Q.; Wu, C.H.; Wang, L.Q. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4362–4368. [Google Scholar] [PubMed]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, K.; Hong, R.; Xu, F.; Xia, W.; Qin, G.; Lee, K.; Zheng, Q.; Lu, Q.; Zhai, Q.; et al. Identification and characterization of critical genes associated with tamoxifen resistance in breast cancer. PeerJ 2020, 8, e10468. [Google Scholar]
- Zhao, Y.; Jin, L.J.; Zhang, X.Y. Exosomal miRNA-205 promotes breast cancer chemoresistance and tumorigenesis through E2F1. Aging 2021, 13, 18498–18514. [Google Scholar]
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Zhao, S.; Pan, T.; Deng, J.; Cao, L.; Vicencio, J.M.; Liu, J.; Zhou, G.; Ng, T.; Zhang, J. Exosomal transfer of miR-181b-5p confers senescence-mediated doxorubicin resistance via modulating BCLAF1 in breast cancer. Br. J. Cancer 2023, 128, 665–677. [Google Scholar] [CrossRef]
- Zhou, H.; He, X.; He, Y.; Ou, C.; Cao, P. Exosomal circRNAs: Emerging Players in Tumor Metastasis. Front. Cell Dev. Biol. 2021, 9, 786224. [Google Scholar] [CrossRef]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.X.; Wang, D.D.; Zhu, B.; Zhu, Y.Z.; Zheng, L.; Feng, Z.Q.; Qin, X.H. Exosomal miR-222 from adriamycin-resistant MCF-7 breast cancer cells promote macrophages M2 polarization via PTEN/Akt to induce tumor progression. Aging 2021, 13, 10415–10430. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.-N.; Zhou, K.-F.; Miao, Z.; Chen, Y.-X.; Cui, J.-X.; Su, S.-W. Exosomes regulate doxorubicin resistance in breast cancer via miR-34a-5p/NOTCH1. Mol. Cell. Probes 2024, 76, 101964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Pan, T.; Jiang, D.; Jin, L.; Geng, Y.; Feng, X.; Shen, A.; Zhang, L. The lncRNA-GAS5/miR-221-3p/DKK2 Axis Modulates ABCB1-Mediated Adriamycin Resistance of Breast Cancer via the Wnt/β-Catenin Signaling Pathway. Mol. Ther. Nucleic Acids 2020, 19, 1434–1448. [Google Scholar] [CrossRef]
- Hong, X.; Pan, X. Exosome-Derived MicroRNA-221-3p Desensitizes Breast Cancer Cells to Adriamycin by Regulating PIK3r1-Mediated Glycose Metabolism. Cancer Biother. Radiopharm. 2024, 39, 463–475. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; Li, Z.; Li, X.; Jin, M.; Jia, N.; Cui, X.; Hu, G.; Tang, T.; Yu, Q. Pan-cancer analysis on the role of PIK3R1 and PIK3R2 in human tumors. Sci. Rep. 2022, 12, 5924. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel's Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014, 76 Pt A, 175–183. [Google Scholar]
- Zhao, T.; Zhang, T.; Zhang, Y.; Zhou, B.; Lu, X. Paclitaxel Resistance Modulated by the Interaction between TRPS1 and AF178030.2 in Triple-Negative Breast Cancer. Evid. Based Complement. Alternat Med. 2022, 2022, 6019975. [Google Scholar] [PubMed]
- Jiang, Y.Z.; Yu, K.D.; Peng, W.T.; Di, G.H.; Wu, J.; Liu, G.Y.; Shao, Z.M. Enriched variations in TEKT4 and breast cancer resistance to paclitaxel. Nat. Commun. 2014, 5, 3802. [Google Scholar] [PubMed]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt B, 1444–1453. [Google Scholar]
- Alalawy, A.I. Key genes and molecular mechanisms related to Paclitaxel Resistance. Cancer Cell Int. 2024, 24, 244. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Chen, W.; Ni, C.; Meng, X.; Wu, J.; Yang, Q.; Tang, H.; Yuan, H.; Fang, S. Chemotherapy-induced exosomal circBACH1 promotes breast cancer resistance and stemness via miR-217/G3BP2 signaling pathway. Breast Cancer Res. 2023, 25, 85. [Google Scholar]
- Yang, Q.; Zhao, S.; Shi, Z.; Cao, L.; Liu, J.; Pan, T.; Zhou, D.; Zhang, J. Chemotherapy-elicited exosomal miR-378a-3p and miR-378d promote breast cancer stemness and chemoresistance via the activation of EZH2/STAT3 signaling. J. Exp. Clin. Cancer Res. 2021, 40, 120. [Google Scholar]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Abak, A.; Abbas Raza, S.H.; Pichler, M.; Taheri, M. Role of non-coding RNAs in modulating the response of cancer cells to paclitaxel treatment. Biomed. Pharmacother. 2021, 134, 111172. [Google Scholar]
- Guo, Z.; Guo, A.; Zhou, C. Breast Cancer Stem Cell-Derived ANXA6-Containing Exosomes Sustain Paclitaxel Resistance and Cancer Aggressiveness in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 718721. [Google Scholar] [CrossRef]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Han, J.; Huang, J.; Li, S.; Pang, H. Hypoxia-Induced Intracellular and Extracellular Heat Shock Protein gp96 Increases Paclitaxel-Resistance and Facilitates Immune Evasion in Breast Cancer. Front. Oncol. 2021, 11, 784777. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.; Ye, M.; Wu, J.; Ma, L.; Chen, H. Cisplatin-resistant MDA-MB-231 Cell-derived Exosomes Increase the Resistance of Recipient Cells in an Exosomal miR-423-5p-dependent Manner. Curr. Drug Metab. 2019, 20, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Hsieh, C.H.; Lin, P.H.; Chen, Y.T.; Hsu, D.S.; Tai, S.K.; Chu, P.Y.; Yang, M.H. Snail-regulated exosomal microRNA-21 suppresses NLRP3 inflammasome activity to enhance cisplatin resistance. J. Immunother. Cancer 2022, 10, e004832. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lin, H.; Liu, A.; Qiu, C.; Rao, Z.; Wang, Z.; Chen, S.; She, X.; Zhu, S.; Li, P.; et al. SOD1-high fibroblasts derived exosomal miR-3960 promotes cisplatin resistance in triple-negative breast cancer by suppressing BRSK2-mediated phosphorylation of PIMREG. Cancer Lett. 2024, 590, 216842. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Liu, C.; Lu, C.; Yixi, L.; Hong, J.; Dong, F.; Ruan, S.; Hu, T.; Zhao, X. Exosomal Linc00969 induces trastuzumab resistance in breast cancer by increasing HER-2 protein expression and mRNA stability by binding to HUR. Breast Cancer Res. 2023, 25, 124. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, C.; Wang, Y. Exosome-mediated transfer of circHIPK3 promotes trastuzumab chemoresistance in breast cancer. J. Drug Target. 2021, 29, 1004–1015. [Google Scholar] [CrossRef]
- Martinez, V.G.; O'Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O'Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Martin, E.C.; Conger, A.K.; Yan, T.J.; Hoang, V.T.; Miller, D.F.; Buechlein, A.; Rusch, D.B.; Nephew, K.P.; Collins-Burow, B.M.; Burow, M.E. MicroRNA-335-5p and -3p synergize to inhibit estrogen receptor alpha expression and promote tamoxifen resistance. FEBS Lett. 2017, 591, 382–392. [Google Scholar] [PubMed]
- Stevic, I.; Müller, V.; Weber, K.; Fasching, P.A.; Karn, T.; Marmé, F.; Schem, C.; Stickeler, E.; Denkert, C.; van Mackelenbergh, M.; et al. Specific microRNA signatures in exosomes of triple-negative and HER2-positive breast cancer patients undergoing neoadjuvant therapy within the GeparSixto trial. BMC Med. 2018, 16, 179. [Google Scholar]
- Wu, C.; Luo, J. Long Non-Coding RNA (lncRNA) Urothelial Carcinoma-Associated 1 (UCA1) Enhances Tamoxifen Resistance in Breast Cancer Cells via Inhibiting mTOR Signaling Pathway. Med. Sci. Monit. 2016, 22, 3860–3867. [Google Scholar]
- Jin, L.; Wessely, O.; Marcusson, E.G.; Ivan, C.; Calin, G.A.; Alahari, S.K. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-α in breast cancer. Cancer Res. 2013, 73, 2884–2896. [Google Scholar] [PubMed]
- Yuan, X.; Qian, N.; Ling, S.; Li, Y.; Sun, W.; Li, J.; Du, R.; Zhong, G.; Liu, C.; Yu, G.; et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 2021, 11, 1429–1445. [Google Scholar] [CrossRef]
- Nicolini, A.; Ferrari, P.; Carpi, A. Immune Checkpoint Inhibitors and Other Immune Therapies in Breast Cancer: A New Paradigm for Prolonged Adjuvant Immunotherapy. Biomedicines 2022, 10, 2511. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Li, H.; Li, R.J.; Yin, L.; Zhang, H.F.; Huang, Z.N.; Cheng, Z.; Li, J.; Wang, Z.H.; Peng, H.L. The roles of exosomal immune checkpoint proteins in tumors. Mil. Med. Res. 2021, 8, 56. [Google Scholar]
- Su, N.; Zhang, J.; Liu, W.; Zheng, H.; Li, M.; Zhao, J.; Gao, M.; Zhang, X. Specific isolation and quantification of PD-L1 positive tumor derived exosomes for accurate breast cancer discrimination via aptamer-functionalized magnetic composites and SERS immunoassay. Talanta 2025, 281, 126956. [Google Scholar] [CrossRef]
- Lu, M.-M.; Yang, Y. Exosomal PD-L1 in cancer and other fields: Recent advances and perspectives. Front. Immunol. 2024, 15, 1395332. [Google Scholar]
- Liu, Y.; Hu, Y.; Xue, J.; Li, J.; Yi, J.; Bu, J.; Zhang, Z.; Qiu, P.; Gu, X. Advances in immunotherapy for triple-negative breast cancer. Mol. Cancer 2023, 22, 145. [Google Scholar] [CrossRef]
- Vautrot, V.; Bentayeb, H.; Causse, S.; Garrido, C.; Gobbo, J. Tumor-Derived Exosomes: Hidden Players in PD-1/PD-L1 Resistance. Cancers 2021, 13, 4537. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.; Ong, R.J.; Lim, J.S. Immune checkpoint inhibitors in breast cancer: Development, mechanisms of resistance and potential management strategies. Cancer Drug Resist. 2023, 6, 768–787. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar]
- Yin, Y.; Cai, X.; Chen, X.; Liang, H.; Zhang, Y.; Li, J.; Wang, Z.; Chen, X.; Zhang, W.; Yokoyama, S.; et al. Tumor-secreted miR-214 induces regulatory T cells: A major link between immune evasion and tumor growth. Cell Res. 2014, 24, 1164–1180. [Google Scholar] [PubMed]
- Basak, U.; Chakraborty, S.; Mukherjee, S.; Pati, S.; Khan, P.; Ghosh, S.; Adhikary, A.; Jana, K.; Sa, G.; Das, T. Breast cancer stem cells convert anti-tumor CD4+ T cells to pro-tumor T regulatory cells: Potential role of exosomal FOXP3. Cell. Immunol. 2025, 409–410, 104931. [Google Scholar]
- Lee, C.H.; Bae, J.H.; Choe, E.J.; Park, J.M.; Park, S.S.; Cho, H.J.; Song, B.J.; Baek, M.C. Macitentan improves antitumor immune responses by inhibiting the secretion of tumor-derived extracellular vesicle PD-L1. Theranostics 2022, 12, 1971–1987. [Google Scholar] [CrossRef]
- Im, E.J.; Lee, C.H.; Moon, P.G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.C.; Jee, J.G.; Bae, J.S.; Kwon, T.K.; et al. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar]
- Kong, J.N.; He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.B.; Zhu, G.; Kim, A.; Spassieva, S.; Bieberich, E. Guggulsterone and bexarotene induce secretion of exosome-associated breast cancer resistance protein and reduce doxorubicin resistance in MDA-MB-231 cells. Int. J. Cancer 2015, 137, 1610–1620. [Google Scholar]
- Chen, W.X.; Xu, L.Y.; Qian, Q.; He, X.; Peng, W.T.; Fan, W.Q.; Zhu, Y.L.; Tang, J.H.; Cheng, L. d Rhamnose β-hederin reverses chemoresistance of breast cancer cells by regulating exosome-mediated resistance transmission. Biosci. Rep. 2018, 38, BSR20180110. [Google Scholar]
- Irep, N.; Inci, K.; Tokgun, P.E.; Tokgun, O. Exosome inhibition improves response to first-line therapy in small cell lung cancer. J. Cell Mol. Med. 2024, 28, e18138. [Google Scholar]
- Norouzi-Barough, L.; Asgari Khosro Shahi, A.; Mohebzadeh, F.; Masoumi, L.; Haddadi, M.R.; Shirian, S. Early diagnosis of breast and ovarian cancers by body fluids circulating tumor-derived exosomes. Cancer Cell Int. 2020, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Almeida, F. Role of Exosomal miRNAs and the Tumor Microenvironment in Drug Resistance. Cells 2020, 9, 1450. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Carollo, E.; Melling, G.E.; Carter, D.R.F. Extracellular Vesicles: Emerging Modulators of Cancer Drug Resistance. Cancers 2021, 13, 749. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Cho, W.C.S. Exosome secretion from hypoxic cancer cells reshapes the tumor microenvironment and mediates drug resistance. Cancer Drug Resist. 2022, 5, 577–594. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Shen, K.; Huang, R.; Wang, Z. Biological Roles and Clinical Applications of Exosomes in Breast Cancer: A Brief Review. Int. J. Mol. Sci. 2024, 25, 4620. [Google Scholar] [CrossRef]

| Exosomal Contents | Mechanism of Action | Consequences of Potential Therapeutic Targeting of Exosomes | Subtype | Ref |
|---|---|---|---|---|
| Circ_UBE2D2 |
|
| Luminal A/B | [63] |
| miR-9-5p |
|
| Luminal A/B | [64] |
| miR-205 |
|
| Luminal A/B | [68] |
| miR-181b-5p |
|
| TNBC | [72] |
| miR-155 |
|
| TNBC | [74] |
| miR-222/miR-1246 |
|
| Luminal A/B | [75] |
| miR-423-5p |
|
| TNBC | [94] |
| miR-21 |
|
| TNBC | [95] |
| miR-3960 |
|
| TNBC | [96] |
| Lnc00969 |
|
| HER2+ | [98] |
| circHIPK |
|
| HER2+ | [99] |
| miR-335-5p/-3p |
|
| Luminal A/B | [101] |
| miR-128 |
|
| Luminal A/B | [102] |
| miR-27a |
|
| HER2+ | [102] |
| UCA1 (lncRNA) |
|
| Luminal A/B | [103] |
| miR-27b |
|
| HER2+ | [104] |
| miR-21 |
|
| TNBC | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malla, R.; Bhamidipati, P.; Samudrala, A.S.; Nuthalapati, Y.; Padmaraju, V.; Malhotra, A.; Rolig, A.S.; Malhotra, S.V. Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer. Cancers 2025, 17, 1167. https://doi.org/10.3390/cancers17071167
Malla R, Bhamidipati P, Samudrala AS, Nuthalapati Y, Padmaraju V, Malhotra A, Rolig AS, Malhotra SV. Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer. Cancers. 2025; 17(7):1167. https://doi.org/10.3390/cancers17071167
Chicago/Turabian StyleMalla, RamaRao, Priyamvada Bhamidipati, Anuveda Sree Samudrala, Yerusha Nuthalapati, Vasudevaraju Padmaraju, Aditya Malhotra, Annah S. Rolig, and Sanjay V. Malhotra. 2025. "Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer" Cancers 17, no. 7: 1167. https://doi.org/10.3390/cancers17071167
APA StyleMalla, R., Bhamidipati, P., Samudrala, A. S., Nuthalapati, Y., Padmaraju, V., Malhotra, A., Rolig, A. S., & Malhotra, S. V. (2025). Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer. Cancers, 17(7), 1167. https://doi.org/10.3390/cancers17071167