
Targeting Hsp70 Immunosuppressive Signaling Axis with Lipid Nanovesicles: A Novel Approach to Treat Pancreatic Cancer

 , , , , and
, , , , and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Generation of Exosome/Microparticle-Free Conditioned Media (EMD-CM) from Cancer Cell Lines
2.4. THP-1 Differentiation Assay
2.5. Quantification of Hsp70 in Cancer Cells Conditioned Media by ELISA
2.6. Preparation of Phospholipid Lipid Vesicles and Treatment of THP-1 Cells
2.7. Preparation of Saposin C-Coupled 1,2-Dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) (SapC-DOPG) Lipid Vesicles (LVs)
2.8. Characterization of Lipid Vesicles (LVs)
2.9. Cell Sorting
2.10. Immunofluorescence Staining
2.11. Western Blotting Analyses
2.12. Cell Viability Assay
2.13. Subcutaneous Cancer Cell Implantation and DOPG Lipid Vesicle (LVs) Treatment
2.14. Generation of Orthotopic PDAC Tumors in Mice
2.15. Phospholipids and SapC-DOPG LVs Binding Assays
2.16. PK/PD and Tissue Distribution of SapC-DOPG LVs
2.17. Toxicity Studies of SapC-DOPG LVs
2.18. Quantitative Analysis of Fluorescence Microscopy Images
2.19. Hematological Analyses
2.20. Statistical Analysis
3. Results
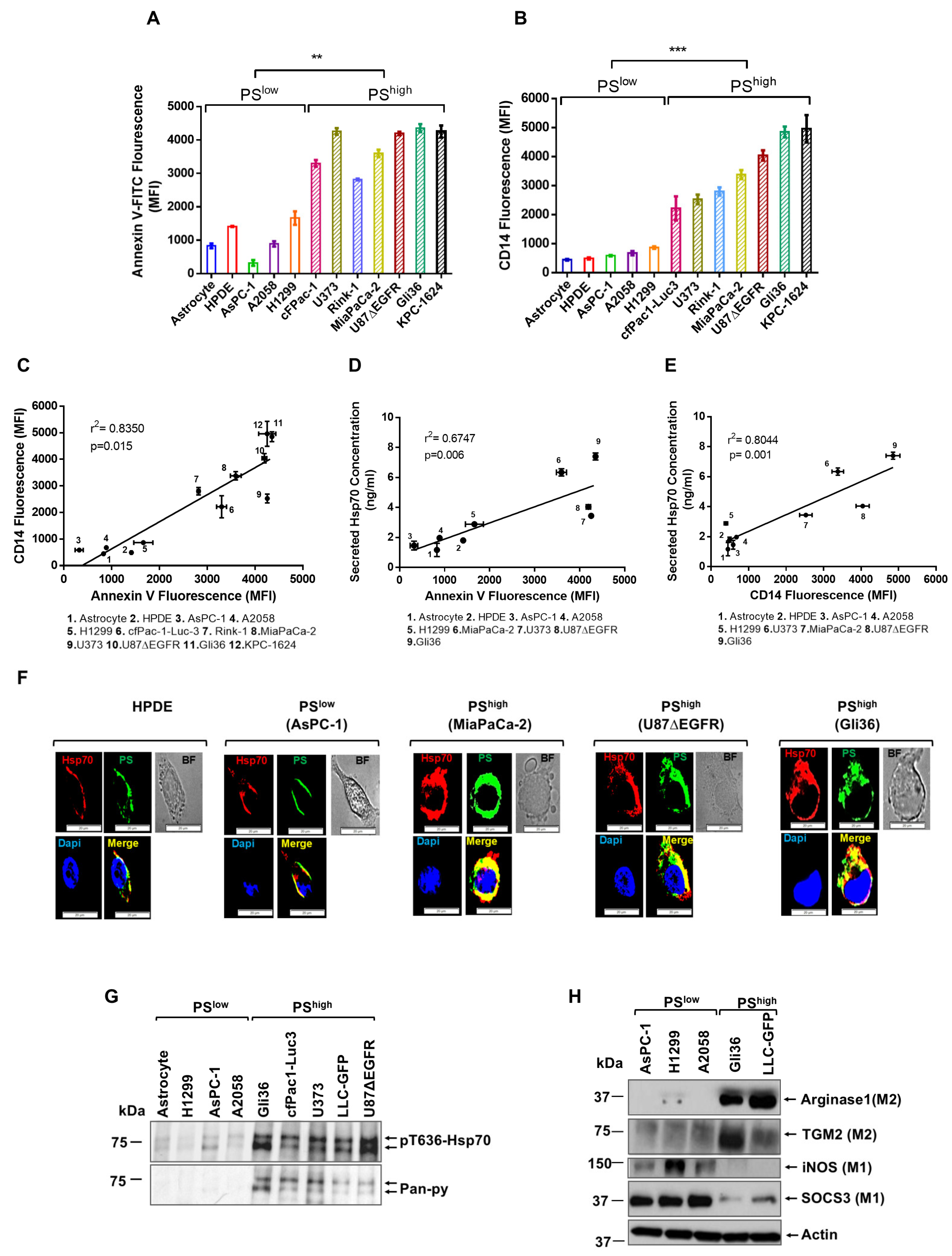
3.1. PShigh Cancer Cells Promote Monocyte Differentiation and M2 Polarization via Secreted-Hsp70
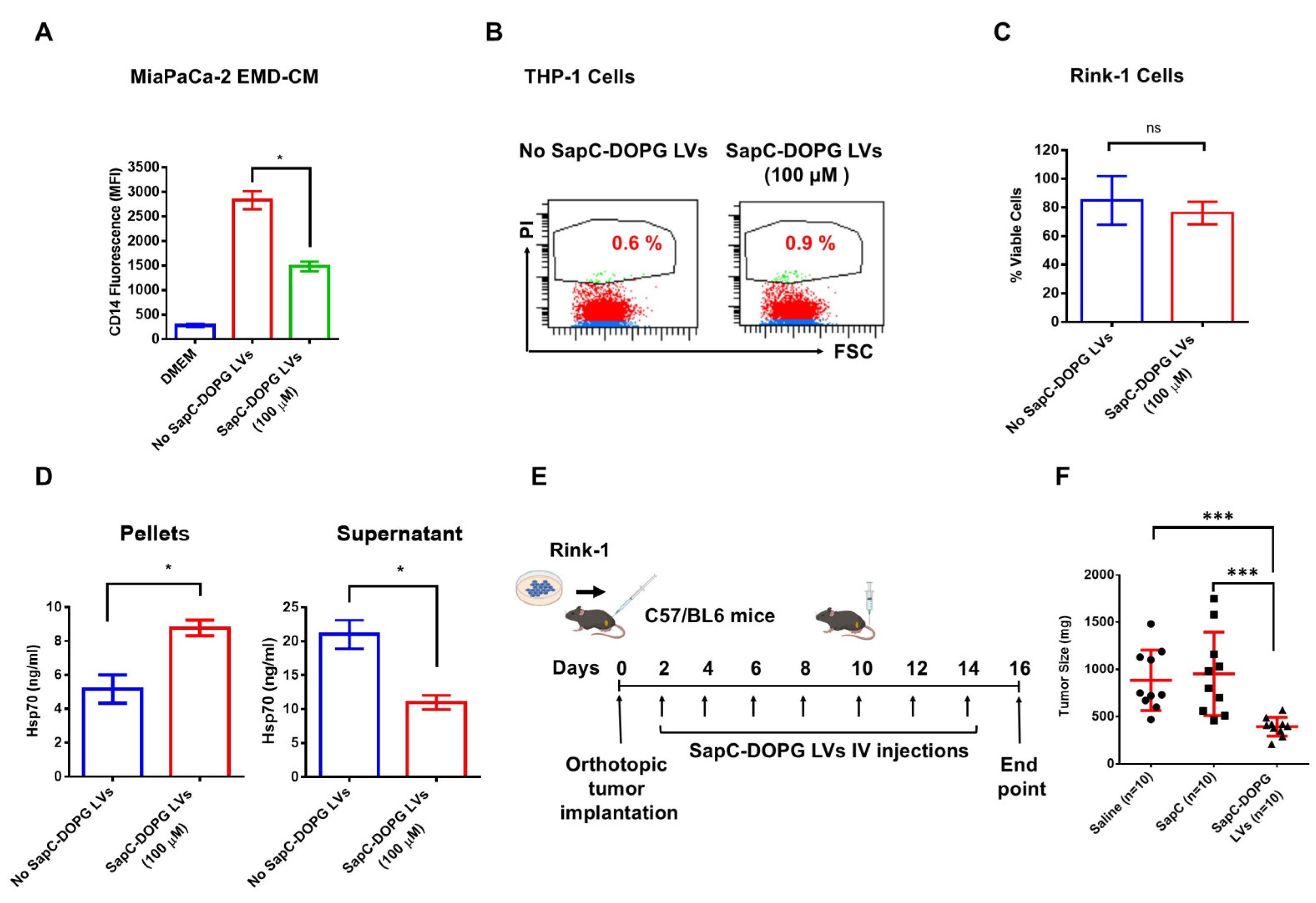
3.2. DOPG Lipid Vesicles (LVs) Block EMD-CM-Induced Monocyte Differentiation and Increase Survival in Mice
3.3. SapC-DOPG LVs Block EMD-CM-Induced Monocyte Differentiation and Inhibit Tumor Growth
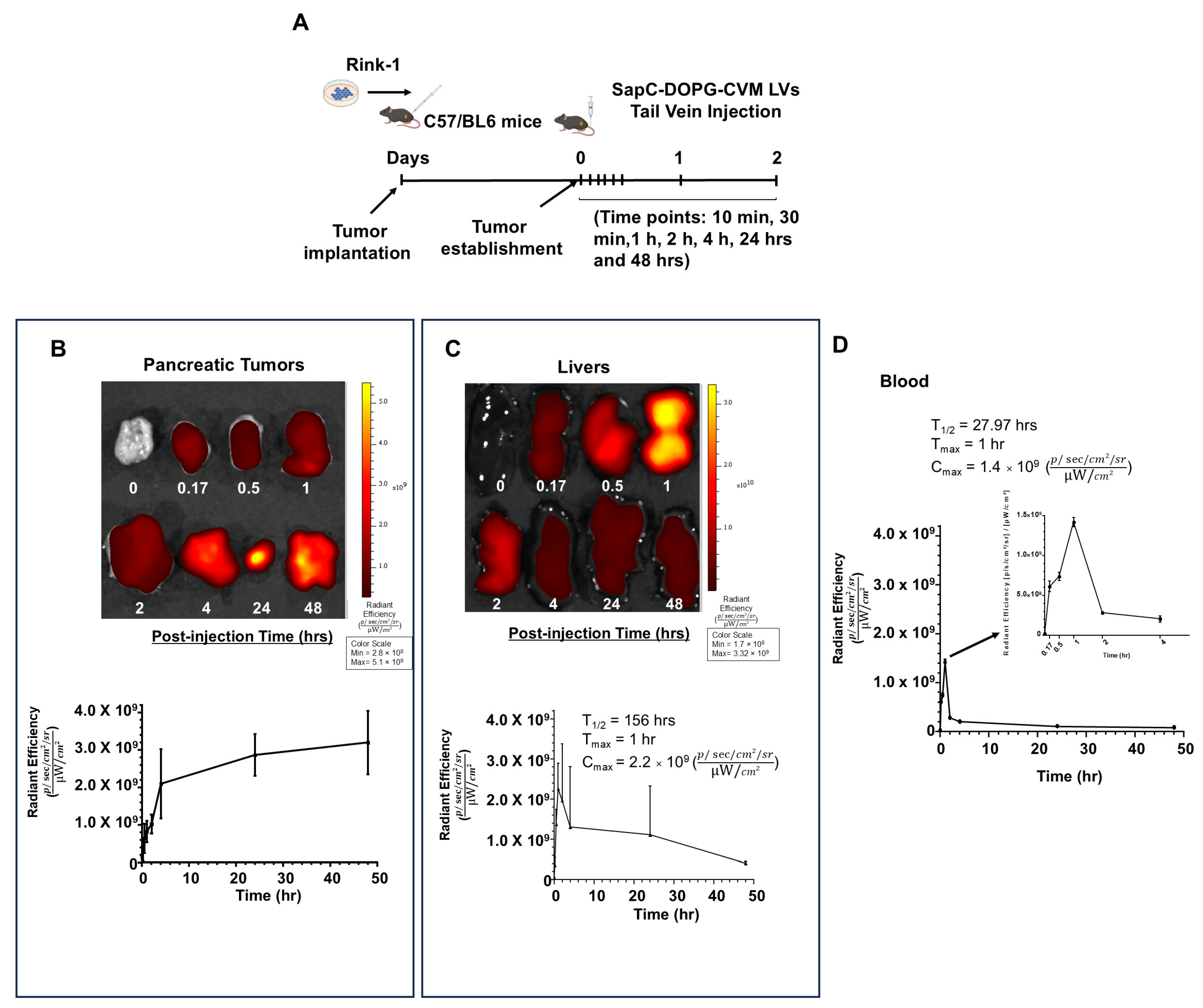
3.4. SapC-DOPG LVs Target Pancreatic Tumors Without Body Weight Loss or Organ Toxicity in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Yao, L.; Wang, M.; Niu, Z.; Liu, Q.; Gao, X.; Zhou, L.; Liao, Q.; Zhao, Y. Interleukin-27 inhibits malignant behaviors of pancreatic cancer cells by targeting M2 polarized tumor associated macrophages. Cytokine 2017, 89, 194–200. [Google Scholar] [CrossRef]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Capurso, G.; Sette, C. Drug resistance in pancreatic cancer: New player caught in act. EBioMedicine 2019, 40, 39–40. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.D.; Blanco, V.M.; Sulaiman, M.K.; Vallabhapurapu, S.L.; Chu, Z.; Franco, R.S.; Qi, X. Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium. Oncotarget 2015, 6, 34375–34388. [Google Scholar] [CrossRef]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 2638–2645. [Google Scholar] [CrossRef]
- N’Guessan, K.F.; Davis, H.W.; Chu, Z.; Vallabhapurapu, S.D.; Lewis, C.S.; Franco, R.S.; Olowokure, O.; Ahmad, S.A.; Yeh, J.J.; Bogdanov, V.Y.; et al. Enhanced Efficacy of Combination of Gemcitabine and Phosphatidylserine-Targeted Nanovesicles against Pancreatic Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1876–1886. [Google Scholar] [CrossRef]
- Phinney, N.Z.; Huang, X.; Toombs, J.E.; Brekken, R.A. Development of betabodies: The next generation of phosphatidylserine targeting agents. J. Biol. Chem. 2024, 300, 107681. [Google Scholar] [CrossRef]
- Hsiehchen, D.; Beg, M.S.; Kainthla, R.; Lohrey, J.; Kazmi, S.M.; Khosama, L.; Maxwell, M.C.; Kline, H.; Katz, C.; Hassan, A.; et al. The phosphatidylserine targeting antibody bavituximab plus pembrolizumab in unresectable hepatocellular carcinoma: A phase 2 trial. Nat. Commun. 2024, 15, 2178. [Google Scholar] [CrossRef]
- Qi, X.; Leonova, T.; Grabowski, G.A. Functional human saposins expressed in Escherichia coli. Evidence for binding and activation properties of saposins C with acid beta-glucosidase. J. Biol. Chem. 1994, 269, 16746–16753. [Google Scholar] [CrossRef]
- Qi, X.; Chu, Z.; Mahller, Y.Y.; Stringer, K.F.; Witte, D.P.; Cripe, T.P. Cancer-selective targeting and cytotoxicity by liposomal-coupled lysosomal saposin C protein. Clin. Cancer Res. 2009, 15, 5840–5851. [Google Scholar] [CrossRef]
- Chu, Z.; Abu-Baker, S.; Palascak, M.B.; Ahmad, S.A.; Franco, R.S.; Qi, X. Targeting and cytotoxicity of SapC-DOPS nanovesicles in pancreatic cancer. PLoS ONE 2013, 8, e75507. [Google Scholar] [CrossRef]
- Sulaiman, M.K.; Chu, Z.; Blanco, V.M.; Vallabhapurapu, S.D.; Franco, R.S.; Qi, X. SapC-DOPS nanovesicles induce Smac- and Bax-dependent apoptosis through mitochondrial activation in neuroblastomas. Mol. Cancer 2015, 14, 78. [Google Scholar] [CrossRef]
- Ellis, J. Proteins as molecular chaperones. Nature 1987, 328, 378–379. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in cancer: Back to the future. Oncogene 2015, 34, 4153–4161. [Google Scholar] [CrossRef]
- Mambula, S.S.; Calderwood, S.K. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef]
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J. Biol. Chem. 2003, 278, 21601–21606. [Google Scholar] [CrossRef]
- Nitika; Truman, A.W. Cracking the Chaperone Code: Cellular Roles for Hsp70 Phosphorylation. Trends Biochem. Sci. 2017, 42, 932–935. [Google Scholar]
- Zemanovic, S.; Ivanov, M.V.; Ivanova, L.V.; Bhatnagar, A.; Michalkiewicz, T.; Teng, R.J.; Kumar, S.; Rathore, R.; Pritchard, K.A., Jr.; Konduri, G.G.; et al. Dynamic Phosphorylation of the C Terminus of Hsp70 Regulates the Mitochondrial Import of SOD2 and Redox Balance. Cell Rep. 2018, 25, 2605–2616. [Google Scholar] [CrossRef]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jaattela, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, K.; Lipp, A.M.; Nimmervoll, B.; Sonnleitner, A.; Hesse, J.; Haselgruebler, T.; Balogi, Z. The complex function of hsp70 in metastatic cancer. Cancers 2013, 6, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Kaynak, A.; Vallabhapurapu, S.D.; Davis, H.W.; Smith, E.P.; Muller, P.; Vojtesek, B.; Franco, R.S.; Shao, W.-H.; Qi, X. TLR2-Bound Cancer-Secreted Hsp70 Induces MerTK-Mediated Immunosuppression and Tumorigenesis in Solid Tumors. Cancers 2025, 17, 450. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Mambula, S.S.; Stevenson, M.A.; Ogawa, K.; Calderwood, S.K. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods 2007, 43, 168–175. [Google Scholar] [CrossRef]
- Liang, X.; Luo, M.; Shao, B.; Yang, J.Y.; Tong, A.; Wang, R.B.; Liu, Y.T.; Jun, R.; Liu, T.; Yi, T.; et al. Phosphatidylserine released from apoptotic cells in tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun. 2022, 42, 205–222. [Google Scholar] [CrossRef]
- Schilling, D.; Gehrmann, M.; Steinem, C.; De Maio, A.; Pockley, A.G.; Abend, M.; Molls, M.; Multhoff, G. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 2467–2477. [Google Scholar] [CrossRef]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- McCallister, C.; Kdeiss, B.; Nikolaidis, N. Biochemical characterization of the interaction between HspA1A and phospholipids. Cell Stress Chaperones 2016, 21, 41–53. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. [Google Scholar] [CrossRef] [PubMed]
- Smulders, L.; Daniels, A.J.; Plescia, C.B.; Berger, D.; Stahelin, R.V.; Nikolaidis, N. Characterization of the Relationship between the Chaperone and Lipid-Binding Functions of the 70-kDa Heat-Shock Protein, HspA1A. Int. J. Mol. Sci. 2020, 21, 5995. [Google Scholar] [CrossRef]
- Kuo, J.H.; Jan, M.S.; Jeng, J.; Chiu, H.W. Induction of apoptosis in macrophages by air oxidation of dioleoylphosphatidylglycerol. J. Control. Release 2005, 108, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Menjivar, R.E.; Nwosu, Z.C.; Du, W.; Donahue, K.L.; Hong, H.S.; Espinoza, C.; Brown, K.; Velez-Delgado, A.; Yan, W.; Lima, F.; et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. eLife 2023, 12, e80721. [Google Scholar] [CrossRef] [PubMed]
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The Biological and Biomechanical Role of Transglutaminase-2 in the Tumour Microenvironment. Cancers 2021, 13, 2788. [Google Scholar] [CrossRef]
- Wojton, J.; Meisen, W.H.; Jacob, N.K.; Thorne, A.H.; Hardcastle, J.; Denton, N.; Chu, Z.; Dmitrieva, N.; Marsh, R.; Van Meir, E.G.; et al. SapC-DOPS-induced lysosomal cell death synergizes with TMZ in glioblastoma. Oncotarget 2014, 5, 9703–9709. [Google Scholar] [CrossRef]
- Zhao, S.; Chu, Z.; Blanco, V.M.; Nie, Y.; Hou, Y.; Qi, X. SapC-DOPS nanovesicles as targeted therapy for lung cancer. Mol. Cancer Ther. 2015, 14, 491–498. [Google Scholar] [CrossRef]
- Abdelbaki, M.S.; DeWire Schottmiller, M.D.; Cripe, T.P.; Curry, R.C.; Cruze, C.A.; Her, L.; Demko, S.; Casey, D.; Setty, B. An open-label multi-center phase 1 safety study of BXQ-350 in children and young adults with relapsed solid tumors, including recurrent malignant brain tumors. Heliyon 2022, 8, e12450. [Google Scholar] [CrossRef]
- Rixe, O.; Villano, J.L.; Wesolowski, R.; Noonan, A.M.; Puduvalli, V.K.; Wise-Draper, T.M.; Curry, R., 3rd; Yilmaz, E.; Cruze, C.; Ogretmen, B.; et al. A First-in-Human Phase I Study of BXQ-350, a First-in-Class Sphingolipid Metabolism Regulator, in Patients with Advanced/Recurrent Solid Tumors or High-Grade Gliomas. Clin. Cancer Res. 2024, 30, 5053–5060. [Google Scholar] [CrossRef]
- Patel, R.A.; Baron, A.D.; Boulmay, B.C.; Flora, D.B.; Gemmill, J.A.L.; Lee, F.C.; Sohal, D.; Williams, G.R.; Curry, R.C.; Gazda, M.; et al. BXQ-350: A phase 1b/2 placebo controlled, double blinded study on the efficacy and safety of BXQ-350 in combination with mFOLFOX7 and bevacizumab in newly diagnosed metastatic colorectal carcinoma (mCRC). J. Clin. Oncol. 2024, 42, TPS224. [Google Scholar] [CrossRef]
- Shin, K.; Kim, J.; Park, S.J.; Lee, M.A.; Park, J.M.; Choi, M.-G.; Kang, D.; Song, K.Y.; Lee, H.H.; Seo, H.S.; et al. Prognostic value of soluble PD-L1 and exosomal PD-L1 in advanced gastric cancer patients receiving systemic chemotherapy. Sci. Rep. 2023, 13, 6952. [Google Scholar] [CrossRef]
- Okuma, Y.; Hosomi, Y.; Nakahara, Y.; Watanabe, K.; Sagawa, Y.; Homma, S. High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer 2017, 104, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Paladini, L.; Fabris, L.; Bottai, G.; Raschioni, C.; Calin, G.A.; Santarpia, L. Targeting microRNAs as key modulators of tumor immune response. J. Exp. Clin. Cancer Res. 2016, 35, 103. [Google Scholar] [CrossRef] [PubMed]
- Khayati, S.; Dehnavi, S.; Sadeghi, M.; Tavakol Afshari, J.; Esmaeili, S.-A.; Mohammadi, M. The potential role of miRNA in regulating macrophage polarization. Heliyon 2023, 9, e21615. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | AUCINF | AUClast | Cmax | Tmax | T1/2 | MRT |
|---|---|---|---|---|---|---|
| Radiant Efficiency [p/s/cm2/sr]*h/[µW/cm2] | Radiant Efficiency [p/s/cm2/sr]*h/[µW/cm2] | Average Radiant Efficiency [p/s/cm2/sr]/[µW/cm2] | h | h | h | |
| Blood (SEM) | 8.622 × 109 (7.947 × 108) | 6.170 × 109 (3.326 × 108) | 1.184 × 109 (2.129 × 108) | 0.83 (0.17) | 29.2 (6.08) | 37.05 (9.01) |
| Tumor (SEM) | N/A | 1.209 × 1011 (1.757 × 1010) | 3.085 × 109 (4.749 × 108) | 40 (8) | N/A | N/A |
| Liver (SEM) | N/A | 4.640 × 1010 (2.693 × 1010) | 2.596 × 109 (6.091 × 108) | 1.33 (0.33) | N/A | N/A |
| Kidney (SEM) | N/A | 9.312 × 109 (3.097 × 109) | 5.821 × 108 (1.301 × 108) | 8.83 (7.6) | N/A | N/A |
| Spleen (SEM) | N/A | 1.739 × 1010 (2.409 × 109) | 5.295 × 108 (8.229 × 107) | 32 (8) | N/A | N/A |
| Brain (SEM) | 8.431 × 109 (1.712 × 108) | 5.360 × 109 (8.997 × 108) | 3.086 × 108 (4.907 × 107) | 8.83 (7.6) | 38.38 (14.95) | 51.7 (23.08) |
| Heart (SEM) | 5.577 × 109 (3.748 × 108) | 4.320 × 109 (5.650 × 108) | 2.480 × 108 (5.119 × 107) | 9.33 (7.33) | 29.37 (4.73) | 42.09 (3.32) |
| Lung (SEM) | N/A | 2.119 × 1010 (8.078 × 109) | 1.051 × 109 (1.821 × 108) | 1.17 (0.44) | N/A | N/A |
| Ovary (SEM) | N/A | 1.464 × 1010 (4.112 × 109) | 4.912 × 108 (1.469 × 108) | 9.33 (7.33) | N/A | N/A |
| Intestine (SEM) | 2.351 × 1010 (8.036 × 109) | 1.302 × 1010 (1.034 × 109) | 7.441 × 108 (2.281 × 108) | 2.33 (0.88) | 30.78 (8.61) | 47.24 (16.14) |
| Bladder (SEM) | N/A | 1.433 × 1010 (7.469 × 109) | 6.028 × 108 (1.992 × 108) | 40 (8) | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaynak, A.; Vallabhapurapu, S.D.; Smith, E.P.; Davis, H.W.; Lewis, C.S.; Ahn, J.; Muller, P.; Vojtesek, B.; Stringer, K.F.; Franco, R.S.; et al. Targeting Hsp70 Immunosuppressive Signaling Axis with Lipid Nanovesicles: A Novel Approach to Treat Pancreatic Cancer. Cancers 2025, 17, 1224. https://doi.org/10.3390/cancers17071224
Kaynak A, Vallabhapurapu SD, Smith EP, Davis HW, Lewis CS, Ahn J, Muller P, Vojtesek B, Stringer KF, Franco RS, et al. Targeting Hsp70 Immunosuppressive Signaling Axis with Lipid Nanovesicles: A Novel Approach to Treat Pancreatic Cancer. Cancers. 2025; 17(7):1224. https://doi.org/10.3390/cancers17071224
Chicago/Turabian StyleKaynak, Ahmet, Subrahmanya D. Vallabhapurapu, Eric P. Smith, Harold W. Davis, Clayton S. Lewis, Joseph Ahn, Petr Muller, Borek Vojtesek, Keith F. Stringer, Robert S. Franco, and et al. 2025. "Targeting Hsp70 Immunosuppressive Signaling Axis with Lipid Nanovesicles: A Novel Approach to Treat Pancreatic Cancer" Cancers 17, no. 7: 1224. https://doi.org/10.3390/cancers17071224
APA StyleKaynak, A., Vallabhapurapu, S. D., Smith, E. P., Davis, H. W., Lewis, C. S., Ahn, J., Muller, P., Vojtesek, B., Stringer, K. F., Franco, R. S., Bogdanov, V. Y., Shao, W.-H., & Qi, X. (2025). Targeting Hsp70 Immunosuppressive Signaling Axis with Lipid Nanovesicles: A Novel Approach to Treat Pancreatic Cancer. Cancers, 17(7), 1224. https://doi.org/10.3390/cancers17071224