
The Persistent Power of the Taxane/Platin Chemotherapy

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Historic Overview and Current State of Cancer Treatment
3. Challenges of Targeted Therapy
4. Resistance of Neoplastic Cells to Programmed Cell Death
5. Nuclear Membrane Rupture Induced by Taxanes
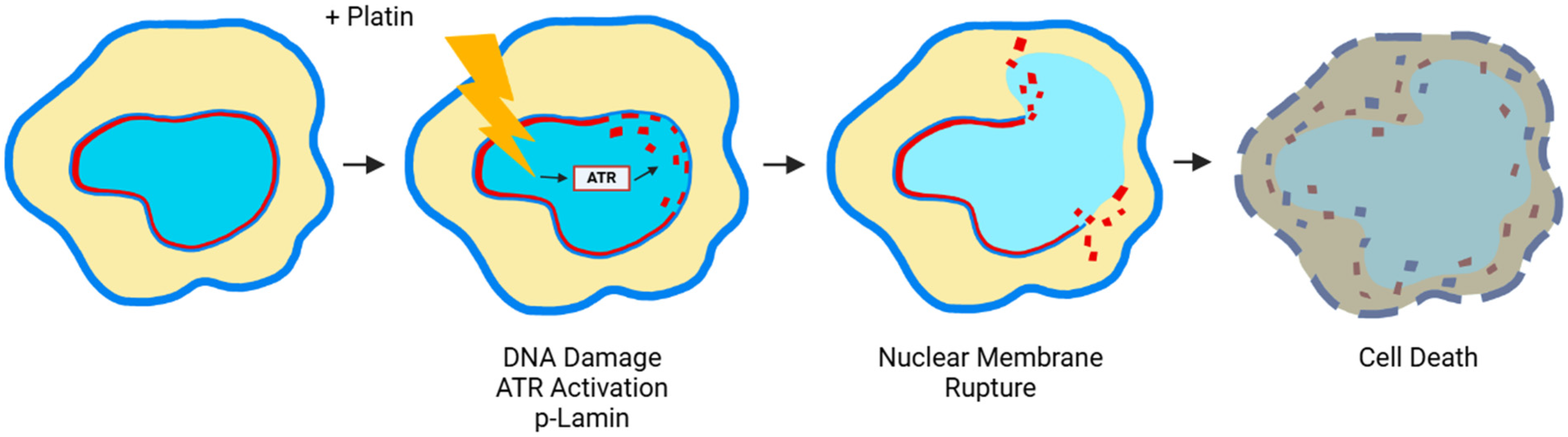
6. Nuclear Membrane Compromising Caused by Platinum Agents
7. Prospective: Cancer Therapy with Non-Programmed Cell Death Mechanisms and Effects to Reduce or Prevent Side Effects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muggia, F. Platinum compounds 30 years after the introduction of cisplatin: Implications for the treatment of ovarian cancer. Gynecol. Oncol. 2009, 112, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Dubashi, B.; Reddy, K.S.; Jain, P. Weekly paclitaxel in ovarian cancer-the latest success story. Curr. Oncol. 2011, 18, 16–17. [Google Scholar] [CrossRef]
- Muggia, F.M.; Bonetti, A.; Hoeschele, J.D.; Rozencweig, M.; Howell, S.B. Platinum Antitumor Complexes: 50 Years Since Barnett Rosenberg’s Discovery. J. Clin. Oncol. 2015, 33, 4219–4226. [Google Scholar] [CrossRef]
- Bookman, M.A. Optimal primary therapy of ovarian cancer. Ann. Oncol. 2016, 27, i58–i62. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A compressive review about taxol: History and future challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Kardinal, C.G. Cancer chemotherapy. Historical aspects and future considerations. Postgrad. Med. 1985, 77, 165–174. [Google Scholar] [CrossRef]
- Wright, J.C. Cancer chemotherapy: Past, present, and future—Part I. J. Natl. Med. Assoc. 1984, 76, 773–784. [Google Scholar] [PubMed]
- Wright, J.C. Cancer chemotherapy: Past, present, and future—Part II. J. Natl. Med. Assoc. 1984, 76, 865–876. [Google Scholar]
- Papac, R.J. Origins of cancer therapy. Yale J. Biol. Med. 2001, 74, 391–398. [Google Scholar]
- Chabner, B.A.; Roberts Jr, T.G. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Burchenal, J.H. The historical development of cancer chemotherapy. Semin. Oncol. 1977, 4, 135–146. [Google Scholar] [PubMed]
- Capizzi, R.L.; Keiser, L.W.; Sartorelli, A.C. Combination chemotherapy—Theory and practice. Semin. Oncol. 1977, 4, 227–253. [Google Scholar]
- Zubrod, C.G.; A Schepartz, S.; Carter, S.K. Historical background of the National Cancer Institute’s drug development thrust. J. Natl. Cancer Inst. Monogr. 1977, 7–11. [Google Scholar]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- DeVita Jr, V.T.; Rosenberg, S.A. Two hundred years of cancer research. N. Engl. J. Med. 2012, 366, 2207–2214. [Google Scholar]
- Zubrod, C.G. Historic milestones in curative chemotherapy. Semin. Oncol. 1979, 6, 490–505. [Google Scholar]
- Zubrod, C.G. The fifth Myron Karon Memorial Lecture. The cure of cancer by chemotherapy—Reflections on how it happened. Med. Pediatr. Oncol. 1980, 8, 107–114. [Google Scholar] [PubMed]
- Ribatti, D. Sidney Farber and the treatment of childhood acute lymphoblastic leukemia with a chemotherapeutic agent. Pediatr. Hematol. Oncol. J. 2012, 29, 299–302. [Google Scholar]
- Morrison, W.B. Cancer chemotherapy: An annotated history. J. Vet. Intern. Med. 2010, 24, 1249–1262. [Google Scholar]
- Hajdu, S.I. 2000 years of chemotherapy of tumors. Cancer 2005, 103, 1097–1102. [Google Scholar]
- Rowinsky, E.K.; Donehower, R.C. Taxol: Twenty years later, the story unfolds. J. Natl. Cancer Inst. 1991, 83, 1778–1781. [Google Scholar] [PubMed]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar]
- Rosenberg, B.; Van Camp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Hoeschele, J.D. In remembrance of Barnett Rosenberg. Dalton Trans. 2009, 12, 10648–10650. [Google Scholar] [CrossRef]
- Hoeschele, J.D. Biography of professor barnett rosenberg: A tribute to his life and his achievements. Anticancer. Res. 2014, 34, 417–421. [Google Scholar] [PubMed]
- Hoeschele, J.D. Dr Barnett Rosenberg—A Personal Perspective. Dalton Trans. 2016, 45, 12966–12969. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- Rosenberg, B.; Van Camp, L. The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res. 1970, 30, 1799–1802. [Google Scholar]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar]
- Ozols, R.F. Combination regimens of paclitaxel and the platinum drugs as first-line regimens for ovarian cancer. Semin. Oncol. 1995, 22, 1–6. [Google Scholar]
- Elmorsy, E.A.; Saber, S.; Hamad, R.S.; Abdel-Reheim, M.A.; El-Kott, A.F.; AlShehri, M.A.; Morsy, K.; Salama, S.A.; Youssef, M.E. Advances in understanding cisplatin-induced toxicity: Molecular mechanisms and protective strategies. Eur. J. Pharm. Sci. 2024, 203, 106939. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, H.-J.; Li, Y.-Y.; Wang, X.; Liu, X.-X.; Mai, J. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Zoń, A.; Bednarek, I. Cisplatin in Ovarian Cancer Treatment-Known Limitations in Therapy Force New Solutions. Int. J. Mol. Sci. 2023, 24, 7585. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Horwitz, S.B. Nature as a remarkable chemist: A personal story of the discovery and development of taxol. Anticancer Drugs 2014, 25, 482–487. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Barbuti, A.M.; Chen, Z.-S. Paclitaxel through the ages of anticancer therapy: Exploring its role in chemoresistance and radiation therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef]
- McGuire, W.P.; Rowinsky, E.K.; Rosenshein, N.B.; Grumbine, F.; Ettinger, D.S.; Armstrong, D.K.; Donehower, R.C. Taxol: A unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann. Intern. Med. 1989, 111, 273–279. [Google Scholar] [CrossRef]
- McGuire, W.P. Taxol: A new drug with significant activity as a salvage therapy in advanced epithelial ovarian carcinoma. Gynecol. Oncol. 1993, 51, 78–85. [Google Scholar] [CrossRef]
- Caldas, C.; McGuire, W.P., 3rd. Taxol in epithelial ovarian cancer. J. Natl. Cancer Inst. Monogr. 1993, 15, 155–159. [Google Scholar]
- Bookman, M.A.; Ozols, R.F. Future directions for paclitaxel (TAXOL) in the treatment of ovarian cancer. Semin. Oncol. Nurs. 1993, 9, 21–29. [Google Scholar] [PubMed]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar]
- Ozols, R.F. Chemotherapy for advanced epithelial ovarian cancer. Hematol. Oncol. Clin. N. Am. 1992, 6, 879–894. [Google Scholar] [PubMed]
- Schilder, R.J.; Ozols, R.F. New therapies for ovarian cancer. Cancer Investig. 1992, 10, 307–315. [Google Scholar]
- Hurwitz, H.I.; McGuire, W.P., 3rd. Primary chemotherapy in epithelial ovarian cancer. Obstet. Gynecol. Clin. N. Am. 1994, 21, 141–154. [Google Scholar]
- Trimble, E.L.; Arbuck, S.G.; McGuire, W.P. Options for primary chemotherapy of epithelial ovarian cancer: Taxanes. Gynecol. Oncol. 1994, 55, S114–S121. [Google Scholar] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar]
- Blagosklonny, M.V.; Fojo, T. Molecular effects of paclitaxel: Myths and reality (a critical review). Int. J. Cancer 1999, 83, 151–156. [Google Scholar]
- Smith, E.R.; Li, Z.; Chen, Z.-S.; Xu, X.-X. Reassessing specificity/selectivity of taxane-based chemotherapy. Cancer Insight 2024, 3, 27. [Google Scholar] [CrossRef]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer; Scribner: New York, NY, USA, 2010. [Google Scholar]
- Guchelaar, H.; Vermes, A.; Vermes, I.; Haanen, C. Apoptosis: Molecular mechanisms and implications for cancer chemotherapy. Pharm. World Sci. 1997, 19, 119–125. [Google Scholar] [CrossRef]
- Kabore, A.F.; Johnston, J.B.; Gibson, S.B. Changes in the apoptotic and survival signaling in cancer cells and their potential therapeutic implications. Curr. Cancer Drug Targets. 2004, 4, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Melet, A.; Song, K.; Bucur, O.; Jagani, Z.; Grassian, A.R.; Khosravi-Far, R. Apoptotic pathways in tumor progression and therapy. Adv. Exp. Med. Biol. 2008, 615, 47–79. [Google Scholar] [CrossRef]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. 2011, 3, 279–296. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Cancer 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Chaudhry, G.-E.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol. Biol. 2022, 2543, 191–210. [Google Scholar] [CrossRef]
- Tian, X.; Srinivasan, P.R.; Tajiknia, V.; Uruchurtu, A.F.S.S.; Seyhan, A.A.; Carneiro, B.A.; De La Cruz, A.; Pinho-Schwermann, M.; George, A.; Zhao, S.; et al. Targeting apoptotic pathways for cancer therapy. J. Clin. Investig. 2024, 134, e179570. [Google Scholar] [CrossRef]
- Røsland, G.V.; Engelsen, A.S.T. Novel points of attack for targeted cancer therapy. Basic Clin. Pharmacol. Toxicol. 2015, 116, 9–18. [Google Scholar]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta 2021, 1876, 188556. [Google Scholar]
- Cox, A.D. Farnesyltransferase inhibitors: Potential role in the treatment of cancer. Drugs 2001, 61, 723–732. [Google Scholar]
- Caponigro, F. Farnesyl transferase inhibitors: A major breakthrough in anticancer therapy? Naples, 12 April 2002. Anticancer Drugs 2002, 13, 891–897. [Google Scholar] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [PubMed]
- Pegram, M.; Slamon, D. Biological rationale for HER2/neu (c-erbB2) as a target for monoclonal antibody therapy. Semin. Oncol. 2000, 27, 13–19. [Google Scholar]
- Shepard, H.M.; Jin, P.; Slamon, D.J.; Pirot, Z.; Maneval, D.C. Herceptin. In The Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; Volume 181, pp. 183–219. [Google Scholar]
- Xia, X.; Gong, C.; Zhang, Y.; Xiong, H. The History and Development of HER2 Inhibitors. Pharmaceuticals 2023, 16, 1450. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [PubMed]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, eabc1495. [Google Scholar]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Zubay, G.; Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef]
- McCormick, F.K. Ras protein as a drug target. J. Mol. Med. 2016, 94, 253–258. [Google Scholar]
- McCormick, F. A brief history of RAS and the RAS Initiative. Adv. Cancer Res. 2022, 153, 1–27. [Google Scholar] [PubMed]
- Aguirre, A.J.; Stanger, B.Z.; Maitra, A. Hope on the Horizon: A Revolution in KRAS Inhibition Is Creating a New Treatment Paradigm for Patients with Pancreatic Cancer. Cancer Res. 2024, 84, 2950–2953. [Google Scholar]
- Wei, D.; Wang, L.; Zuo, X.; Maitra, A.; Bresalier, R.S. A Small Molecule with Big Impact: MRTX1133 Targets the KRASG12D Mutation in Pancreatic Cancer. Clin. Cancer Res. 2024, 30, 655–662. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [Google Scholar] [PubMed]
- Tang, D.; Kroemer, G.; Kang, R. Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 2021, 20, 128. [Google Scholar]
- Perurena, N.; Situ, L.; Cichowski, K. Combinatorial strategies to target RAS-driven cancers. Nat. Rev. Cancer 2024, 24, 316–337. [Google Scholar] [PubMed]
- Dilly, J.; Hoffman, M.T.; Abbassi, L.; Li, Z.; Paradiso, F.; Parent, B.D.; Hennessey, C.J.; Jordan, A.C.; Morgado, M.; Dasgupta, S.; et al. Mechanisms of Resistance to Oncogenic KRAS Inhibition in Pancreatic Cancer. Cancer Discov. 2024, 14, 2135–2161. [Google Scholar]
- Reynolds, S. Can Chemo Help KRAS Inhibitors Work Better Against Pancreatic Cancer? NCI News & Events, Cancer Currents Blog. 20 August 2024. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2024/pancreatic-cancer-kras-inhibitors-chemotherapy?cid=eb_govdel (accessed on 10 February 2025).
- Singhal, A.; Styers, H.C.; Rub, J.; Li, Z.; Torborg, S.R.; Kim, J.Y.; Grbovic-Huezo, O.; Feng, H.; Tarcan, Z.C. Classical Epithelial State Drives Acute Resistance to KRAS Inhibition in Pancreatic Cancer. Cancer Discov. 2024, 14, 2122–2134. [Google Scholar]
- Dana, H.; Chalbatani, G.M.; Jalali, S.A.; Mirzaei, H.R.; Grupp, S.A.; Suarez, E.R.; Rapôso, C.; Webster, T.J. CAR-T cells: Early successes in blood cancer and challenges in solid tumors. Acta Pharm. Sin. B 2021, 11, 1129–1147. [Google Scholar]
- Chen, Q.; Lu, L.; Ma, W. Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors. Cancers 2022, 14, 5983. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, M.; Chen, Y.; Liu, Y. Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer Cell Int. 2024, 24, 133. [Google Scholar] [CrossRef]
- Guha, P.; Heatherton, K.R.; O’connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar]
- Sorkhabi, A.D.; Khosroshahi, L.M.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef]
- Leaf, C. The Truth in Small Doses: Why We’re Losing the War on Cancer—And How to Win It; Simon & Schuster: New York, NY, USA, 2013. [Google Scholar]
- Gerl, R.; Vaux, D.L. Apoptosis in the development and treatment of cancer. Carcinogenesis 2005, 26, 263–270. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Modulation of apoptosis signaling for cancer therapy. Arch. Immunol. Ther. Exp. 2006, 54, 173–175. [Google Scholar]
- Ziegler, D.S.; Kung, A.L. Therapeutic targeting of apoptosis pathways in cancer. Curr. Opin. Oncol. 2008, 20, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef]
- Hersey, P.; Zhang, X.D. Overcoming resistance of cancer cells to apoptosis. J. Cell Physiol. 2003, 196, 9–18. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [PubMed]
- Igney, F.H.; Krammer, P.H. Immune escape of tumors: Apoptosis resistance and tumor counterattack. J. Leukoc. Biol. 2002, 71, 907–920. [Google Scholar]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is cisplatin-induced cell death always produced by apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Huisman, C.; Ferreira, C.G.; Bröker, L.E.; Rodriguez, J.A.; Smit, E.F.; Postmus, P.E.; Kruyt, F.A.E.; Giaccone, G. Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin. Cancer Res. 2002, 8, 596–606. [Google Scholar]
- Bhalla, K.N. Microtubule-targeted anticancer agents and apoptosis. Oncogene 2003, 22, 9075–9086. [Google Scholar] [CrossRef]
- Ikuta, K.; Takemura, K.; Kihara, M.; Naito, S.; Lee, E.; Shimizu, E.; Yamauchi, A. Defects in apoptotic signal transduction in cisplatin-resistant non-small cell lung cancer cells. Oncol. Rep. 2005, 13, 1229–1234. [Google Scholar]
- Smolka, M.B.; Lammerding, J. ATR takes a crack at the nuclear envelope. Mol. Cell 2023, 83, 3588–3590. [Google Scholar] [PubMed]
- Xu, A.P.; Xu, L.B.; Smith, E.R.; Fleishman, J.S.; Chen, Z.-S.; Xu, X.-X. Cell death in cancer chemotherapy using taxanes. Front. Pharmacol. 2024, 14, 1338633. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5, S3–S6. [Google Scholar] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells. J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Ganansia-Leymarie, V.; Bischoff, P.; Bergerat, J.-P.; Holl, V. Signal transduction pathways of taxanes-induced apoptosis. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef]
- Komlodi-Pasztor, E.; Sackett, D.L.; Fojo, A.T. Inhibitors targeting mitosis: Tales of how great drugs against a promising target were brought down by a flawed rationale. Clin. Cancer Res. 2012, 18, 51–63. [Google Scholar] [CrossRef]
- Mitchison, T.J. The proliferation rate paradox in antimitotic chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yan, V.C.; Butterfield, H.E.; Poral, A.H.; Yan, M.J.; Yang, K.L.; Pham, C.-D.; Muller, F.L. Why great mitotic inhibitors make poor cancer drugs. Trends Cancer 2020, 6, 924–941. [Google Scholar] [CrossRef]
- Fürst, R.; Vollmar, A.M. A new perspective on old drugs: Non-mitotic actions of tubulin-binding drugs play a major role in cancer treatment. Pharmzie 2013, 68, 478–483. [Google Scholar]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar]
- Mitchison, T.J.; Pineda, J.; Shi, J.; Florian, S. Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol. 2017, 7, 170182. [Google Scholar]
- Smith, E.R.; Leal, J.; Amaya, C.; Li, B.; Xu, X.-X. Nuclear Lamin A/C Expression Is a Key Determinant of Paclitaxel Sensitivity. Mol. Cell Biol. 2021, 41, e00648-20. [Google Scholar]
- Smith, E.R.; Xu, X.-X. Breaking malignant nuclei as a non-mitotic mechanism of taxol/paclitaxel. J. Cancer Biol. 2021, 2, 86–93. [Google Scholar]
- Panvichian, R.; Orth, K.; Day, M.L.; Day, K.C.; Pilat, M.J.; Pienta, K.J. Paclitaxel-associated multimininucleation is permitted by the inhibition of caspase activation: A potential early step in drug resistance. Cancer Res. 1998, 58, 4667–4672. [Google Scholar]
- Merlin, J.-L.; Bour-Dill, C.; Marchal, S.; Bastien, L.; Gramain, M.-P. Resistance to paclitaxel induces time-delayed multinucleation and DNA fragmentation into large fragments in MCF-7 human breast adenocarcinoma cells. Anti-Cancer Drugs 2000, 11, 295–302. [Google Scholar] [PubMed]
- Zhu, Y.; Zhou, Y.; Shi, J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 2014, 13, 1756–1764. [Google Scholar] [PubMed]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Wang, J.-Q.; Yang, D.-H.; Xu, X.-X. Paclitaxel resistance related to nuclear envelope structural sturdiness. Drug Resist. Updates 2022, 65, 100881. [Google Scholar]
- Smith, E.R.; George, S.H.; Kobetz, E.; Xu, X. New biological research and understanding of Papanicolaou’s test. Diagn. Cytopathol. 2018, 46, 507–515. [Google Scholar]
- Capo-chichi, C.D.; Cai, K.Q.; Smedberg, J.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.X. Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin. J. Cancer 2011, 30, 415–425. [Google Scholar]
- Capo-Chichi, C.D.; Cai, K.Q.; Simpkins, F.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.-X. Nuclear envelope structural defects cause chromosomal numerical instability and aneuploidy in ovarian cancer. BMC Med. 2011, 9, 28. [Google Scholar] [PubMed]
- Xu, X.-X.; Smith, E.R. The Second Selectivity of Taxanes to Malignant Cells—Nuclear Envelope Malleability. J. Cancer 2025, 16, 1051–1053. [Google Scholar] [CrossRef]
- Vargas, J.D.; Hatch, E.M.; Anderson, D.J.; Hetzer, M.W. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 2012, 3, 88–100. [Google Scholar]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [PubMed]
- Blagosklonny, M.V.; Robey, R.; Sheikh, M.S.; Fojo, T. Paclitaxel-induced FasL-independent apoptosis and slow (non-apoptotic) cell death. Cancer Biol. Ther. 2002, 1, 113–117. [Google Scholar] [PubMed]
- Schimming, R.; Mason, K.A.; Hunter, N.; Weil, M.; Kishi, K.; Milas, L. Lack of correlation between mitotic arrest or apoptosis and antitumor effect of docetaxel. Cancer Chemother. Pharmacol. 1999, 43, 165–172. [Google Scholar] [CrossRef]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef]
- Kovacs, M.T.; Vallette, M.; Wiertsema, P.; Dingli, F.; Loew, D.; Nader, G.P.d.F.; Piel, M.; Ceccaldi, R. DNA damage induces nuclear envelope rupture through ATR-mediated phosphorylation of lamin A/C. Mol. Cell 2023, 83, 3659–3668. [Google Scholar]
- Joo, Y.K.; Black, E.M.; Trier, I.; Haakma, W.; Zou, L.; Kabeche, L. ATR promotes clearance of damaged DNA and damaged cells by rupturing micronuclei. Mol. Cell 2023, 83, 3642–3658. [Google Scholar] [CrossRef]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Sazonova, E.V.; Kopeina, G.S.; Imyanitov, E.N.; Zhivotovsky, B. Platinum drugs and taxanes: Can we overcome resistance? Cell Death Discov. 2021, 7, 155. [Google Scholar] [PubMed]
- Das, T.; Anand, U.; Pandey, S.K.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.-S.; Dey, A. Therapeutic strategies to overcome taxane resistance in cancer. Drug Resist. Updates 2021, 55, 100754. [Google Scholar]
- Marupudi, N.I.; E Han, J.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [PubMed]
- Visconti, R.; Grieco, D. Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocr. Relat. Cancer 2017, 24, T107–T117. [Google Scholar]
- Legha, S.S.; Tenney, D.M.; Krakoff, I.R. Phase I study of taxol using a 5-day intermittent schedule. J. Clin. Oncol. 1986, 4, 762–766. [Google Scholar] [PubMed]
- Amaya, C.; Luo, S.; Baigorri, J.; Baucells, R.; Smith, E.R.; Xu, X.-X. Exposure to low intensity ultrasound removes paclitaxel cytotoxicity in breast and ovarian cancer cells. BMC Cancer 2021, 21, 981. [Google Scholar]
- Amaya, C.; Smith, E.R.; Xu, X.-X. Low Intensity Ultrasound as an Antidote to Taxane/Paclitaxel-induced Cytotoxicity. J. Cancer 2022, 13, 2362–2373. [Google Scholar] [CrossRef]
- Zhao, Y.; Mu, X.; Du, G. Microtubule-stabilizing agents: New drug discovery and cancer therapy. Pharmacol. Ther. 2016, 162, 134–143. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.B.; Smith, E.R.; Koutouratsas, V.; Chen, Z.-S.; Xu, X.-X. The Persistent Power of the Taxane/Platin Chemotherapy. Cancers 2025, 17, 1208. https://doi.org/10.3390/cancers17071208
Xu LB, Smith ER, Koutouratsas V, Chen Z-S, Xu X-X. The Persistent Power of the Taxane/Platin Chemotherapy. Cancers. 2025; 17(7):1208. https://doi.org/10.3390/cancers17071208
Chicago/Turabian StyleXu, Lucy B., Elizabeth R. Smith, Vasili Koutouratsas, Zhe-Sheng Chen, and Xiang-Xi Xu. 2025. "The Persistent Power of the Taxane/Platin Chemotherapy" Cancers 17, no. 7: 1208. https://doi.org/10.3390/cancers17071208
APA StyleXu, L. B., Smith, E. R., Koutouratsas, V., Chen, Z.-S., & Xu, X.-X. (2025). The Persistent Power of the Taxane/Platin Chemotherapy. Cancers, 17(7), 1208. https://doi.org/10.3390/cancers17071208