
Inflammatory Pathways to Carcinogenesis: Deciphering the Rheumatoid Arthritis–Lung Cancer Connection
 , , and
, , and
Simple Summary
Abstract
1. Introduction
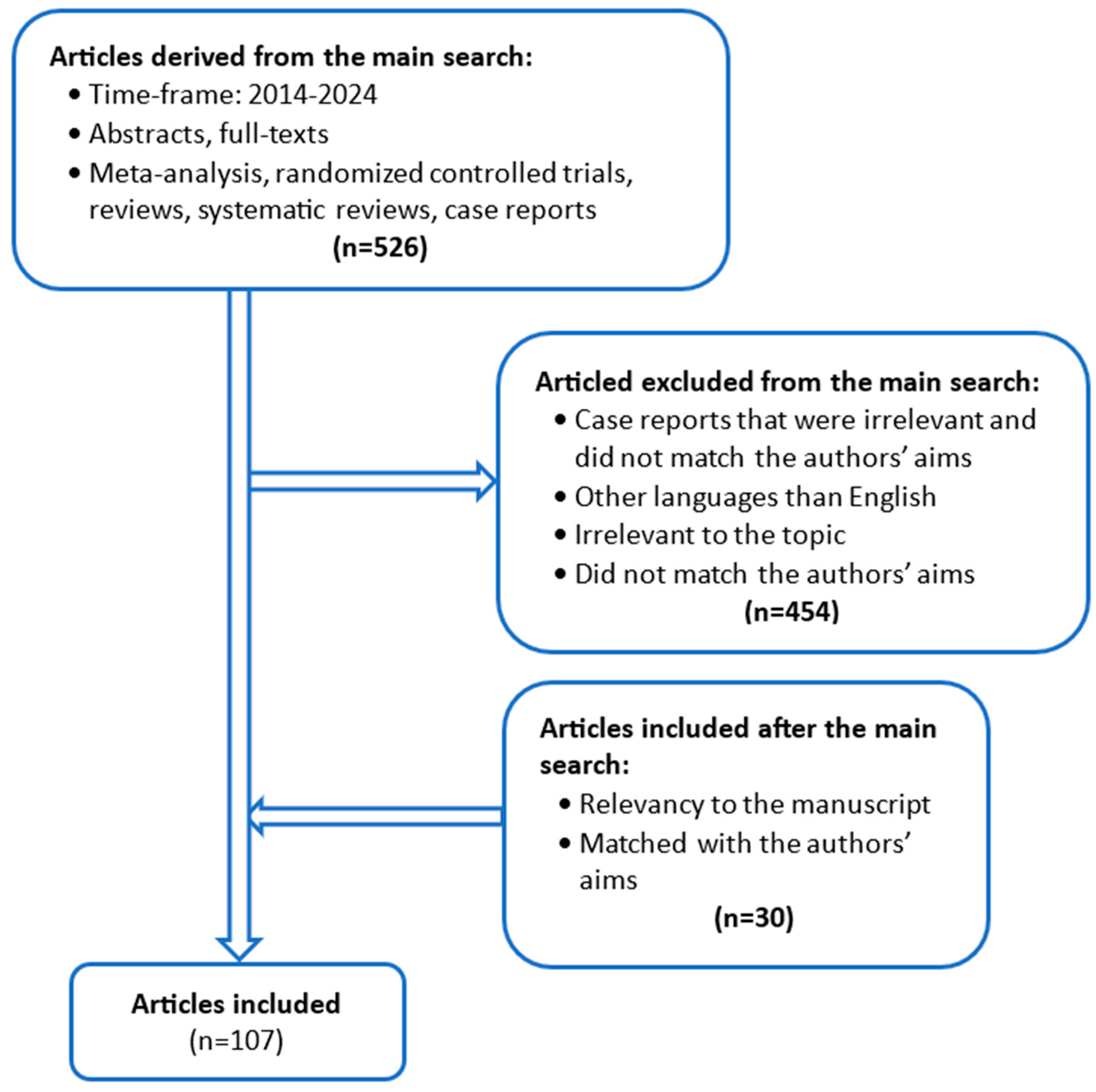
2. Materials and Methods
3. Epidemiology
4. Molecular Mechanism of Lung Cancer Development in RA Patients
5. Drug-Induced Lung Cancer in RA Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lung Cancer Risk in RA Patients During Synthetic and Biological Drug Therapy | |||||
|---|---|---|---|---|---|
| Synthetic DMARD | Medication | Drug | Analyzed Group with RA | Number of Patients with Lung Cancer/Calculated Risk | Ref. Number |
| conventional | Methotrexate(MTX) |
|
| [21] | |
| Leflunomide(LF) |
|
| [50] | ||
| Sulfasalazine (SFN) | N/A | N/A | N/A | ||
| Chloroquine (CQ)/Hydroxychloroquine (HCQ) |
|
| [51] | ||
| target | Tofacitinib (TOFA) |
|
| [52] | |
| Baricitinib (BARI) |
|
| [53] | ||
| Upadacitinib (UPA) |
|
| [54] | ||
| biologic | anti-cytokine | Adalimumab(ADA) |
|
| [54] |
| Anakinra |
|
| [55] | ||
| Certolizumab(CERTO) |
|
| [56] | ||
| Etanercept(ETA) |
|
| [57] | ||
| Golimumab (GLM) |
|
| [58] | ||
| Infliximab(IFX) |
|
| [59] | ||
| Whole-group anti-TNFα inhibitors |
|
| [59] | ||
| TNF inhibitors (ADA, CERTO, ETA, GLM, IFX) |
|
| [60] | ||
| Tocilizumab |
|
| [60] | ||
| Sarilumab |
|
| N/A | ||
| non-anti-cytokine | Abatacept |
|
| [61] | |
| Rituximab |
|
| [62] | ||
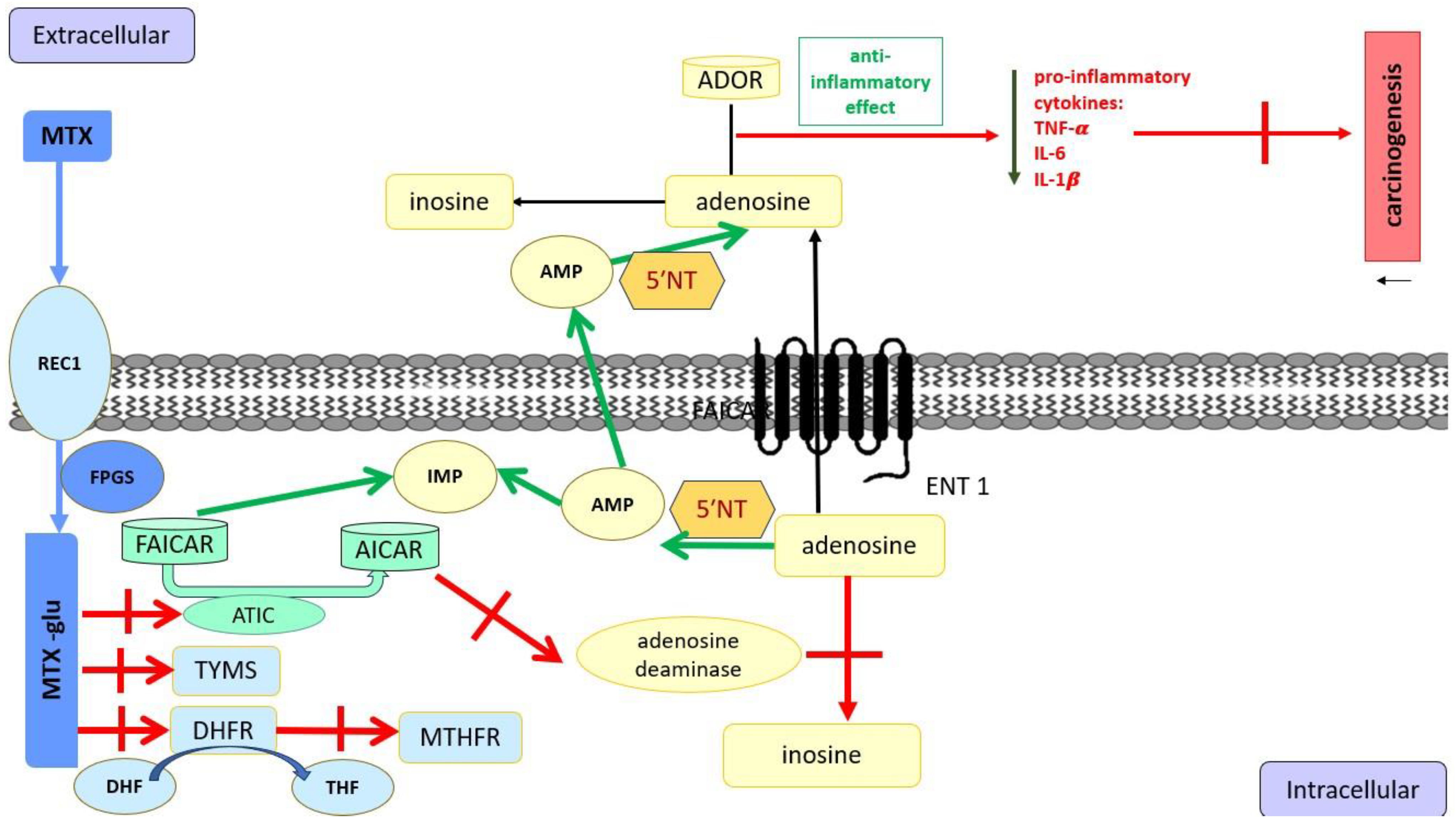
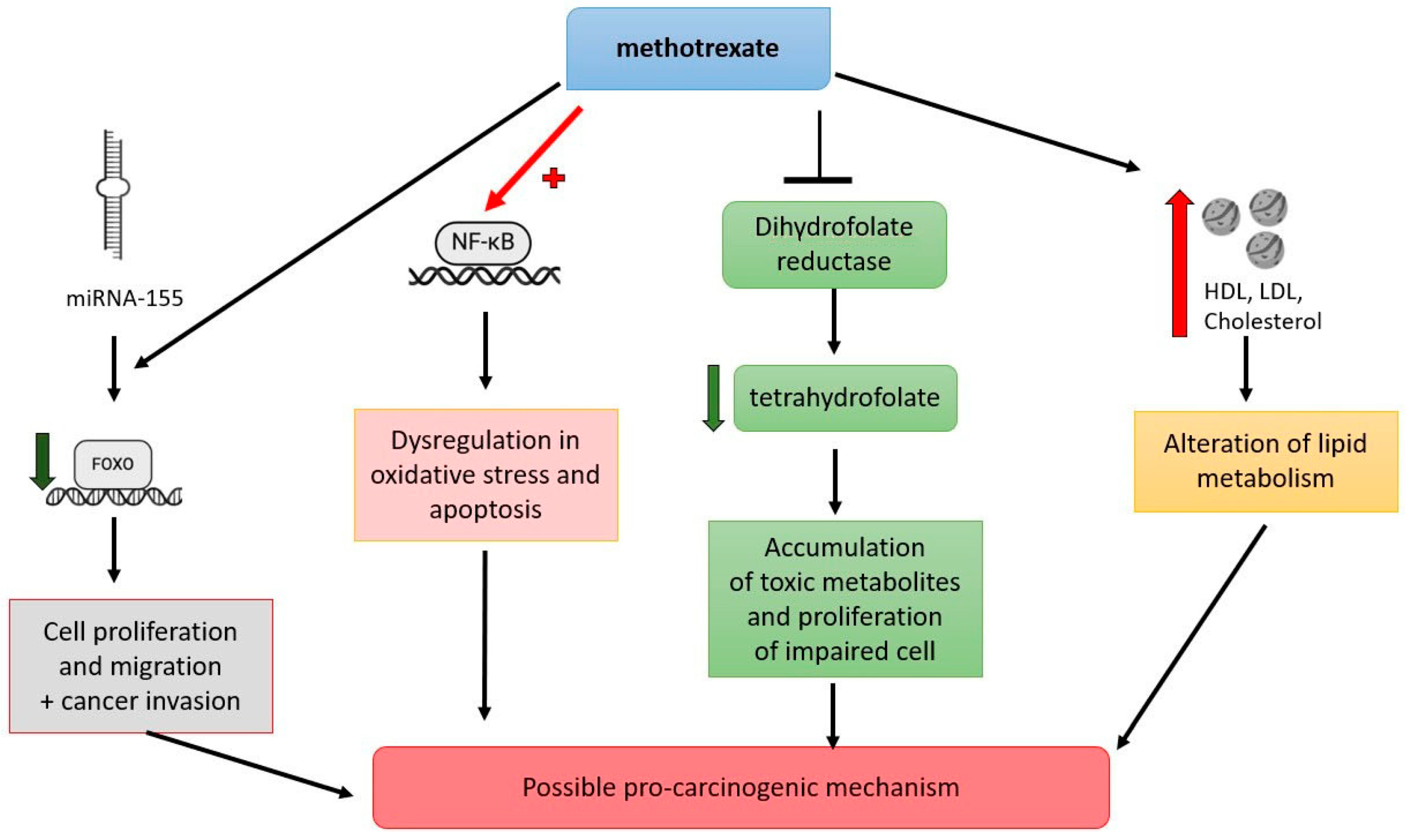
5.1. Methotrexate
5.2. Leflunomide
5.3. Sulfasalazine
5.4. Antimalarial Medications
5.5. JAK Inhibitors
6. Biologic Therapy
TNFα Inhibitors
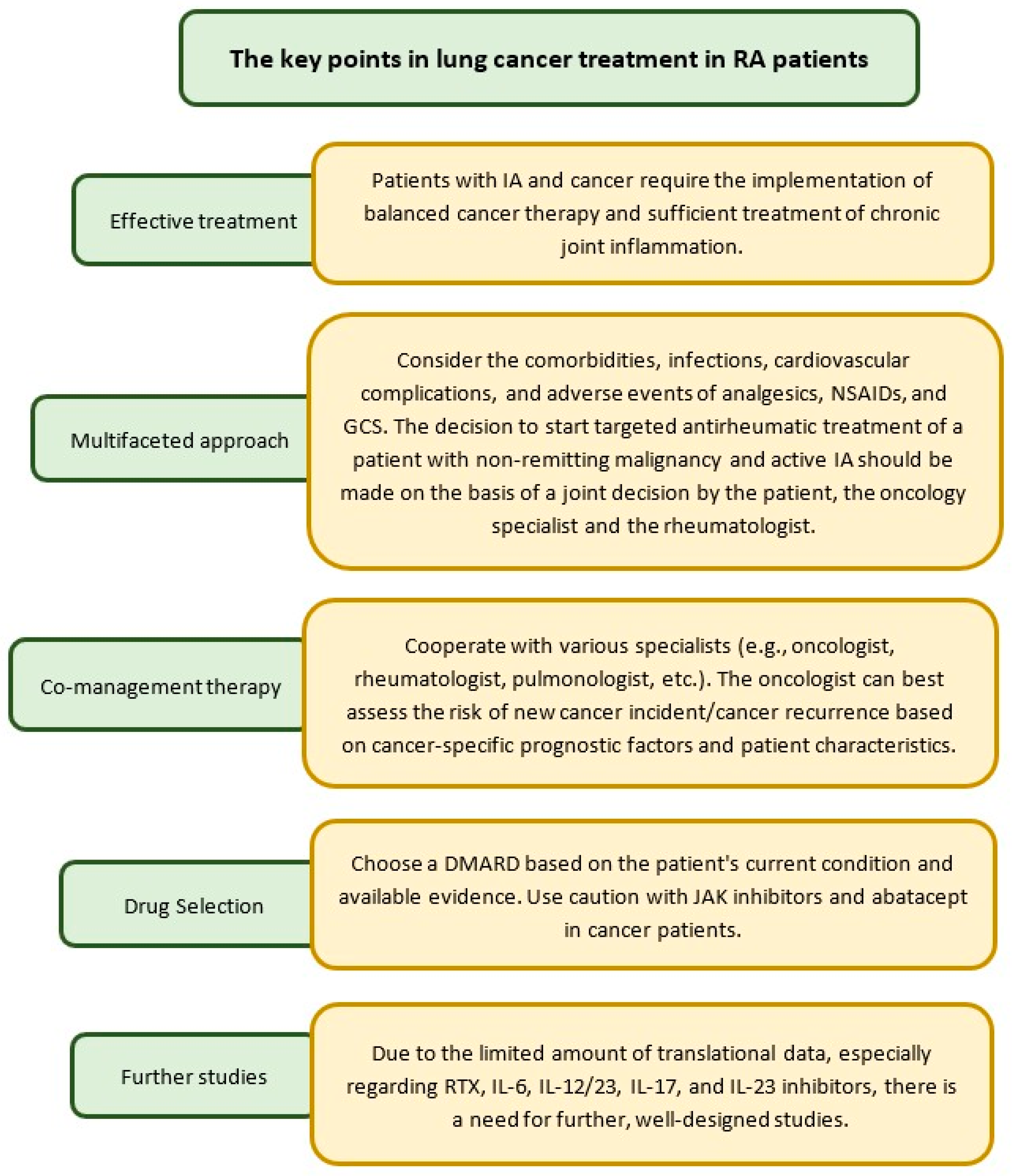
7. Treatment of Lung Cancer in RA Patients
8. Prevention of Lung Cancer in RA Patients Treated with Conventional DMARDs and Biological DMARDs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADOR | 4 G-protein coupled receptor |
| aHR | Adjusted hazard ratio |
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| cAMP | Cyclic adenosine monophosphate |
| cHR | Crude hazard ratio |
| CI | Confidence interval |
| CIR | Crude incidence ratio |
| csDMARDs | Conventional synthetic DMARDs |
| CQ | Chloroquine |
| DMARDs | Disease-modifying anti-rheumatic drugs |
| EAIR | Exposure-adjusted incidence rate |
| ENT1 | Equilibrative nucleoside transporter 1 |
| FAICAR | 5-form-aminoimidazole-4-carboxamide ribonucleotide |
| FOXO | Forkhead box O |
| NF-kB | Nuclear factor kappa B |
| GCS | Glucocorticosteroid |
| GLM | Golimumab |
| GLM-IV | Golimumab intravenous |
| HCQ | Hydroxychloroquine |
| HDL | High-density lipoprotein |
| HDMTX | Methotrexate is used in high doses |
| i.v. | Intravenously |
| IA | Inflammatory arthritis |
| IFN-γ | Interferon-gamma |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin -6 |
| IR | Incidence ratio |
| JAK | Janus kinase |
| JAKi | Janus kinase inhibitor |
| JIA | Juvenile idiopathic arthritis |
| LDL | Low-density lipoprotein |
| LEF | Leflunomide |
| LORA | Late-onset rheumatoid arthritis |
| miRNA155 | Micro RNA 155 |
| MTX | Methotrexate |
| N/A | Not available or not reported |
| NF-kB | Nuclear factor kappa B |
| NSAIDs | Non-steroidal anti-inflammatory arthritis |
| NSLC | Non-small lung cancer |
| RA | Rheumatoid arthritis |
| RCTs | Randomized control trials |
| RTX | Rituximab |
| s.c. | Subcutaneously |
| SCC | Squamous cell carcinoma |
| SCLC | Small cell lung cancer |
| SIR | Standardized incidence ratio |
| SN | Sulfasalazine |
| SR-B1 | Scavenger receptor class B type 1 |
| TAAs | Tumor-associated antigens |
| TNF-α | Tumor necrosis factor-alpha |
| tsDMARD | Targeted synthetic DMARD |
| VEGF | Vascular endothelial growth factor |
References
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid Arthritis: Pathological Mechanisms and Modern Pharmacologic Therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Shah, A.A. The Relationships Between Cancer and Autoimmune Rheumatic Diseases. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101472. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of Lung Cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, J.; You, Z.; Tu, H.; He, P.; Li, J.; Gao, R.; Liu, Z.; Xi, Z.; Li, Z.; et al. Cancer Risks in Rheumatoid Arthritis Patients Who Received Immunosuppressive Therapies: Will Immunosuppressants Work? Front. Immunol. 2022, 13, 1050876. [Google Scholar] [CrossRef]
- Chatzidionysiou, K.; di Giuseppe, D.; Soderling, J.; Catrina, A.; Askling, J. Risk of Lung Cancer in Rheumatoid Arthritis and in Relation to Autoantibody Positivity and Smoking. RMD Open 2022, 8, e002465. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Gomez, I.; Soós, B.; Bodoki, L.; Szamosi, S.; András, C.; Juhász, B.; Váróczy, L.; Antal-Szalmás, P.; Szodoray, P.; et al. Eight Pillars of Oncorheumatology: Crossroads Between Malignancies and Musculoskeletal Diseases. Autoimmun. Rev. 2020, 19, 102658. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung Cancer: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The International Epidemiology of Lung Cancer: Geographical Distribution and Secular Trends. J. Thorac. Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The Epidemiology of Lung Cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung Cancer Epidemiology: Contemporary and Future Challenges Worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Pathology of Lung Cancer. Clin. Chest Med. 2011, 32, 669–692. [Google Scholar] [CrossRef]
- Abásolo, L.; Júdez, E.; Descalzo, M.A.; González-Alvaro, I.; Jover, J.A.; Carmona, L.; EMECAR Study Group. Cancer in Rheumatoid Arthritis: Occurrence, Mortality, and Associated Factors in a South European Population. Semin. Arthritis Rheum. 2008, 37, 388–397. [Google Scholar] [CrossRef]
- Khurana, R.; Wolf, R.; Berney, S.; Caldito, G.; Hayat, S.; Berney, S.M. Risk of Development of Lung Cancer Is Increased in Patients with Rheumatoid Arthritis: A Large Case Control Study in US Veterans. J. Rheumatol. 2008, 35, 1704–1708. [Google Scholar]
- Simon, T.A.; Thompson, A.; Gandhi, K.K.; Hochberg, M.C.; Suissa, S. Incidence of Malignancy in Adult Patients with Rheumatoid Arthritis: A Meta-Analysis. Arthritis Res. Ther. 2015, 17, 212. [Google Scholar] [CrossRef]
- De Cock, D.; Hyrich, K. Malignancy and Rheumatoid Arthritis: Epidemiology, Risk Factors and Management. Best Pract. Res. Clin. Rheumatol. 2018, 32, 869–886. [Google Scholar] [CrossRef]
- Morand, S.; Staats, H.; Creeden, J.F.; Iqbal, A.; Kahaleh, B.; Stanbery, L.; Dworkin, L.; Nemunaitis, J. Molecular Mechanisms Underlying Rheumatoid Arthritis and Cancer Development and Treatment. Future Oncol. 2020, 16, 483–495. [Google Scholar] [CrossRef]
- Smitten, A.L.; Simon, T.A.; Hochberg, M.C.; Suissa, S. A Meta-Analysis of the Incidence of Malignancy in Adult Patients with Rheumatoid Arthritis. Arthritis Res. Ther. 2008, 10, R45. [Google Scholar] [CrossRef]
- Buchbinder, R.; Barber, M.; Heuzenroeder, L.; Wluka, A.E.; Giles, G.; Hall, S.; Harkness, A.; Lewis, D.; Littlejohn, G.; Miller, M.H.; et al. Incidence of Melanoma and Other Malignancies Among Rheumatoid Arthritis Patients Treated with Methotrexate. Arthritis Rheum. 2008, 59, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Hazlett, J.; Roberts, R.L.; Frampton, C.; Highton, J.; Hessian, P.A. Adenosine Receptor Expression in Rheumatoid Synovium: A Basis for Methotrexate Action. Arthritis Res. Ther. 2012, 14, R138. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Geng, W.; Han, L.; Song, R.; Qu, Q.; Chen, X.; Luo, X. Pro-Carcinogenic Actions of miR-155/FOXO3a in Colorectal Cancer Development. Cell Mol. Biol. 2023, 69, 160–165. [Google Scholar] [CrossRef]
- Wani, F.A.; Ibrahim, M.A.; Ameen, S.H.; Farage, A.E.; Ali, Z.A.-E.; Saleh, K.; Farag, M.M.; Sayeed, M.U.; Alruwaili, M.A.Y.; Alruwaili, A.H.F.; et al. Platelet Rich Plasma and Adipose-Derived Mesenchymal Stem Cells Mitigate Methotrexate-Induced Nephrotoxicity in Rat via Nrf2/Pparγ/HO-1 and NF-Κb/Keap1/Caspase-3 Signaling Pathways: Oxidative Stress and Apoptosis Interplay. Toxics 2023, 11, 398. [Google Scholar] [CrossRef]
- Østergaard, A.; Bohnstedt, C.; Grell, K.; Degn, M.; Zeller, B.; Taskinen, M.; Hafsteinsdottir, S.; Björgvinsdóttir, H.; Heyman, M.; Hoogerbrugge, P.; et al. Acute Lymphoblastic Leukemia and down Syndrome: 6-Mercaptopurine and Methotrexate Metabolites During Maintenance Therapy. Leukemia 2021, 35, 863–866. [Google Scholar] [CrossRef]
- Wang, B.; Deng, H.; Hu, Y.; Han, L.; Huang, Q.; Fang, X.; Yang, K.; Wu, S.; Zheng, Z.; Yawalkar, N.; et al. The Difference of Lipid Profiles Between Psoriasis with Arthritis and Psoriasis Without Arthritis and Sex-Specific Downregulation of Methotrexate on the Apolipoprotein B/Apolipoprotein A-1 Ratio. Arthritis Res. Ther. 2022, 24, 17. [Google Scholar] [CrossRef]
- Leger, D.Y.; Liagre, B.; Beneytout, J.L. Low Dose Leflunomide Activates PI3K/Akt Signalling in Erythroleukemia Cells and Reduces Apoptosis Induced by Anticancer Agents. Apoptosis 2006, 11, 1747–1760. [Google Scholar] [CrossRef]
- Spodnik, J.H.; Wozniak, M.; Budzko, D.; Teranishi, M.-A.; Karbowski, M.; Nishizawa, Y.; Usukura, J.; Wakabayashi, T. Mechanism of Leflunomide-Induced Proliferation of Mitochondria in Mammalian Cells. Mitochondrion 2002, 2, 163–179. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Ponneri Babuharisankar, A.; Lin, Y.-C.; Lien, H.-W.; Lo, Y.K.; Chou, H.-Y.; Tangeda, V.; Cheng, L.-C.; Cheng, A.N.; Lee, A.Y.-L. Mitochondrial Oxidative Stress in the Tumor Microenvironment and Cancer Immunoescape: Foe or Friend? J. Biomed. Sci. 2022, 29, 74. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Koul, A.M.; Wani, U.M.; Farooq, F.; Amin, B.; Wani, Z.; Lone, A.; Qadri, A.; Qadri, R.A. Dissection of Paracrine/Autocrine Interplay in Lung Tumor Microenvironment Mimicking Cancer Cell-Monocyte Co-Culture Models Reveals Proteins That Promote Inflammation and Metastasis. BMC Cancer 2023, 23, 926. [Google Scholar] [CrossRef] [PubMed]
- Daghestani, M.H.; Hakami, H.H.; Ambreen, K.; Saleem, A.M.; Aleisa, N.A.; Al Neghery, L.M.; Amin, M.H. Assessment of Tumor-Suppressive Inflammatory Tendency of Echis coloratus Venom Against Colon Carcinogenesis, via up-Regulation of Anti-Tumorigenic Inflammatory Cells and down-Regulation of pro-Tumorigenic Inflammatory Cells, in Colon Cancer Cell Lines. Toxin Rev. 2022, 41, 11–24. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Baldwin, A.S. Series Introduction: The Transcription Factor NF-κB and Human Disease. J. Clin. Investig. 2001, 107, 3–6. [Google Scholar] [CrossRef]
- Robles-Escajeda, E.; Lerma, D.; Nyakeriga, A.M.; Ross, J.A.; Kirken, R.A.; Aguilera, R.J.; Varela-Ramirez, A. Searching in Mother Nature for Anti-Cancer Activity: Anti-Proliferative and Pro-Apoptotic Effect Elicited by Green Barley on Leukemia/Lymphoma Cells. PLoS ONE 2013, 8, e73508. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef]
- Klein, K.; Stoiber, D.; Sexl, V.; Witalisz-Siepracka, A. Untwining Anti-Tumor and Immunosuppressive Effects of JAK Inhibitors—A Strategy for Hematological Malignancies? Cancers 2021, 13, 2611. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Buch, M.H.; Charles-Schoeman, C.; Galloway, J.; Karpouzas, G.A.; Kristensen, L.E.; Ytterberg, S.R.; Hamar, A.; Fleischmann, R. Efficacy and Safety of JAK Inhibitors in Rheumatoid Arthritis: Update for the Practising Clinician. Nat. Rev. Rheumatol. 2024, 20, 101–115. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. STAT3-Induced WNT5A Signaling Loop in Embryonic Stem Cells, Adult Normal Tissues, Chronic Persistent Inflammation, Rheumatoid Arthritis and Cancer (Review). Int. J. Mol. Med. 2007, 19, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Cici, D.; Corrado, A.; Rotondo, C.; Cantatore, F.P. Wnt Signaling and Biological Therapy in Rheumatoid Arthritis and Spondyloarthritis. Int. J. Mol. Sci. 2019, 20, 5552. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2022 Update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef]
- Howard, S.C.; McCormick, J.; Pui, C.-H.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef]
- Widemann, B.C.; Adamson, P.C. Understanding and Managing Methotrexate Nephrotoxicity. Oncologist 2006, 11, 694–703. [Google Scholar] [CrossRef]
- Linker, C.; Damon, L.; Ries, C.; Navarro, W. Intensified and Shortened Cyclical Chemotherapy for Adult Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2002, 20, 2464–2471. [Google Scholar] [CrossRef]
- Thomas, D.A.; O’Brien, S.; Cortes, J.; Giles, F.J.; Faderl, S.; Verstovsek, S.; Ferrajoli, A.; Koller, C.; Beran, M.; Pierce, S.; et al. Outcome with the Hyper-CVAD Regimens in Lymphoblastic Lymphoma. Blood 2004, 104, 1624–1630. [Google Scholar] [CrossRef]
- Kvacskay, P.; Blank, N.; Lorenz, H.-M.; Merkt, W. Leflunomide in Combination with JAK Inhibitors in the Treatment of Rheumatoid Arthritis: A Case Series. Rheumatology 2022, 61, e280–e281. [Google Scholar] [CrossRef]
- Mao, I.-C.; Lin, C.-Y.; Wu, C.-L.; Kor, C.-T.; Chang, C.-C. Hydroxychloroquine and Risk of Development of Cancers: A Nationwide Population-Based Cohort Study. Ther. Clin. Risk Manag. 2018, 14, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Yamaoka, K.; Chen, Y.-H.; Bhatt, D.L.; Gunay, L.M.; Sugiyama, N.; Connell, C.A.; Wang, C.; Wu, J.; Menon, S.; et al. Malignancy Risk with Tofacitinib versus TNF Inhibitors in Rheumatoid Arthritis: Results from the Open-Label, Randomised Controlled ORAL Surveillance Trial. Ann. Rheum. Dis. 2023, 82, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Takeuchi, T.; Burmester, G.R.; Durez, P.; Smolen, J.S.; Deberdt, W.; Issa, M.; Terres, J.R.; Bello, N.; Winthrop, K.L. Safety of Baricitinib for the Treatment of Rheumatoid Arthritis over a Median of 4.6 and up to 9.3 Years of Treatment: Final Results from Long-Term Extension Study and Integrated Database. Ann. Rheum. Dis. 2022, 81, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Mysler, E.; Bessette, L.; Peterfy, C.G.; Durez, P.; Tanaka, Y.; Swierkot, J.; Khan, N.; Bu, X.; Li, Y.; et al. Long-Term Safety and Efficacy of Upadacitinib or Adalimumab in Patients with Rheumatoid Arthritis: Results through 3 Years from the SELECT-COMPARE Study. RMD Open 2022, 8, e002012. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Tesser, J.; Schiff, M.H.; Schechtman, J.; Burmester, G.-R.; Bennett, R.; Modafferi, D.; Zhou, L.; Bell, D.; Appleton, B. Safety of Extended Treatment with Anakinra in Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2006, 65, 1006–1012. [Google Scholar] [CrossRef]
- Harrold, L.R.; Litman, H.J.; Saunders, K.C.; Dandreo, K.J.; Gershenson, B.; Greenberg, J.D.; Low, R.; Stark, J.; Suruki, R.; Jaganathan, S.; et al. One-Year Risk of Serious Infection in Patients Treated with Certolizumab Pegol as Compared with Other TNF Inhibitors in a Real-World Setting: Data from a National U.S. Rheumatoid Arthritis Registry. Arthritis Res. Ther. 2018, 20, 2. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Bathon, J.M.; Kremer, J.M.; Fleischmann, R.M.; Schiff, M.H.; Martin, R.W.; Baumgartner, S.W.; Park, G.S.; Mancini, E.L.; Genovese, M.C. Safety and Efficacy of Etanercept beyond 10 Years of Therapy in North American Patients with Early and Longstanding Rheumatoid Arthritis. Arthritis Care Res. 2011, 63, 373–382. [Google Scholar] [CrossRef]
- Rahman, P.; Baer, P.; Keystone, E.; Choquette, D.; Thorne, C.; Haraoui, B.; Chow, A.; Faraawi, R.; Olszynski, W.; Kelsall, J.; et al. Long-Term Effectiveness and Safety of Infliximab, Golimumab and Golimumab-IV in Rheumatoid Arthritis Patients from a Canadian Prospective Observational Registry. BMC Rheumatol. 2020, 4, 46. [Google Scholar] [CrossRef]
- Mercer, L.K.; Lunt, M.; Low, A.L.S.; Dixon, W.G.; Watson, K.D.; Symmons, D.P.M.; Hyrich, K.L.; Consortium, B.C.C. Risk of Solid Cancer in Patients Exposed to Anti-Tumour Necrosis Factor Therapy: Results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Ann. Rheum. Dis. 2015, 74, 1087–1093. [Google Scholar] [CrossRef]
- Huss, V.; Bower, H.; Wadström, H.; Frisell, T.; Askling, J.; The ARTIS Group. Short- and Longer-Term Cancer Risks with Biologic and Targeted Synthetic Disease-Modifying Antirheumatic Drugs as Used Against Rheumatoid Arthritis in Clinical Practice. Rheumatology 2022, 61, 1810–1818. [Google Scholar] [CrossRef]
- Simon, T.A.; Smitten, A.L.; Franklin, J.; Askling, J.; Lacaille, D.; Wolfe, F.; Hochberg, M.C.; Qi, K.; Suissa, S. Malignancies in the Rheumatoid Arthritis Abatacept Clinical Development Programme: An Epidemiological Assessment. Ann. Rheum. Dis. 2009, 68, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Furst, D.E.; Kirchner, P.; Melega, S.; Lacey, S.; Lehane, P.B. Risk of Malignancies in Patients with Rheumatoid Arthritis Treated with Rituximab: Analyses of Global Postmarketing Safety Data and Long-Term Clinical Trial Data. Rheumatol. Ther. 2019, 7, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Ronda, N.; Greco, D.; Adorni, M.P.; Zimetti, F.; Favari, E.; Hjeltnes, G.; Mikkelsen, K.; Borghi, M.O.; Favalli, E.G.; Gatti, R.; et al. Newly Identified Antiatherosclerotic Activity of Methotrexate and Adalimumab: Complementary Effects on Lipoprotein Function and Macrophage Cholesterol Metabolism. Arthritis Rheumatol. 2015, 67, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Jimenez, N.A.; Garcia-Gonzalez, C.E.; Ayala-Lopez, K.P.; Trujillo-Hernandez, B.; Aguilar-Chavez, E.A.; Rocha-Muñoz, A.D.; Vasquez-Jimenez, J.C.; Olivas-Flores, E.; Salazar-Paramo, M.; Corona-Sanchez, E.G.; et al. Modifications in Lipid Levels Are Independent of Serum TNF-α in Rheumatoid Arthritis: Results of an Observational 24-Week Cohort Study Comparing Patients Receiving Etanercept Plus Methotrexate or Methotrexate as Monotherapy. BioMed Res. Int. 2014, 2014, 510305. [Google Scholar] [CrossRef]
- Navarro-Millán, I.; Charles-Schoeman, C.; Yang, S.; Bathon, J.M.; Bridges, S.L.; Chen, L.; Cofield, S.S.; Dell’Italia, L.J.; Moreland, L.W.; O’Dell, J.R.; et al. Changes in Lipoproteins Associated with Treatment with Methotrexate or Combination Therapy in Early Rheumatoid Arthritis: Results from the TEAR Trial. Arthritis Rheum. 2013, 65, 1430–1438. [Google Scholar] [CrossRef]
- Borgquist, S.; Butt, T.; Almgren, P.; Shiffman, D.; Stocks, T.; Orho-Melander, M.; Manjer, J.; Melander, O. Apolipoproteins, Lipids and Risk of Cancer. Int. J. Cancer 2016, 138, 2648–2656. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid Metabolism and Cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Feng, H.; Wang, M.; Wu, C.; Yu, J.; Wang, D.; Ma, J.; Han, J. High Scavenger Receptor Class B Type I Expression Is Related to Tumor Aggressiveness and Poor Prognosis in Lung Adenocarcinoma: A STROBE Compliant Article. Medicine 2018, 97, e0203. [Google Scholar] [CrossRef]
- Mooberry, L.K.; Sabnis, N.A.; Panchoo, M.; Nagarajan, B.; Lacko, A.G. Targeting the SR-B1 Receptor as a Gateway for Cancer Therapy and Imaging. Front. Pharmacol. 2016, 7, 466. [Google Scholar] [CrossRef]
- Kremer, J.M. Is Methotrexate Oncogenic in Patientswith Rheumatoid Arthritis? Semin. Arthritis Rheum. 1997, 26, 785–787. [Google Scholar] [CrossRef]
- Solomon, D.H.; Kremer, J.M.; Fisher, M.; Curtis, J.R.; Furer, V.; Harrold, L.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Greenberg, J.D. Comparative Cancer Risk Associated with Methotrexate, Other Non-Biologic and Biologic Disease-Modifying Anti-Rheumatic Drugs. Semin. Arthritis Rheum. 2014, 43, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Sekimura, A.; Iwai, S.; Funasaki, A.; Motono, N.; Usuda, K.; Uramoto, H. Lung Cancer Combined with Methotrexate-Associated Lymphoproliferative Disorder: A Case Report. Int. J. Surg. Case Rep. 2019, 59, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Chehade, L.; Jaafar, Z.A.; El Masri, D.; Zmerly, H.; Kreidieh, D.; Tannir, H.; Itani, L.; El Ghoch, M. Lifestyle Modification in Rheumatoid Arthritis: Dietary and Physical Activity Recommendations Based on Evidence. Curr. Rheumatol. Rev. 2019, 15, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Cannon, G.W.; Kremer, J.M. Leflunomide. Rheum. Dis. Clin. N. Am. 2004, 30, 295–309. [Google Scholar] [CrossRef]
- Price, S. The Threat of Leflunomide-Induced Lung Disease. Nat. Rev. Rheumatol. 2009, 5, 294. [Google Scholar] [CrossRef]
- Behrens, F.; Koehm, M.; Burkhardt, H. Update 2011: Leflunomide in Rheumatoid Arthritis—Strengths and Weaknesses. Curr. Opin. Rheumatol. 2011, 23, 282–287. [Google Scholar] [CrossRef]
- Maskey-Warzęchowska, M.; Karwat, K.; Szczepankiewicz, B.; Langfort, R.; Szołkowska, M.; Chazan, R. Leflunomide-Induced Acute Interstitial Pneumonia in a Patient Treated for Rheumatoid Arthritis. Adv. Respir. Med. 2015, 83, 50–54. [Google Scholar] [CrossRef]
- Yoshikawa, G.T.; Dias, G.A.d.S.; Fujihara, S.; Silva, L.F.E.; Cruz, L.D.B.P.; Fuzii, H.T.; Koyama, R.V.L. Formation of Multiple Pulmonary Nodules During Treatment with Leflunomide. J. Bras. Pneumol. 2015, 41, 281–284. [Google Scholar] [CrossRef]
- Plosker, G.L.; Croom, K.F. Sulfasalazine: A Review of Its Use in the Management of Rheumatoid Arthritis. Drugs 2005, 65, 1825–1849. [Google Scholar] [CrossRef]
- Bullock, J.; Rizvi, S.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Parry, S.D.; Barbatzas, C.; Peel, E.T.; Barton, J.R. Sulphasalazine and Lung Toxicity. Eur. Respir. J. 2002, 19, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Kerget, B.; Araz, O.; Ucar, E.Y.; Aydin, O.; Akgun, M.; Saglam, L. Sulfasalazine-Induced Interstitial Fibrosis. Eurasian J. Med. 2018, 50, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Nirk, E.L.; Reggiori, F.; Mauthe, M. Hydroxychloroquine in Rheumatic Autoimmune Disorders and Beyond. EMBO Mol. Med. 2020, 12, e12476. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Qiang, L.; Sample, A.; Shah, P.; He, Y.-Y. NF-κB Signaling Activation Induced by Chloroquine Requires Autophagosome, P62 Protein, and c-Jun N-Terminal Kinase (JNK) Signaling and Promotes Tumor Cell Resistance. J. Biol. Chem. 2017, 292, 3379–3388. [Google Scholar] [CrossRef]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting Pharmacological Effects of Chloroquine in Cancer Treatment: Interference with Inflammatory Signaling Pathways. Immunology 2020, 159, 257–278. [Google Scholar] [CrossRef]
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and Future Status of JAK Inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Shimaoka, H.; Takeno, S.; Maki, K.; Sasaki, T.; Hasegawa, S.; Yamashita, Y. A Cytokine Signal Inhibitor for Rheumatoid Arthritis Enhances Cancer Metastasis via Depletion of NK Cells in an Experimental Lung Metastasis Mouse Model of Colon Cancer. Oncol. Lett. 2017, 14, 3019–3027. [Google Scholar] [CrossRef]
- Russell, M.D.; Stovin, C.; Alveyn, E.; Adeyemi, O.; Chan, C.K.D.; Patel, V.; Adas, M.A.; Atzeni, F.; Ng, K.K.H.; Rutherford, A.I.; et al. JAK Inhibitors and the Risk of Malignancy: A Meta-Analysis across Disease Indications. Ann. Rheum. Dis. 2023, 82, 1059–1067. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef]
- Sebbag, E.; Lauper, K.; Molina-Collada, J.; Aletaha, D.; Askling, J.; Gente, K.; Bertheussen, H.; Bitoun, S.; Bolek, E.C.; Burmester, G.R.; et al. 2024 EULAR Points to Consider on the Initiation of Targeted Therapies in Patients with Inflammatory Arthritis and a History of Cancer. Ann. Rheum. Dis. 2024, 84, 388–397. [Google Scholar] [CrossRef]
- Benjamin, O.; Goyal, A.; Lappin, S.L. Disease-Modifying Antirheumatic Drugs (DMARD). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Burke, R.A.; White, N.D. Biologic Disease-Modifying Antirheumatic Drugs; American College of Clinical Pharmacy: Lenexa, KS, USA, 2014. [Google Scholar]
- Joseph, D.; Tintinger, G.R.; Ker, J.A.; Pannell, N. Adverse Effects of Biologic Anti-Inflammatory Agents on the Respiratory System: A Review. Afr. J. Thorac. Crit. Care Med. 2021, 27, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wadström, H.; Frisell, T.; Askling, J.; Anti-Rheumatic Therapy in Sweden (ARTIS) Study Group. Malignant Neoplasms in Patients with Rheumatoid Arthritis Treated with Tumor Necrosis Factor Inhibitors, Tocilizumab, Abatacept, or Rituximab in Clinical Practice: A Nationwide Cohort Study From Sweden. JAMA Intern. Med. 2017, 177, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Montastruc, F.; Renoux, C.; Dell’Aniello, S.; Simon, T.A.; Azoulay, L.; Hudson, M.; Suissa, S. Abatacept Initiation in Rheumatoid Arthritis and the Risk of Cancer: A Population-Based Comparative Cohort Study. Rheumatology 2019, 58, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Muir, V.S.; Jones, B.E.; Wall, V.Z.; Ylescupidez, A.; Hocking, A.M.; Pribitzer, S.; Thorpe, J.; Fuchs, B.; Wiedeman, A.E.; et al. Abatacept Increases T Cell Exhaustion in Early RA Individuals Who Carry HLA Risk Alleles. Front. Immunol. 2024, 15, 1383110. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour Necrosis Factor and Cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Leone, G.M.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; Fagone, P. Past, Present and (Foreseeable) Future of Biological Anti-TNF Alpha Therapy. J. Clin. Med. 2023, 12, 1630. [Google Scholar] [CrossRef]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular Mechanism of Action of Anti-Tumor Necrosis Factor Antibodies in Inflammatory Bowel Diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF Antibody Therapy in Rheumatoid Arthritis and the Risk of Serious Infections and Malignancies: Systematic Review and Meta-Analysis of Rare Harmful Effects in Randomized Controlled Trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-Alpha Therapies: The next Generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear Factor-kappaB in Cancer Development and Progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Zogala, R.J.; Goutsouliak, K.; Suarez-Almazor, M.E. Management Considerations in Cancer Patients with Rheumatoid Arthritis. Oncology 2017, 31, 374–380. [Google Scholar] [PubMed]
- Sebbag, E.; Molina-Collada, J.; Ndoye, R.; Aletaha, D.; Askling, J.; Gente, K.; Bertheussen, H.; Bitoun, S.; Bolek, E.C.; Buch, M.H.; et al. Systematic Literature Review and Meta-Analysis Informing the EULAR Points to Consider on the Initiation of Targeted Therapies in Patients with Inflammatory Arthritis and a History of Cancer. Ann. Rheum. Dis. 2024, 84, 643–652. [Google Scholar] [CrossRef]
- Lopez-Olivo, M.A.; Colmegna, I.; Karpes Matusevich, A.R.; Qi, S.R.; Zamora, N.V.; Sharma, R.; Pratt, G.; Suarez-Almazor, M.E. Systematic Review of Recommendations on the Use of Disease-Modifying Antirheumatic Drugs in Patients with Rheumatoid Arthritis and Cancer. Arthritis Care Res. 2020, 72, 309–318. [Google Scholar] [CrossRef]
- Suarez-Almazor, M.E.; Ruiz, J.I.; Lei, X.; Wu, C.-F.; Zhao, H.; Rajan, S.S.; Giordano, S.H. Trends in the Use of Biologic Disease-Modifying Antirheumatic Drugs in Patients with Rheumatoid Arthritis and Recently Diagnosed Colorectal, Lung, or Prostate Cancer. Clin. Rheumatol. 2024, 43, 3301–3312. [Google Scholar] [CrossRef]
- Wu, C.-F.; Fu, J.-Y.; Yeh, C.-J.; Liu, Y.-H.; Hsieh, M.-J.; Wu, Y.-C.; Wu, C.-Y.; Tsai, Y.-H.; Chou, W.-C. Recurrence Risk Factors Analysis for Stage I Non-Small Cell Lung Cancer. Medicine 2015, 94, e1337. [Google Scholar] [CrossRef]
- Vedamurthy, A.; Gangasani, N.; Ananthakrishnan, A.N. Vedolizumab or Tumor Necrosis Factor Antagonist Use and Risk of New or Recurrent Cancer in Patients with Inflammatory Bowel Disease with Prior Malignancy: A Retrospective Cohort Study. Clin. Gastroenterol. Hepatol. 2022, 20, 88–95. [Google Scholar] [CrossRef]
- Raaschou, P.; Söderling, J.; Turesson, C.; Askling, J.; ARTIS Study Group. Tumor Necrosis Factor Inhibitors and Cancer Recurrence in Swedish Patients with Rheumatoid Arthritis: A Nationwide Population-Based Cohort Study. Ann. Intern. Med. 2018, 169, 291–299. [Google Scholar] [CrossRef]
- von Itzstein, M.S.; Gonugunta, A.S.; Mayo, H.G.; Minna, J.D.; Gerber, D.E. Immunotherapy Use in Patients with Lung Cancer and Comorbidities. Cancer J. 2020, 26, 525–536. [Google Scholar] [CrossRef]
- Kostine, M.; Finckh, A.; Bingham, C.O.; Visser, K.; Leipe, J.; Schulze-Koops, H.; Choy, E.H.; Benesova, K.; Radstake, T.R.D.J.; Cope, A.P.; et al. EULAR Points to Consider for the Diagnosis and Management of Rheumatic Immune-Related Adverse Events Due to Cancer Immunotherapy with Checkpoint Inhibitors. Ann. Rheum. Dis. 2021, 80, 36–48. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Hjeily, B.; Nevaneeth, B.C.; Samborski, W.; Szekanecz, Z.; Grygiel-Górniak, B. Inflammatory Pathways to Carcinogenesis: Deciphering the Rheumatoid Arthritis–Lung Cancer Connection. Cancers 2025, 17, 1330. https://doi.org/10.3390/cancers17081330
Abou Hjeily B, Nevaneeth BC, Samborski W, Szekanecz Z, Grygiel-Górniak B. Inflammatory Pathways to Carcinogenesis: Deciphering the Rheumatoid Arthritis–Lung Cancer Connection. Cancers. 2025; 17():1330. https://doi.org/10.3390/cancers17081330
Chicago/Turabian StyleAbou Hjeily, Boushra, Briana Candace Nevaneeth, Włodzimierz Samborski, Zoltán Szekanecz, and Bogna Grygiel-Górniak. 2025. "Inflammatory Pathways to Carcinogenesis: Deciphering the Rheumatoid Arthritis–Lung Cancer Connection" Cancers 17, no. : 1330. https://doi.org/10.3390/cancers17081330
APA StyleAbou Hjeily, B., Nevaneeth, B. C., Samborski, W., Szekanecz, Z., & Grygiel-Górniak, B. (2025). Inflammatory Pathways to Carcinogenesis: Deciphering the Rheumatoid Arthritis–Lung Cancer Connection. Cancers, 17(), 1330. https://doi.org/10.3390/cancers17081330