
Immune-Based Strategies for Pancreatic Cancer in the Adjuvant Setting



Simple Summary
Abstract
1. Introduction
1.1. Unmet Need for Improved Adjuvant Treatments for Pancreatic Cancer
1.2. Obstacles for Adjuvant Immunotherapy
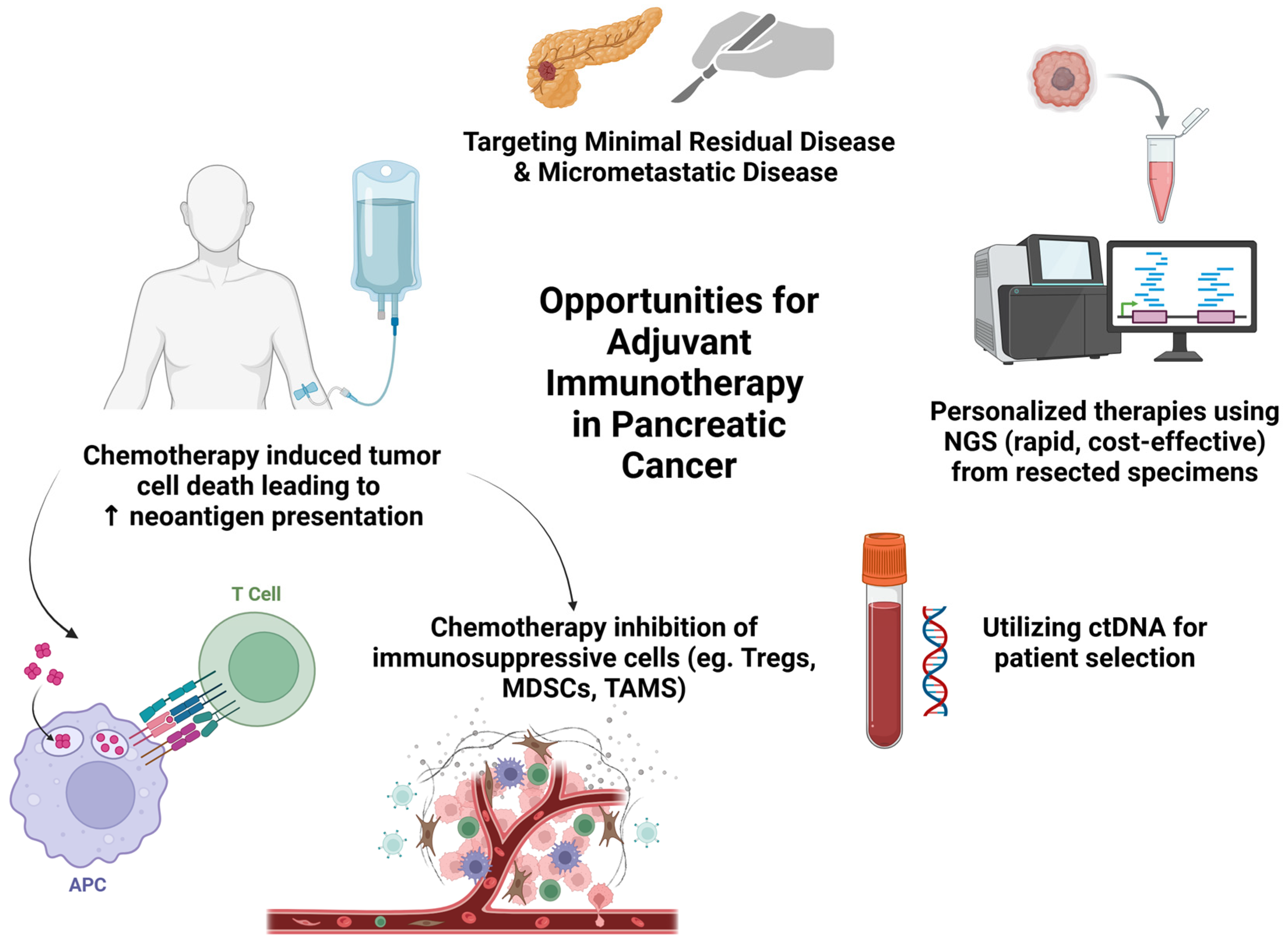
1.3. Opportunities for Adjuvant Immunotherapy
2. Vaccine Therapies
2.1. Rationale for Vaccine-Based Immunotherapy
2.2. Whole Cancer Cell Vaccines
2.2.1. GVAX Vaccine
2.2.2. Algenpantucel-L
2.3. Peptide Vaccines
2.3.1. KRAS Targeting Peptide Vaccines
2.3.2. Targovax TG01
2.3.3. GI-4000
2.3.4. ELI-002
2.3.5. Pooled KRAS Peptide Vaccine
2.3.6. Personalized Neoantigen Peptide Vaccines
2.3.7. HSP-Peptide Complex Vaccines
2.4. Dendritic Cell Vaccines
2.5. mRNA Vaccines
3. Adoptive Cellular Therapies
3.1. CAR T Cell Therapy
3.2. Allogenic Natural Killer Cell Therapy
3.3. Cytokine-Induced Killer (CIK) Cell Therapy
4. Future Directions in Immune-Based Strategies in the Adjuvant Setting
4.1. Tumor Microenvironment Remodeling
4.2. Leveraging Novel Agents
4.3. Optimization of Patient Selection and Trial Design
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.; Walsh, R.M.; Ramanathan, R.K.; Khorana, A.A. Pancreatic adenocarcinoma: Treating a systemic disease with systemic therapy. J. Natl. Cancer Inst. 2014, 106, dju011. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.C.; Nathan, H.; Cameron, J.L.; Olino, K.; Edil, B.H.; Herman, J.M.; Hirose, K.; Schulick, R.D.; Choti, M.A.; Wolfgang, C.L.; et al. Conditional survival in patients with pancreatic ductal adenocarcinoma resected with curative intent. Cancer 2012, 118, 2674–2681. [Google Scholar] [CrossRef]
- Conroy, T.; Castan, F.; Lopez, A.; Turpin, A.; Ben Abdelghani, M.; Wei, A.C.; Mitry, E.; Biagi, J.J.; Evesque, L.; Artru, P.; et al. Five-Year Outcomes of FOLFIRINOX vs Gemcitabine as Adjuvant Therapy for Pancreatic Cancer: A Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1571–1578. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef]
- Tempero, M.A.; Pelzer, U.; O’Reilly, E.M.; Winter, J.; Oh, D.Y.; Li, C.P.; Tortora, G.; Chang, H.M.; Lopez, C.D.; Bekaii-Saab, T.; et al. Adjuvant nab-Paclitaxel + Gemcitabine in Resected Pancreatic Ductal Adenocarcinoma: Results From a Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2023, 41, 2007–2019. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- O’Brien, M.; Paz-Ares, L.; Marreaud, S.; Dafni, U.; Oselin, K.; Havel, L.; Esteban, E.; Isla, D.; Martinez-Marti, A.; Faehling, M.; et al. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB-IIIA non-small-cell lung cancer (PEARLS/KEYNOTE-091): An interim analysis of a randomised, triple-blind, phase 3 trial. Lancet Oncol. 2022, 23, 1274–1286. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Bajorin, D.F.; Witjes, J.A.; Gschwend, J.E.; Schenker, M.; Valderrama, B.P.; Tomita, Y.; Bamias, A.; Lebret, T.; Shariat, S.F.; Park, S.H.; et al. Adjuvant Nivolumab versus Placebo in Muscle-Invasive Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Awad, M.M.; Spicer, J.D.; He, J.; Lu, S.; Sepesi, B.; Tanaka, F.; Taube, J.M.; Cornelissen, R.; Havel, L.; et al. Perioperative Nivolumab in Resectable Lung Cancer. N. Engl. J. Med. 2024, 390, 1756–1769. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Lennard, T.W.; Shenton, B.K.; Borzotta, A.; Donnelly, P.K.; White, M.; Gerrie, L.M.; Proud, G.; Taylor, R.M. The influence of surgical operations on components of the human immune system. Br. J. Surg. 1985, 72, 771–776. [Google Scholar] [CrossRef]
- Ogawa, K.; Hirai, M.; Katsube, T.; Murayama, M.; Hamaguchi, K.; Shimakawa, T.; Naritake, Y.; Hosokawa, T.; Kajiwara, T. Suppression of cellular immunity by surgical stress. Surgery 2000, 127, 329–336. [Google Scholar] [CrossRef]
- Gulley, J.L.; Madan, R.A.; Schlom, J. Impact of tumour volume on the potential efficacy of therapeutic vaccines. Curr. Oncol. 2011, 18, e150–e157. [Google Scholar] [CrossRef]
- Cali Daylan, A.E.; Halmos, B. Long-term benefit of immunotherapy in metastatic non-small cell lung cancer: The tale of the tail. Transl. Lung Cancer Res. 2023, 12, 1636–1642. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin. Cancer Res. 2005, 11, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef]
- Le, D.T.; Jaffee, E.M. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: A current perspective. Cancer Res. 2012, 72, 3439–3444. [Google Scholar] [CrossRef]
- Lutsiak, M.E.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Forget, M.A.; Lucas, F.A.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ding, D.; Edil, B.H.; Judkins, C.; Durham, J.N.; Thomas, D.L., 2nd; Bever, K.M.; Mo, G.; Solt, S.E.; Hoare, J.A.; et al. Vaccine-Induced Intratumoral Lymphoid Aggregates Correlate with Survival Following Treatment with a Neoadjuvant and Adjuvant Vaccine in Patients with Resectable Pancreatic Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Heumann, T.; Judkins, C.; Li, K.; Lim, S.J.; Hoare, J.; Parkinson, R.; Cao, H.; Zhang, T.; Gai, J.; Celiker, B.; et al. A platform trial of neoadjuvant and adjuvant antitumor vaccination alone or in combination with PD-1 antagonist and CD137 agonist antibodies in patients with resectable pancreatic adenocarcinoma. Nat. Commun. 2023, 14, 3650. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Parkinson, R.; Jaffee, E.M.; Sugar, E.; Zheng, L.; Onners, B.; Weiss, M.J.; Wolfgang, C.L.; Cameron, J.L.; Pawlik, T.M.; et al. Phase 1 Study of Adjuvant Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor-Transduced Pancreatic Tumor Cell Vaccine, Low-Dose Cyclophosphamide, and Stereotactic Body Radiation Therapy Followed by FOLFIRINOX in High-Risk Resected Pancreatic Ductal Adenocarcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2024, 121, 930–941. [Google Scholar] [CrossRef]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: A phase 2 study. J. Gastrointest. Surg. 2013, 17, 94–100. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.F.; Capanu, M.; Gansukh, B.; Jacobs, G.; Levin, A.; Neville, D.; Kelsen, D.P.; et al. Targeting mutated K-ras in pancreatic adenocarcinoma using an adjuvant vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef]
- Palmer, D.H.; Valle, J.W.; Ma, Y.T.; Faluyi, O.; Neoptolemos, J.P.; Jensen Gjertsen, T.; Iversen, B.; Amund Eriksen, J.; Møller, A.S.; Aksnes, A.K.; et al. TG01/GM-CSF and adjuvant gemcitabine in patients with resected RAS-mutant adenocarcinoma of the pancreas (CT TG01-01): A single-arm, phase 1/2 trial. Br. J. Cancer 2020, 122, 971–977. [Google Scholar] [CrossRef]
- Kasi, A.; Diaz, F.; Al-Rajabi, R.M.d.T.; Baranda, J.C.; Carroll, E.; Belcher, C.; Olyaee, M.; Rastogi, A.; Schmitt, T.; Kumer, S.; et al. Targeting minimal residual disease (MRD) in resected RAS mutated pancreatic cancer with vaccine TG01/QS-21 +/− PD-1 inhibitor, balstilimab: A randomized phase II study (TESLA). J. Clin. Oncol. 2023, 41, TPS4205. [Google Scholar] [CrossRef]
- Muscarella, P.; Bekaii-Saab, T.; McIntyre, K.; Rosemurgy, A.; Ross, S.B.; Richards, D.A.; Fisher, W.E.; Flynn, P.J.; Mattson, A.; Coeshott, C.; et al. A Phase 2 Randomized Placebo-Controlled Adjuvant Trial of GI-4000, a Recombinant Yeast Expressing Mutated RAS Proteins in Patients with Resected Pancreas Cancer. J. Pancreat Cancer 2021, 7, 8–19. [Google Scholar]
- Pant, S.; Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Basturk, O.; VanWyk, H.; et al. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: The phase 1 AMPLIFY-201 trial. Nat. Med. 2024, 30, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Huff, A.; Heumann, T.; Berg, M.; Ferguson, A.; Lim, S.; Wang, H.; Thomas, A.; Nauroth, J.; Baretti, M.; et al. Abstract CT036: Safety and immunogenicity of a first-in-human mutant KRAS long peptide vaccine combined with ipilimumab/nivolumab in resected pancreatic cancer: Preliminary analysis from a phase I study. Cancer Res. 2023, 83, CT036. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, Y.; Kong, X.; Wu, Z.; Wang, H. Impact of personalized peptide neoantigen vaccines on immunologic responses in patients with pancreatic cancer. J. Clin. Oncol. 2024, 42, e16351. [Google Scholar] [CrossRef]
- Maki, R.G.; Livingston, P.O.; Lewis, J.J.; Janetzki, S.; Klimstra, D.; Desantis, D.; Srivastava, P.K.; Brennan, M.F. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig. Dis. Sci. 2007, 52, 1964–1972. [Google Scholar] [CrossRef]
- Lepisto, A.J.; Moser, A.J.; Zeh, H.; Lee, K.; Bartlett, D.; McKolanis, J.R.; Geller, B.A.; Schmotzer, A.; Potter, D.P.; Whiteside, T.; et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008, 6, 955–964. [Google Scholar]
- Lau, S.P.; Klaase, L.; Vink, M.; Dumas, J.; Bezemer, K.; van Krimpen, A.; van der Breggen, R.; Wismans, L.V.; Doukas, M.; de Koning, W.; et al. Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: A phase I study. Eur. J. Cancer 2022, 169, 20–31. [Google Scholar] [CrossRef]
- Lau, S.P.; van’t Land, F.R.; van der Burg, S.H.; Homs, M.Y.V.; Lolkema, M.P.; Aerts, J.; van Eijck, C.H.J. Safety and tumour-specific immunological responses of combined dendritic cell vaccination and anti-CD40 agonistic antibody treatment for patients with metastatic pancreatic cancer: Protocol for a phase I, open-label, single-arm, dose-escalation study (REACtiVe-2 trial). BMJ Open 2022, 12, e060431. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma-A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Bear, A.S.; Nadler, R.B.; O’Hara, M.H.; Stanton, K.L.; Xu, C.; Saporito, R.J.; Rech, A.J.; Baroja, M.L.; Blanchard, T.; Elliott, M.H.; et al. Natural TCRs targeting KRASG12V display fine specificity and sensitivity to human solid tumors. J. Clin. Investig. 2024, 134, e175790. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.C.; Antoine, M.; Danel, C.; Heudes, D.; Wislez, M.; Poulot, V.; Rabbe, N.; Laurans, L.; Tartour, E.; de Chaisemartin, L.; et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 2008, 26, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, M.; Ren, Y.; Ba, Y.; Liu, S.; Zuo, A.; Xu, H.; Weng, S.; Han, X.; Liu, Z. Tertiary lymphoid structural heterogeneity determines tumour immunity and prospects for clinical application. Mol. Cancer 2024, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.T.; Herman, J.M.; Dholakia, A.S.; Moningi, S.; Lu, Y.; Rosati, L.M.; Hacker-Prietz, A.; Assadi, R.K.; Saeed, A.M.; Pawlik, T.M.; et al. Lymphocyte-Sparing Effect of Stereotactic Body Radiation Therapy in Patients with Unresectable Pancreatic Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 571–579. [Google Scholar] [CrossRef]
- Hewitt, D.B.; Nissen, N.; Hatoum, H.; Musher, B.; Seng, J.; Coveler, A.L.; Al-Rajabi, R.; Yeo, C.J.; Leiby, B.; Banks, J.; et al. A Phase 3 Randomized Clinical Trial of Chemotherapy with or Without Algenpantucel-L (HyperAcute-Pancreas) Immunotherapy in Subjects with Borderline Resectable or Locally Advanced Unresectable Pancreatic Cancer. Ann. Surg. 2022, 275, 45–53. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Nusrat, F.; Khanna, A.; Jain, A.; Jiang, W.; Lavu, H.; Yeo, C.J.; Bowne, W.; Nevler, A. The Clinical Implications of KRAS Mutations and Variant Allele Frequencies in Pancreatic Ductal Adenocarcinoma. J. Clin. Med. 2024, 13, 2103. [Google Scholar] [CrossRef]
- Coelho, M.A.; de Carné Trécesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e1086. [Google Scholar] [CrossRef]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.J.; Schlechter, B.L.; Fakih, M.G.; Tsimberidou, A.M.; Grossman, J.E.; Gordon, M.S.; Wilky, B.A.; Pimentel, A.; Mahadevan, D.; Balmanoukian, A.S.; et al. Botensilimab plus balstilimab in relapsed/refractory microsatellite stable metastatic colorectal cancer: A phase 1 trial. Nat. Med. 2024, 30, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Heumann, T.; Berg, M.; Ferguson, A.; Lim, S.; Wang, H.; Nauroth, J.; Laheru, D.; Jaffee, E.; Azad, N.; et al. A phase I study of a mutant KRAS-targeted long peptide vaccine combined with ipilimumab/nivolumab in resected pancreatic cancer and MMR-proficient metastatic colorectal cancer. J. Clin. Oncol. 2023, 41, TPS814. [Google Scholar] [CrossRef]
- Sultan, H.; Salazar, A.M.; Celis, E. Poly-ICLC, a multi-functional immune modulator for treating cancer. Semin. Immunol. 2020, 49, 101414. [Google Scholar] [CrossRef]
- Niemi, J.V.L.; Sokolov, A.V.; Schiöth, H.B. Neoantigen Vaccines; Clinical Trials, Classes, Indications, Adjuvants and Combinatorial Treatments. Cancers 2022, 14, 5163. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e289. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef]
- Diakonos Oncology Receives FDA Fast Track Designation for Pancreatic Cancer Dendritic Cell Vaccine; Names Daniel D. Von Hoff, M.D. to Scientific Advisory Board. Press Releases. Available online: https://www.diakonosoncology.com/news/diakonos-oncology-receives-fda-fast-track-designation-for-pancreatic-cancer-dendritic-cell-vaccine-names-daniel-d-von-hoff-m-d-to-scientific-advisory-board/ (accessed on 15 January 2025).
- Swift, M.D.; Breeher, L.E.; Tande, A.J.; Tommaso, C.P.; Hainy, C.M.; Chu, H.; Murad, M.H.; Berbari, E.F.; Virk, A. Effectiveness of Messenger RNA Coronavirus Disease 2019 (COVID-19) Vaccines Against Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in a Cohort of Healthcare Personnel. Clin. Infect. Dis. 2021, 73, e1376–e1379. [Google Scholar] [CrossRef]
- Gote, V.; Bolla, P.K.; Kommineni, N.; Butreddy, A.; Nukala, P.K.; Palakurthi, S.S.; Khan, W. A Comprehensive Review of mRNA Vaccines. Int. J. Mol. Sci. 2023, 24, 2700. [Google Scholar] [CrossRef]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Guo, Y.J.; Bin, L.; Sun, S.H. Hepatitis C virus single-stranded RNA induces innate immunity via Toll-like receptor 7. J. Hepatol. 2009, 51, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Diorio, C.; Maude, S.L. CAR T cells vs allogeneic HSCT for poor-risk ALL. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 501–507. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-Specific T-Cell Activation Independently of the MHC: Chimeric Antigen Receptor-Redirected T Cells. Front. Immunol. 2013, 4, 371. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J. Surg. Oncol. 2017, 116, 63–74. [Google Scholar] [CrossRef]
- Wöll, S.; Schlitter, A.M.; Dhaene, K.; Roller, M.; Esposito, I.; Sahin, U.; Türeci, Ö. Claudin 18.2 is a target for IMAB362 antibody in pancreatic neoplasms. Int. J. Cancer 2014, 134, 731–739. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, P.; Liu, C.; Zhang, J.; Zhou, J.; Yuan, J.; Liu, D.; Zhang, M.; Gong, J.; Wang, X.; et al. Safety and Efficacy of CT041 in Patients with Refractory Metastatic Pancreatic Cancer: A Pooled Analysis of Two Early-Phase Trials. J. Clin. Oncol. 2024, 42, 2565–2577. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Jacobs, M.T.; Fehniger, T.A. Allogeneic natural killer cell therapy. Blood 2023, 141, 856–868. [Google Scholar] [CrossRef]
- Chester, C.; Fritsch, K.; Kohrt, H.E. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front. Immunol. 2015, 6, 601. [Google Scholar] [CrossRef]
- Kumar, V.; Mahato, R.I. Natural killer cells for pancreatic cancer immunotherapy: Role of nanoparticles. Cancer Lett. 2023, 579, 216462. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Sommaggio, R.; Zanovello, P.; Rosato, A. Cytokines for the induction of antitumor effectors: The paradigm of Cytokine-Induced Killer (CIK) cells. Cytokine Growth Factor Rev. 2017, 36, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Sangiolo, D. Cytokine induced killer cells as promising immunotherapy for solid tumors. J. Cancer 2011, 2, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.; Qiang, L.; Jian, W.; Ti, Z.; Da-Lu, K. A randomized, controlled trial of postoperative adjuvant cytokine-induced killer cells immunotherapy after radical resection of hepatocellular carcinoma. Dig. Liver Dis. 2009, 41, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Kim, Y.; Shuang, Z.Y.; Zhang, Y.J.; Lao, X.M.; Li, Y.Q.; Chen, M.S.; Pawlik, T.M.; Xia, J.C.; et al. A randomized controlled trial on patients with or without adjuvant autologous cytokine-induced killer cells after curative resection for hepatocellular carcinoma. Oncoimmunology 2016, 5, e1083671. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: A systematic review and meta-analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Agostini, A.; Delfino, P.; De Sanctis, F.; Nasca, V.; Spallotta, F.; Sette, C.; Martini, M.; Ugel, S.; et al. Intratumoral injection of TLR9 agonist promotes an immunopermissive microenvironment transition and causes cooperative antitumor activity in combination with anti-PD1 in pancreatic cancer. J. Immunother. Cancer 2021, 9, e002876. [Google Scholar] [CrossRef]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Lim, K.H.; McWilliams, R.; Suresh, R.; Lockhart, A.C.; Brown, A.; Breden, M.; Belle, J.I.; Herndon, J.; Bogner, S.J.; et al. Defactinib, Pembrolizumab, and Gemcitabine in Patients with Advanced Treatment Refractory Pancreatic Cancer: A Phase I Dose Escalation and Expansion Study. Clin. Cancer Res. 2022, 28, 5254–5262. [Google Scholar] [CrossRef]
- Musiu, C.; Adamo, A.; Caligola, S.; Agostini, A.; Frusteri, C.; Lupo, F.; Boschi, F.; Busato, A.; Poffe, O.; Anselmi, C.; et al. Local ablation disrupts immune evasion in pancreatic cancer. Cancer Lett. 2025, 609, 217327. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Yaeger, R.; Spira, A.I.; Pelster, M.S.; Sabari, J.K.; Hafez, N.; Barve, M.; Velastegui, K.; Yan, X.; Shetty, A.; et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. J. Clin. Oncol. 2023, 41, 4097–4106. [Google Scholar] [CrossRef]
- Hamarsheh, S.a.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- Mahadevan, K.K.; LeBleu, V.S.; Ramirez, E.V.; Chen, Y.; Li, B.; Sockwell, A.M.; Gagea, M.; Sugimoto, H.; Sthanam, L.K.; Tampe, D.; et al. Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8(+) T cells. Dev. Cell 2023, 58, 1562–1577.e1568. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, J.; Hsu, W.H.; LaBella, K.A.; Deng, P.; Shang, X.; Tallón de Lara, P.; Cai, L.; Jiang, S.; DePinho, R.A. Combined KRAS Inhibition and Immune Therapy Generates Durable Complete Responses in an Autochthonous PDAC Model. Cancer Discov. 2025, 15, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Tie, J.; Wang, Y.; Cohen, J.D.; Shapiro, J.D.; Wong, R.; Aghmesheh, M.; Kiberu, A.D.; Francesconi, A.; Burge, M.E.; et al. The potential role of serial circulating tumor DNA (ctDNA) testing after upfront surgery to guide adjuvant chemotherapy for early stage pancreatic cancer: The AGITG DYNAMIC-Pancreas trial. J. Clin. Oncol. 2024, 42, 107. [Google Scholar] [CrossRef]
- Botta, G.P.; Abdelrahim, M.; Drengler, R.L.; Aushev, V.N.; Esmail, A.; Laliotis, G.; Brewer, C.M.; George, G.V.; Abbate, S.M.; Chandana, S.R.; et al. Association of personalized and tumor-informed ctDNA with patient survival outcomes in pancreatic adenocarcinoma. Oncologist 2024, 29, 859–869. [Google Scholar] [CrossRef]
- Mirza, M.; Goerke, L.; Anderson, A.; Wilsdon, T. Assessing the Cost-Effectiveness of Next-Generation Sequencing as a Biomarker Testing Approach in Oncology and Policy Implications: A Literature Review. Value Health 2024, 27, 1300–1309. [Google Scholar] [CrossRef]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-Time Genomic Profiling of Pancreatic Ductal Adenocarcinoma: Potential Actionability and Correlation with Clinical Phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef]
- Haldar, S.D.; Vilar, E.; Maitra, A.; Zaidi, N. Worth a Pound of Cure? Emerging Strategies and Challenges in Cancer Immunoprevention. Cancer Prev. Res. 2023, 16, 483–495. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpää, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef]
- Chick, R.C.; Gunderson, A.J.; Rahman, S.; Cloyd, J.M. Neoadjuvant Immunotherapy for Localized Pancreatic Cancer: Challenges and Early Results. Cancers 2023, 15, 3967. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients with Advanced BRAF-Mutant Melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; McArthur, H.; Pusztai, L.; Kümmel, S.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer. N. Engl. J. Med. 2024, 391, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; Altorki, N.; Zhou, C.; Csőszi, T.; Vynnychenko, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet 2021, 398, 1344–1357. [Google Scholar] [CrossRef]
- Oh, D.Y.; Ruth He, A.; Qin, S.; Chen, L.T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Fang, Q.; Qian, Y.; Xie, Z.; Zhao, H.; Zheng, Y.; Li, D. Predictors of severity and onset timing of immune-related adverse events in cancer patients receiving immune checkpoint inhibitors: A retrospective analysis. Front. Immunol. 2025, 16, 1508512. [Google Scholar] [CrossRef]
- Rached, L.; Laparra, A.; Sakkal, M.; Danlos, F.-X.; Barlesi, F.; Carbonnel, F.; De Martin, E.; Ducreux, M.; Even, C.; Le Pavec, J.; et al. Toxicity of immunotherapy combinations with chemotherapy across tumor indications: Current knowledge and practical recommendations. Cancer Treat. Rev. 2024, 127, 102751. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| NCT ID/Authors | Title | Phase | Status | Treatment | Clinical and Translational Outcomes | Publications |
|---|---|---|---|---|---|---|
| Whole Cell Vaccines | ||||||
| GVAX | ||||||
| Jaffee et al., 2001 | Novel Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor–Secreting Tumor Vaccine for Pancreatic Cancer: A Phase I Trial of Safety and Immune Activation | I | Completed | GVAX vaccine | 3/14 patients experienced delayed type hypersensitivity (DTH) responses; DFS at least 25 mo after diagnosis | [39] |
| NCT00389610 | A Safety and Efficacy Trial of Vaccine Boosting With Lethally Irradiated Allogeneic Pancreatic Tumor Cells Transfected With the GM-CSF Gene for the Treatment of Pancreatic Adenocarcinoma | IIb | Completed | GVAX vaccine | N/A | N/A |
| NCT00084383 | A Safety and Efficacy Trial of Lethally Irradiated Allogeneic Pancreatic Tumor Cells Transfected With the GM-CSF Gene in Combination With Adjuvant Chemoradiotherapy for Treatment of Adenocarcinoma of the Pancreas | II | Completed | GVAX vaccine, 5-FU Chemoradiotherapy | 60 patients; mDFS 17.3 mo; mOS 24.8 mo. Induction of mesothelin-specific CD8+ T cells with HLA-A1+ and HLA-A2+ patients correlated with disease-free survival. Resected pancreatic cancer tissue revealed the common presence of tumor-infiltrating Tregs, which in previous PDAC studies and other solid tumors had been associated with shorter patient survival. Upregulation of PD-1/PD-L1 | [40] |
| NCT00727441 | A Randomized Three-arm Neoadjuvant and Adjuvant Feasibility and Toxicity Study of a GM-CSF Secreting Allogeneic Pancreatic Cancer Vaccine Administered Either Alone or in Combination With Either a Single Intravenous Dose or Daily Metronomic Oral Doses of Cyclophosphamide for the Treatment of Patients With Surgically Resected Adenocarcinoma of the Pancreas | II | Completed | GVAX vaccine, Cyclophosphamide | Patients who received neoadjuvant and adjuvant GVAX alone (Arm A) had trend toward longer mOS 35.0 mo vs. historical controls who received adjuvant GVAX alone 24.8 mo. 33/39 patients developed intratumoral tertiary lymphoid aggregates (TLAs). Decreased Tregs within the TLAs associated with increased intratumoral Teffector/Treg ratios and improved patient survival | [41,42] |
| NCT02451982 | A Platform Study of Combination Immunotherapy for the Neoadjuvant and Adjuvant Treatment of Patients With Surgically Resectable Adenocarcinoma of the Pancreas | II | Recruiting | GVAX vaccine, Cyclophosphamide, Nivolumab (PD-1), Urelumab (CD137), BMS-986253 (Anti-IL-8) | Underpowered to reach statistical significance GVAX + nivolumab + urelumab (Arm C) compared to GVAX + nivolumab (Arm B) had improved mDFS (33.5 mo vs. 15.0 mo) and mOS (35.6 mo vs. 27.0 mo). | [43] |
| NCT01595321 | Pilot Study Evaluating Allogeneic GM-CSF-Transduced Pancreatic Tumor Cell Vaccine (GVAX) and Low Dose Cyclophosphamide With Fractionated Stereotactic Body Radiation Therapy (SBRT) and FOLFIRINOX Chemotherapy in Patients With Resected Adenocarcinoma of the Pancreas | II | Completed | GVAX vaccine, Cyclophosphamide, Stereotactic Body Radiation, FOLFIRINOX | SBRT + mFOLFIRINOX + GVAX (Cohort 3) mDFS 24.1 mo and mOS 61.3 mo and which was numerically superior to overall cohort mDFS 18.2 mo and mOS 36.2 mo | [44] |
| Algenpantucel-L | ||||||
| NCT00569387 | A Phase III Study of Chemotherapy and Chemoradiotherapy With or Without Algenpantucel-L (HyperAcute®-Pancreas) Immunotherapy in Subjects With Surgically Resected Pancreatic Cancer | II | Completed | HyperAcute-Pancreas Immunotherapy, Gemcitabine, 5-FU Chemoradiation | 1-year DFS 62%; 12-month OS 86% | [45] |
| Peptide Vaccines | ||||||
| Ras/KRAS Targeted | ||||||
| Gjertsen et al., 2001 | Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma | I/II | Completed | Mutant ras peptide, GM-CSF | 25/43 patients (58%) peptide specific immunity was induced. Improved mOS among responders vs. non-responders (148 days vs. 61 days). | [46] |
| Abou-Alfa et al., 2011 | Targeting Mutated K-ras in Pancreatic Adenocarcinoma Using an Adjuvant Vaccine | I | Completed | KRAS peptide vaccine, GM-CSF | 24 patients resected PDAC; median RFS 8.6 mo; median OS 20.3 mo | [47] |
| NCT02261714 | A Phase I/II Trial of TG01 and Gemcitabine as Adjuvant Therapy for Treating Patients With Resected Adenocarcinoma of the Pancreas | I/II | Completed | KRAS vaccine, Gemcitabine | 4/19 patients in main cohort (vaccine during gemcitabine with serious adverse reactions. In main cohort mOS 33.1 mo and mDFS 13.9 mo. Modified cohort (no vaccine during gemcitabine) with mOS 34.3 mo and median DFS 19.5 mo. | [48] |
| NCT05638698 | Phase II Randomized Trial Combining Tg01 Vaccine/Qs-21 Stimulon™ With Or Without Balstilimab As Maintenance Therapy Following Adjuvant Chemotherapy In Patients With Resected Pancreatic Cancer(TESLA) | II | Not yet recruiting | KRAS Vaccine, Balstilimab (PD-L1) | N/A | [49] |
| NCT00300950 | A Phase 2 Double-Blind, Placebo Controlled, Multi-center Adjuvant Trial of the Efficacy, Immunogenicity, and Safety of GI-4000; an Inactivated Recombinant Saccharomyces Cerevisiae Expressing Mutant Ras Protein Combined With a Gemcitabine Regimen Versus a Gemcitabine Regimen With Placebo, in Patients With Post-resection R0/R1 Pancreatic Cancer With Tumor Sequence Confirmation of Ras Mutations. | II | Completed | GI-4000, Gemcitabine | Similar RFS for the GI-4000 and placebo groups 354 and 357 days, respectively (HR = 1.01 [95% CI 0.73–1.41], p = 0.936). Reduction in Tregs was observed in R0/R1 subjects treated with GI-4000 compared to placebo (p = 0.033) | [50] |
| NCT04853017 | First in Human Phase 1 Trial of ELI-002 Immunotherapy as Treatment for Subjects With Kirsten Rat Sarcoma (KRAS) Mutated Pancreatic Ductal Adenocarcinoma and Other Solid Tumors | I | Active, not recruiting | KRAS peptide vaccine | 25 patients (20 PDAC; 5 CRC). 21/25 patients with mKRAS-specific T cell responses; 21/25 patients with tumor biomarker responses; biomarker clearance 6/25; mRFS 16.33 mo | [51] |
| NCT05726864 | First in Human Phase 1/2 Trial of ELI-002 7P Immunotherapy as Treatment for Subjects With Kirsten Rat Sarcoma (KRAS)/Neuroblastoma RAS Viral Oncogene Homolog (NRAS) Mutated Pancreatic Ductal Adenocarcinoma (PDAC) and Other Solid Tumors | I/II | Recruiting | KRAS peptide vaccine | N/A | N/A |
| NCT04117087 | Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined With Nivolumab and Ipilimumab for Patients With Resected MMR-p Colorectal and Pancreatic Cancer | I | Active, not recruiting | KRAS peptide vaccine, Nivolumab (PD-1), Ipilimumab (CTLA-4) | 8/11 patients mounted a >5–fold increase in IFNγ-producing mKRAS-specific T cells post vaccination; Improved DFS compared to non-responders (not reached vs. 2.8 mo; p = 0.045). | [52] |
| Neoantigen Peptide Vaccines | ||||||
| NCT03558945 | Clinical Trial to Evaluate Safety and Effect of Personalized Neoantigen Vaccine for Pancreatic Tumor Following Surgical Resection and Adjuvant Chemotherapy | Ib | Recruiting | Neoantigen peptide vaccine | 3-year RFS rate 56%; 3-year OS rate 74% Expansion of cytotoxic CD8+ T cells at the priming phase with CD4+ T cells during boosting phase. Helper B-cell subtype which interacted with T cells in patients associated with prognosis | [53] |
| NCT04810910 | Clinical Study of a Personalized Neoantigen Vaccine in Pancreatic Cancer Patients Following Surgical Resection and Adjuvant Chemotherapy | I | Recruiting | Neoantigen peptide vaccine, GM-CSF | N/A | N/A |
| NCT06344156 | Adjuvant Therapy of Neoantigen Vaccine Plus Anti-PD-1 and Chemotherapy in Patients With Resected Pancreatic Cancer | I | Recruiting | Neoantigen peptide vaccine, Gemcitabine, Capecitabine, Tislelizumab (PD-1) | N/A | N/A |
| Heat Shock Protein Peptide Complex | ||||||
| Maki et al., 2007 | A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma | I | Completed | Autologous HSPPC-96 (gp96, Oncophage) | Median OS 2.2 years. Autologous-HSPPC-96 ELISpot reactivity increased significantly in 1/5 patients, no observed correlation between immune response and prognosis | [54] |
| Dendritic Cell Vaccines | ||||||
| Lepisto et al., 2008 | A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors | I/II | Completed | MUC1 peptide-loaded dendritic cell vaccine | 4/12 patients alive without evidence of recurrence >4 years | [55] |
| Lau et al., 2022 | Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: A phase I study | I | Completed | Allogeneic tumor lysate-loaded autologous monocyte-derived dendritic cell vaccine | 7/10 patients without disease recurrence of progression at median follow up of 25 months. Following vaccination peripheral blood with increased memory CD4+ T cells expressing Ki67+PD-1+ (phenotype seen in patients with improved survival following anti-PD-1 in NSCLC) | [56,57] |
| NCT04157127 | Phase I Study of Th-1 Dendritic Cell Immunotherapy in Combination with Standard Chemotherapy for the Adjuvant Treatment of Pancreatic Adenocarcinoma (DECIST) | I | Active, not recruiting | Autologous DC vaccine | N/A | N/A |
| NCT04627246 | A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine and Standard Of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma | Ib | Recruiting | Autologous Dendritic Cell Vaccine Loaded with Personalized Peptides (PEP-DC vaccine) | N/A | [58] |
| NCT03592888 | Pilot Study of Mature Dendritic Cell Vaccination Against Mutated KRAS in Patients With Resectable Pancreatic Cancer | I | Completed | Mature dendritic cell (mDC3/8) vaccine (primer and booster) | Median time of follow up of 25.3 months, 5 patients were without evidence of tumor recurrence. 6/9 (67%) patients generated mutant KRAS specific T cell responses | [59] |
| mRNA Vaccines | ||||||
| NCT04161755 | Phase 1 Clinical Trial of Personalized Neoantigen Tumor Vaccines and Programmed Death-Ligand I (PD-L1) Blockade in Patients With Surgically Resected Pancreatic Cancer | I | Active, not recruiting | mRNA neoantigen vaccine, Atezolizumab (PD-L1), mFOLFIRINOX | Responders vs. non-responders (mRFS not reached vs. 13.4 months (p = 0.003). In responders, vaccination expanded multiple clones (median 7.5 clones) from undetectable levels to up to 10% (median 2.8%). | [60] |
| NCT05968326 | A Phase II, Open-Label, Multicenter, Randomized Study of the Efficacy and Safety of Adjuvant Autogene Cevumeran Plus Atezolizumab and mFOLFIRINOX Versus mFOLFIRINOX Alone in Patients With Resected Pancreatic Ductal Adenocarcinoma | II | Recruiting | mRNA neoantigen vaccine, Atezolizumab (PD-L1), mFOLFIRINOX | N/A | N/A |
| NCT06496373 | Clinical Study of XP-004 Personalized mRNA Tumor Vaccine Combined With PD-1 Inhibitor for Postoperative Adjuvant Therapy for Pancreatic Cancer in Patients With Advanced Solid Tumors | I | Recruiting | mRNA vaccine, PD-1 inhibitor | N/A | N/A |
| NCT06353646 | Efficacy and Safety Trial of XH001 (Neoantigen Cancer Vaccine) Sequential Combination With Ipilimumab and Chemotherapy for Patients With Resected Pancreatic Cancer | N/A | Not yet recruiting | mRNA neoantigen vaccine, Ipilimumab (CTLA-4), Gemcitabine + Capecitabine | N/A | N/A |
| Adoptive Cellular Therapies | ||||||
| NCT05911217 | An Open-label, Single-arm, Multicenter, Phase Ib Clinical Trial to Evaluate the Efficacy and Safety of CT041 Autologous CAR T Cell Injection After Adjuvant Chemotherapy in Subjects With Pancreatic Cancer | I | Recruiting | CT041 autologous CAR T-cell injection | N/A | N/A |
| NCT06730009 | A Dose-Finding Phase I Followed by a Phase II Study to Evaluate the Safety and Efficacy of Allogeneic NK-cell Combined with Chemotherapy in Patients with PDA or Cholangiocarcinoma After Surgery | I/II | Recruiting | S-1, leucovorin, oxaliplatin, and gemcitabine (SLOG) + Allogeneic NK cell | N/A | N/A |
| NCT04969731 | An Open-label, Randomized, Multi-center, Parallel, Phase III Clinical Trial to Evaluate the Efficacy and Safety of Adjuvant Immuncell-LC Therapy Combined With Gemcitabine Versus Adjuvant Gemcitabine Single Therapy After Resection in Patients With Pancreatic Ductal Adenocarcinoma | III | Recruiting | Autologous Cytokine Induced Killer Cells, Gemcitabine | N/A | N/A |
| Stromal-targeting Therapies | ||||||
| NCT03727880 | A Randomized Phase II Study of Pembrolizumab With or Without Defactinib, a Focal Adhesion Kinase Inhibitor Following Chemotherapy as a Neoadjuvant and Adjuvant Treatment for Resectable Pancreatic Ductal Adenocarcinoma (PDAC) | II | Recruiting | Pembrolizumab (PD-1), Defactinib (FAK) | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, K.-L.; Azad, N.S. Immune-Based Strategies for Pancreatic Cancer in the Adjuvant Setting. Cancers 2025, 17, 1246. https://doi.org/10.3390/cancers17071246
Liang K-L, Azad NS. Immune-Based Strategies for Pancreatic Cancer in the Adjuvant Setting. Cancers. 2025; 17(7):1246. https://doi.org/10.3390/cancers17071246
Chicago/Turabian StyleLiang, Kai-Li, and Nilofer S. Azad. 2025. "Immune-Based Strategies for Pancreatic Cancer in the Adjuvant Setting" Cancers 17, no. 7: 1246. https://doi.org/10.3390/cancers17071246
APA StyleLiang, K.-L., & Azad, N. S. (2025). Immune-Based Strategies for Pancreatic Cancer in the Adjuvant Setting. Cancers, 17(7), 1246. https://doi.org/10.3390/cancers17071246