

Alternative Splicing in Lung Adenocarcinoma: From Bench to Bedside


Simple Summary
Abstract
1. Introduction
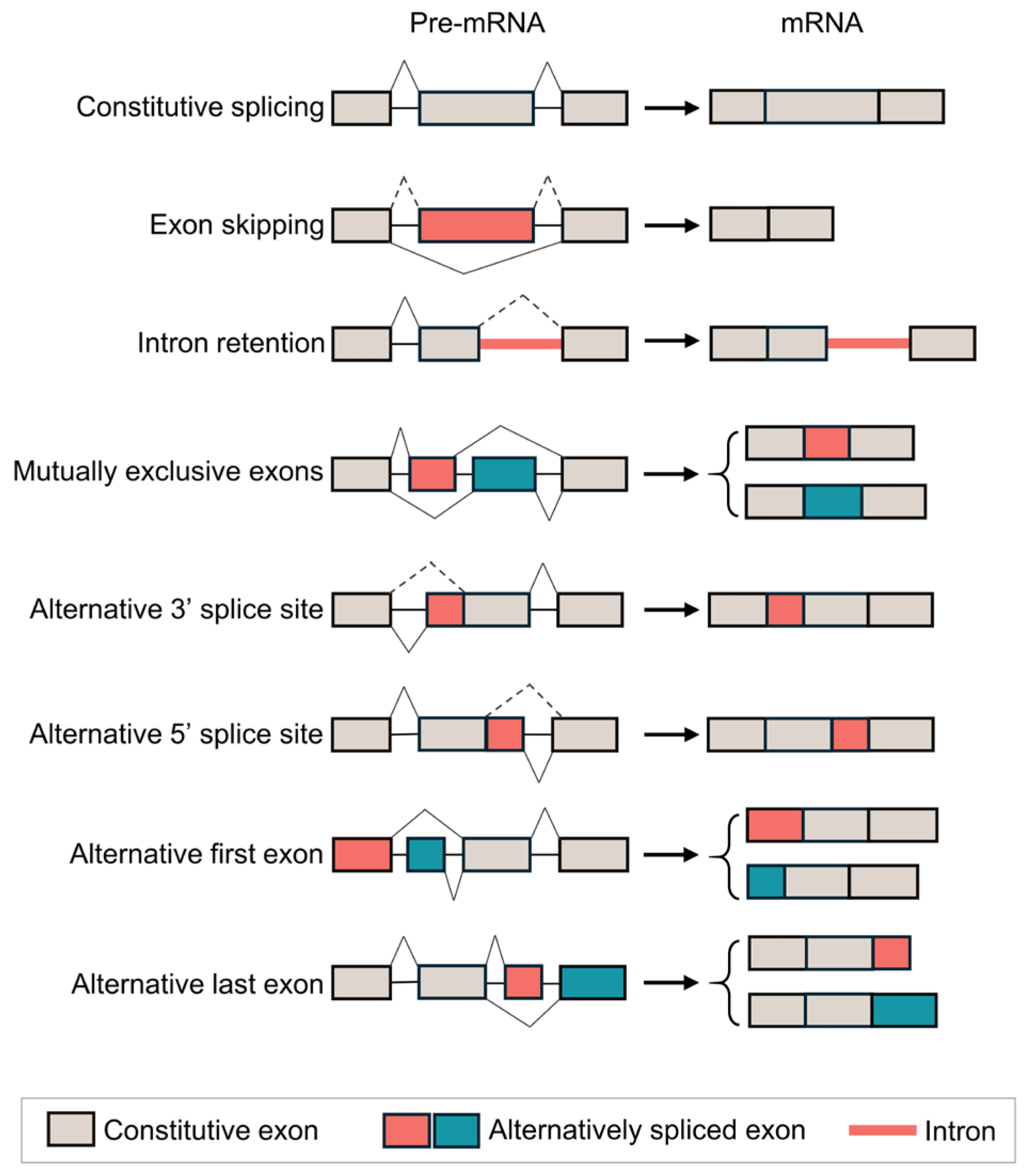
2. The Mechanism of AS in LUAD
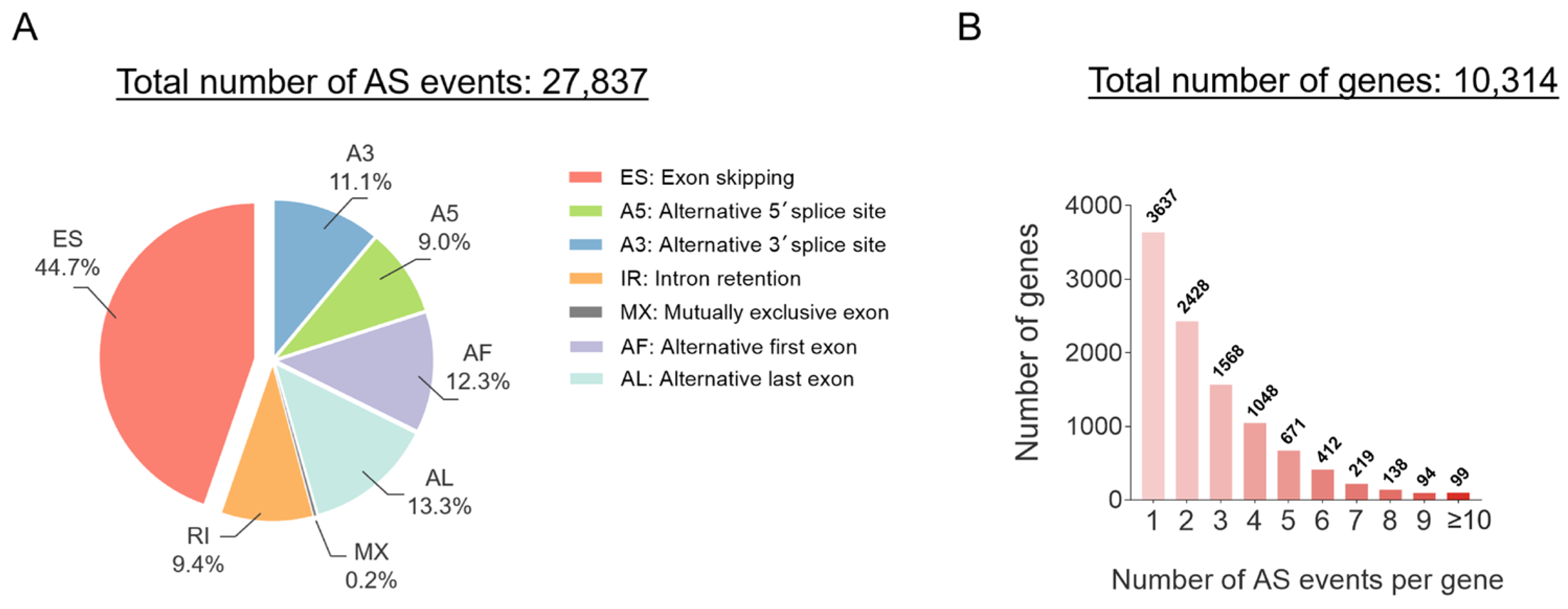
3. Emerging Technologies to Assess AS in LUAD
4. The AS Events Involving Driver Genes of LUAD
4.1. KRAS
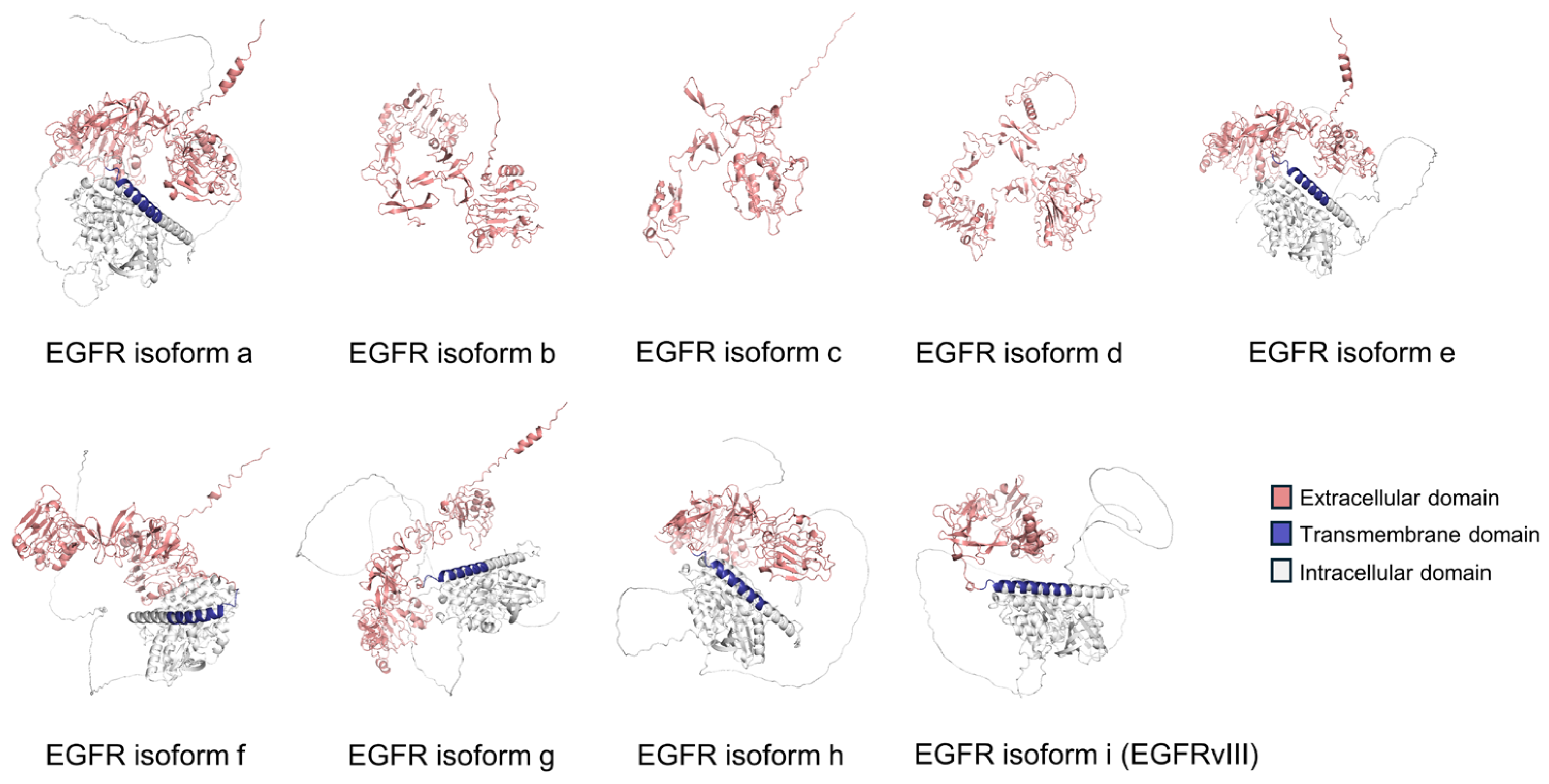
4.2. EGFR
4.3. C-Met
4.4. CD44
4.5. PD-1/PD-L1
5. Therapeutic Strategies Targeting AS in LUAD
5.1. Small-Molecule Inhibitors
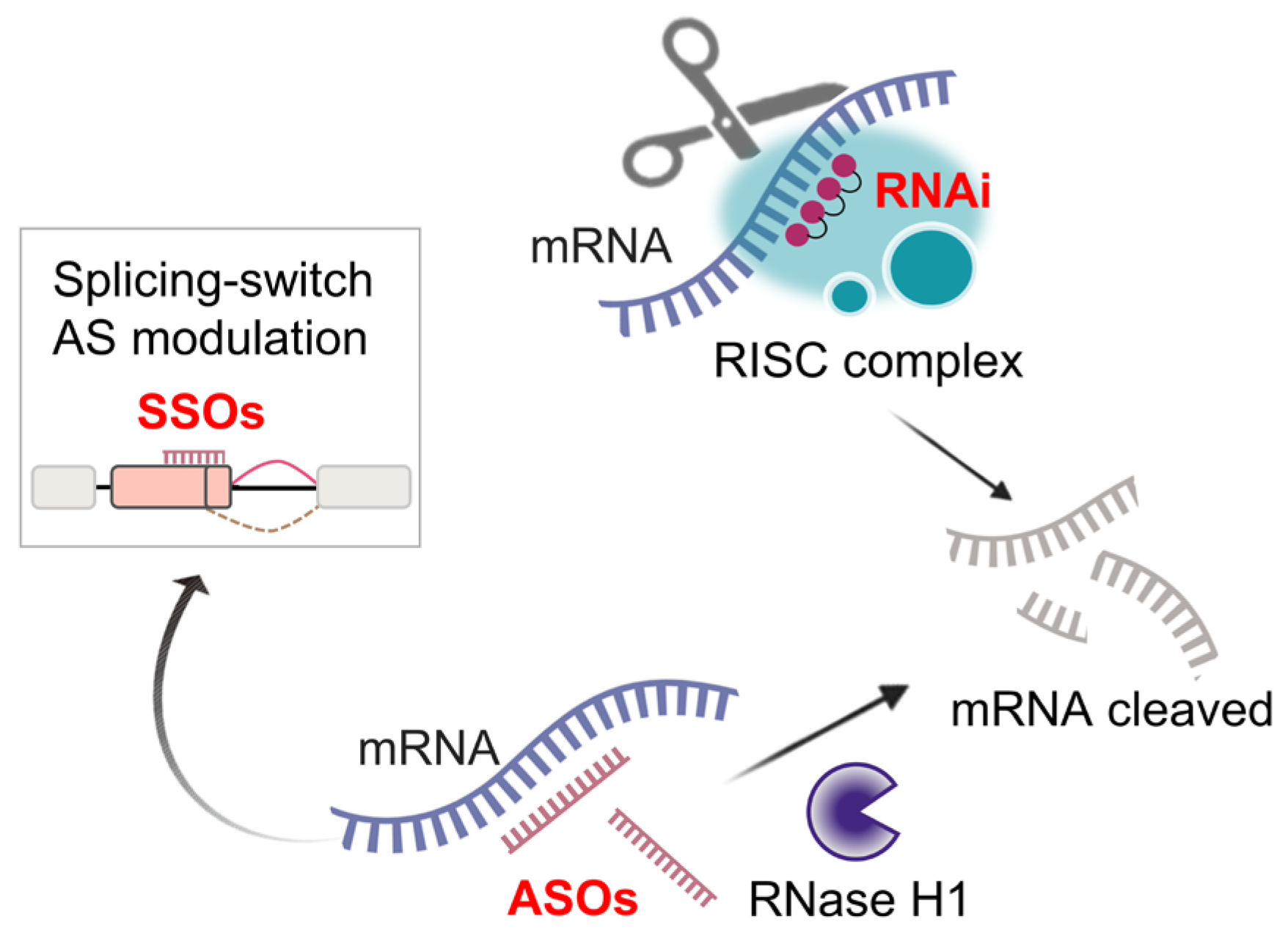
5.2. RNA-Targeted Therapies
5.3. Emerging Gene Therapies
5.4. Combining Splicing-Targeted Strategies with Standard Treatments
5.5. Upcoming AS-Based Immunotherapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Sinjab, A.; Rahal, Z.; Lynch, A.M.; Treekitkarnmongkol, W.; Liu, Y.; Serrano, A.G.; Feng, J.; Liang, K.; Khan, K.; et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. Nature 2024, 627, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Chen, H.; Gao, F.; He, M.; Ding, X.F.; Wong, A.M.; Sze, S.C.; Yu, A.C.; Sun, T.; Chan, A.W.; Wang, X.; et al. Long-Read RNA Sequencing Identifies Alternative Splice Variants in Hepatocellular Carcinoma and Tumor-Specific Isoforms. Hepatology 2019, 70, 1011–1025. [Google Scholar] [CrossRef]
- Whibley, A.; Kelley, J.L.; Narum, S.R. The changing face of genome assemblies: Guidance on achieving high-quality reference genomes. Mol. Ecol. Resour. 2021, 21, 641–652. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Miyake, S.; Oka, M.; Kanai, A.; Kawai, Y.; Nagasawa, S.; Shiraishi, Y.; Tokunaga, K.; Kohno, T.; Seki, M.; et al. Phasing analysis of lung cancer genomes using a long read sequencer. Nat. Commun. 2022, 13, 3464. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Wright, C.J.; Smith, C.W.J.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Marasco, L.E.; Kornblihtt, A.R. The physiology of alternative splicing. Nat. Rev. Mol. Cell. Biol. 2023, 24, 242–254. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Feng, L.; Wang, Y.; Mao, Y.; Di, X.; Zhang, K.; Cheng, S.; Xiao, T. Multi-omics analysis reveals RNA splicing alterations and their biological and clinical implications in lung adenocarcinoma. Signal Transduct. Target. Ther. 2022, 7, 270. [Google Scholar] [CrossRef]
- Robinson, T.J.; Freedman, J.A.; Al Abo, M.; Deveaux, A.E.; LaCroix, B.; Patierno, B.M.; George, D.J.; Patierno, S.R. Alternative RNA Splicing as a Potential Major Source of Untapped Molecular Targets in Precision Oncology and Cancer Disparities. Clin. Cancer Res. 2019, 25, 2963–2968. [Google Scholar] [CrossRef]
- Pan, Y.; Phillips, J.W.; Zhang, B.D.; Noguchi, M.; Kutschera, E.; McLaughlin, J.; Nesterenko, P.A.; Mao, Z.; Bangayan, N.J.; Wang, R.; et al. IRIS: Discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. Proc. Natl. Acad. Sci. USA 2023, 120, e2221116120. [Google Scholar] [CrossRef]
- Pan, Y.; Kadash-Edmondson, K.E.; Wang, R.; Phillips, J.; Liu, S.; Ribas, A.; Aplenc, R.; Witte, O.N.; Xing, Y. RNA Dysregulation: An Expanding Source of Cancer Immunotherapy Targets. Trends Pharmacol. Sci. 2021, 42, 268–282. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e216. [Google Scholar] [CrossRef]
- Hoyos, L.E.; Abdel-Wahab, O. Cancer-Specific Splicing Changes and the Potential for Splicing-Derived Neoantigens. Cancer Cell 2018, 34, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- El Zarif, T.; Meador, C.B.; Qiu, X.; Seo, J.H.; Davidsohn, M.P.; Savignano, H.; Lakshminarayanan, G.; McClure, H.M.; Canniff, J.; Fortunato, B.; et al. Detecting Small Cell Transformation in Patients with Advanced EGFR Mutant Lung Adenocarcinoma through Epigenomic cfDNA Profiling. Clin. Cancer Res. 2024, 30, 3798–3811. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Ryan, M.; Wong, W.C.; Brown, R.; Akbani, R.; Su, X.; Broom, B.; Melott, J.; Weinstein, J. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 2016, 44, D1018–D1022. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, E.; Zou, M.; Lv, C.; Cui, Y.; Zhai, S.; Sang, S.; Xiong, K.; Yang, X.; Zhuang, S.; et al. Unraveling immune heterogeneity across pan-cancer and deep insights in lung adenocarcinoma based on alternative splicing. J. Leukoc. Biol. 2025, 117, qiae104. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Zhao, X.; Wang, K.; Wang, C.; Du, J. Prognostic alternative splicing events related splicing factors define the tumor microenvironment and pharmacogenomic landscape in lung adenocarcinoma. Aging 2022, 14, 6689–6715. [Google Scholar] [CrossRef]
- Shao, M.M.; Zhai, K.; Huang, Z.Y.; Yi, F.S.; Zheng, S.C.; Liu, Y.L.; Qiao, X.; Chen, Q.Y.; Wang, Z.; Shi, H.Z. Characterization of the alternative splicing landscape in lung adenocarcinoma reveals novel prognosis signature associated with B cells. PLoS ONE 2023, 18, e0279018. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Salmen, F.; De Jonghe, J.; Kaminski, T.S.; Alemany, A.; Parada, G.E.; Verity-Legg, J.; Yanagida, A.; Kohler, T.N.; Battich, N.; van den Brekel, F.; et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 2022, 40, 1780–1793. [Google Scholar] [CrossRef]
- Salcher, S.; Sturm, G.; Horvath, L.; Untergasser, G.; Kuempers, C.; Fotakis, G.; Panizzolo, E.; Martowicz, A.; Trebo, M.; Pall, G.; et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell 2022, 40, 1503–1520.e1508. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e213. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Penter, L.; Borji, M.; Nagler, A.; Lyu, H.; Lu, W.S.; Cieri, N.; Maurer, K.; Oliveira, G.; Al’Khafaji, A.M.; Garimella, K.V.; et al. Integrative genotyping of cancer and immune phenotypes by long-read sequencing. Nat. Commun. 2024, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Dondi, A.; Lischetti, U.; Jacob, F.; Singer, F.; Borgsmüller, N.; Coelho, R.; Heinzelmann-Schwarz, V.; Beisel, C.; Beerenwinkel, N. Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing in ovarian cancer. Nat. Commun. 2023, 14, 7780. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Liu, S.; Wang, C.; Zhang, E.; Chen, C.; Zhu, M.; Zhang, J.; Zhu, C.; Ji, M.; et al. Integrative splicing-quantitative-trait-locus analysis reveals risk loci for non-small-cell lung cancer. Am. J. Hum. Genet. 2023, 110, 1574–1589. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- De Zuani, M.; Xue, H.; Park, J.S.; Dentro, S.C.; Seferbekova, Z.; Tessier, J.; Curras-Alonso, S.; Hadjipanayis, A.; Athanasiadis, E.I.; Gerstung, M.; et al. Single-cell and spatial transcriptomics analysis of non-small cell lung cancer. Nat. Commun. 2024, 15, 4388. [Google Scholar] [CrossRef] [PubMed]
- Belchikov, N.; Hsu, J.; Li, X.J.; Jarroux, J.; Hu, W.; Joglekar, A.; Tilgner, H.U. Understanding isoform expression by pairing long-read sequencing with single-cell and spatial transcriptomics. Genome Res. 2024, 34, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Lebrigand, K.; Bergenstråhle, J.; Thrane, K.; Mollbrink, A.; Meletis, K.; Barbry, P.; Waldmann, R.; Lundeberg, J. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 2023, 51, e47. [Google Scholar] [CrossRef]
- Paavola, K.J.; Roda, J.M.; Lin, V.Y.; Chen, P.; O’Hollaren, K.P.; Ventura, R.; Crawley, S.C.; Li, B.; Chen, H.H.; Malmersjö, S.; et al. The Fibronectin-ILT3 Interaction Functions as a Stromal Checkpoint that Suppresses Myeloid Cells. Cancer Immunol. Res. 2021, 9, 1283–1297. [Google Scholar] [CrossRef]
- Di Modugno, F.; Di Carlo, A.; Spada, S.; Palermo, B.; D’Ambrosio, L.; D’Andrea, D.; Morello, G.; Belmonte, B.; Sperduti, I.; Balzano, V.; et al. Tumoral and stromal hMENA isoforms impact tertiary lymphoid structure localization in lung cancer and predict immune checkpoint blockade response in patients with cancer. EBioMedicine 2024, 101, 105003. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Song, J.Y.; Kwon, M.C.; Proost, N.; Zevenhoven, J.; Berns, A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 4952–4957. [Google Scholar] [CrossRef]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.Q.; Varmus, H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. eLife 2015, 4, e06907. [Google Scholar] [CrossRef]
- Salmón, M.; Paniagua, G.; Lechuga, C.G.; Fernández-García, F.; Zarzuela, E.; Álvarez-Díaz, R.; Musteanu, M.; Guerra, C.; Caleiras, E.; Muñoz, J.; et al. KRAS4A induces metastatic lung adenocarcinomas in vivo in the absence of the KRAS4B isoform. Proc. Natl. Acad. Sci. USA 2021, 118, e2023112118. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar] [CrossRef]
- Tran, T.T.T.; Phung, C.D.; Yeo, B.Z.J.; Prajogo, R.C.; Jayasinghe, M.K.; Yuan, J.; Tan, D.S.W.; Yeo, E.Y.M.; Goh, B.C.; Tam, W.L.; et al. Customised design of antisense oligonucleotides targeting EGFR driver mutants for personalised treatment of non-small cell lung cancer. EBioMedicine 2024, 108, 105356. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Giovannetti, E.; Pauwels, P.; Peters, G.J.; Hong, D.S.; Cappuzzo, F.; Hirsch, F.R.; Rolfo, C. cMET Exon 14 Skipping: From the Structure to the Clinic. J. Thorac. Oncol. 2016, 11, 1423–1432. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Xie, Z.; Gao, Y.; Ho, C.; Li, L.; Jin, C.; Wang, X.; Zou, C.; Mao, Y.; Wang, X.; Li, Q.; et al. Exosome-delivered CD44v6/C1QBP complex drives pancreatic cancer liver metastasis by promoting fibrotic liver microenvironment. Gut 2022, 71, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Asperti, C.; Corna, S.; Cicoria, E.; Valtolina, V.; Stornaiuolo, A.; Valentinis, B.; Bordignon, C.; Traversari, C. CAR T Cells Redirected to CD44v6 Control Tumor Growth in Lung and Ovary Adenocarcinoma Bearing Mice. Front. Immunol. 2020, 11, 99. [Google Scholar] [CrossRef]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef]
- Sun, J.; Bai, J.; Jiang, T.; Gao, Y.; Hua, Y. Modulation of PDCD1 exon 3 splicing. RNA Biol. 2019, 16, 1794–1805. [Google Scholar] [CrossRef]
- Bodac, A.; Mayet, A.; Rana, S.; Pascual, J.; Bowler, A.D.; Roh, V.; Fournier, N.; Craciun, L.; Demetter, P.; Radtke, F.; et al. Bcl-xL targeting eliminates ageing tumor-promoting neutrophils and inhibits lung tumor growth. EMBO Mol. Med. 2024, 16, 158–184. [Google Scholar] [CrossRef] [PubMed]
- Bertino, E.M.; Gentzler, R.D.; Clifford, S.; Kolesar, J.; Muzikansky, A.; Haura, E.B.; Piotrowska, Z.; Camidge, D.R.; Stinchcombe, T.E.; Hann, C.; et al. Phase IB Study of Osimertinib in Combination with Navitoclax in EGFR-mutant NSCLC Following Resistance to Initial EGFR Therapy (ETCTN 9903). Clin. Cancer Res. 2021, 27, 1604–1611. [Google Scholar] [CrossRef]
- Xu, N.; Ren, Y.; Bao, Y.; Shen, X.; Kang, J.; Wang, N.; Wang, Z.; Han, X.; Li, Z.; Zuo, J.; et al. PUF60 promotes cell cycle and lung cancer progression by regulating alternative splicing of CDC25C. Cell Rep. 2023, 42, 113041. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Han, B.; Hua, M.; Wang, C.; Li, T. SF3B4 downregulation restrains lung adenocarcinoma tumorigenesis via 5′ alternative splicing of KAT2A. Sci. Rep. 2024, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bao, Y.; Shen, X.; Pan, Y.; Sun, Y.; Xiao, M.; Chen, K.; Wei, H.; Zuo, J.; Saffen, D.; et al. RNA binding motif protein 10 suppresses lung cancer progression by controlling alternative splicing of eukaryotic translation initiation factor 4H. EBioMedicine 2020, 61, 103067. [Google Scholar] [CrossRef]
- Chen, Q.W.; Cai, Q.Q.; Yang, Y.; Dong, S.; Liu, Y.Y.; Chen, Z.Y.; Kang, C.L.; Qi, B.; Dong, Y.W.; Wu, W.; et al. LncRNA BC promotes lung adenocarcinoma progression by modulating IMPAD1 alternative splicing. Clin. Transl. Med. 2023, 13, e1129. [Google Scholar] [CrossRef]
- Klomp, J.A.; Klomp, J.E.; Stalnecker, C.A.; Bryant, K.L.; Edwards, A.C.; Drizyte-Miller, K.; Hibshman, P.S.; Diehl, J.N.; Lee, Y.S.; Morales, A.J.; et al. Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers. Science 2024, 384, eadk0775. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Mitsudomi, T. Biological and clinical significance of KRAS mutations in lung cancer: An oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010, 29, 49–60. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Aran, V.; Masson Domingues, P.; Carvalho de Macedo, F.; Moreira de Sousa, C.A.; Caldas Montella, T.; de Souza Accioly, M.T.; Ferreira, C.G. A cross-sectional study examining the expression of splice variants K-RAS4A and K-RAS4B in advanced non-small-cell lung cancer patients. Lung Cancer 2018, 116, 7–14. [Google Scholar] [CrossRef]
- Yang, I.S.; Kim, S. Isoform specific gene expression analysis of KRAS in the prognosis of lung adenocarcinoma patients. BMC Bioinformatics 2018, 19, 40. [Google Scholar] [CrossRef]
- Newlaczyl, A.U.; Coulson, J.M.; Prior, I.A. Quantification of spatiotemporal patterns of Ras isoform expression during development. Sci. Rep. 2017, 7, 41297. [Google Scholar] [CrossRef] [PubMed]
- Omerovic, J.; Hammond, D.E.; Clague, M.J.; Prior, I.A. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 2008, 27, 2754–2762. [Google Scholar] [CrossRef]
- Chen, W.C.; To, M.D.; Westcott, P.M.K.; Delrosario, R.; Kim, I.J.; Philips, M.; Tran, Q.; Bollam, S.R.; Goodarzi, H.; Bayani, N.; et al. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat. Commun. 2021, 12, 4288. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lv, Q.; Xu, Y.; Cai, Z.; Zheng, J.; Cheng, X.; Dai, Y.; Jänne, P.A.; Ambrogio, C.; Köhler, J. An integrative pharmacogenomics analysis identifies therapeutic targets in KRAS-mutant lung cancer. EBioMedicine 2019, 49, 106–117. [Google Scholar] [CrossRef]
- Chiari, R.; Palladino, S.; Emili, R.; De Lisa, M.; Sarti, D.; Catalano, V.; Magnani, M.; Graziano, F.; Ruzzo, A. KRAS4A and KRAS4B in liquid biopsy of metastatic lung adenocarcinoma patients treated with Pembrolizumab or chemotherapy plus Pembrolizumab. Sci. Rep. 2023, 13, 21036. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- Iamaroon, A.; Tait, B.; Diewert, V.M. Cell proliferation and expression of EGF, TGF-alpha, and EGF receptor in the developing primary palate. J. Dent. Res. 1996, 75, 1534–1539. [Google Scholar] [CrossRef]
- Ovrevik, J.; Refsnes, M.; Totlandsdal, A.I.; Holme, J.A.; Schwarze, P.E.; Låg, M. TACE/TGF-α/EGFR regulates CXCL8 in bronchial epithelial cells exposed to particulate matter components. Eur. Respir. J. 2011, 38, 1189–1199. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Chen, L.; Chen, H.Y.; Hu, L.; Li, L.; Sun, H.Y.; Jiang, F.; Zhao, J.; Liu, G.M.; Tang, J.; et al. MUC15 inhibits dimerization of EGFR and PI3K-AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology 2013, 145, 1436–1448.e1–12. [Google Scholar] [CrossRef] [PubMed]
- Gala, K.; Chandarlapaty, S. Molecular pathways: HER3 targeted therapy. Clin. Cancer Res. 2014, 20, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Nakakuki, T.; Yumoto, N.; Naka, T.; Shirouzu, M.; Yokoyama, S.; Hatakeyama, M. Topological analysis of MAPK cascade for kinetic ErbB signaling. PLoS ONE 2008, 3, e1782. [Google Scholar] [CrossRef]
- Zhang, R.; Fruhwirth, G.O.; Coban, O.; Barrett, J.E.; Burgoyne, T.; Lee, S.H.; Simonson, P.D.; Baday, M.; Kholodenko, B.N.; Futter, C.E.; et al. Probing the Heterogeneity of Protein Kinase Activation in Cells by Super-resolution Microscopy. ACS Nano 2017, 11, 249–257. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Ohnishi, H.; Furuyashiki, G.; Nogami, H.; Koshiishi, Y.; Ooide, A.; Matsushima, S.; Watanabe, T.; Goya, T. Clinico-pathological and biological significance of tyrosine kinase domain gene mutations and overexpression of epidermal growth factor receptor for lung adenocarcinoma. J. Thorac. Oncol. 2006, 1, 787–795. [Google Scholar] [CrossRef]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Selvaggi, G.; Novello, S.; Hirsch, F.R. The biology of epidermal growth factor receptor in lung cancer. Clin. Cancer Res. 2004, 10, 4227s–4232s. [Google Scholar] [CrossRef]
- Lee, V.H.; Tin, V.P.; Choy, T.S.; Lam, K.O.; Choi, C.W.; Chung, L.P.; Tsang, J.W.; Ho, P.P.; Leung, D.K.; Ma, E.S.; et al. Association of exon 19 and 21 EGFR mutation patterns with treatment outcome after first-line tyrosine kinase inhibitor in metastatic non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 1148–1155. [Google Scholar] [CrossRef]
- Chung, K.P.; Wu, S.G.; Wu, J.Y.; Yang, J.C.; Yu, C.J.; Wei, P.F.; Shih, J.Y.; Yang, P.C. Clinical outcomes in non-small cell lung cancers harboring different exon 19 deletions in EGFR. Clin. Cancer Res. 2012, 18, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kawano, O.; Endo, K.; Yukiue, H.; Yano, M.; Fujii, Y. EGFRvIII mutation in lung cancer correlates with increased EGFR copy number. Oncol. Rep. 2007, 17, 319–323. [Google Scholar] [CrossRef]
- Okamoto, I.; Kenyon, L.C.; Emlet, D.R.; Mori, T.; Sasaki, J.; Hirosako, S.; Ichikawa, Y.; Kishi, H.; Godwin, A.K.; Yoshioka, M.; et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003, 94, 50–56. [Google Scholar] [CrossRef]
- Piccione, E.C.; Lieu, T.J.; Gentile, C.F.; Williams, T.R.; Connolly, A.J.; Godwin, A.K.; Koong, A.C.; Wong, A.J. A novel epidermal growth factor receptor variant lacking multiple domains directly activates transcription and is overexpressed in tumors. Oncogene 2012, 31, 2953–2967. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours—Molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 188–202. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Che, J.; Jänne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef]
- Remon, J.; Hendriks, L.E.L.; Mountzios, G.; García-Campelo, R.; Saw, S.P.L.; Uprety, D.; Recondo, G.; Villacampa, G.; Reck, M. MET alterations in NSCLC-Current Perspectives and Future Challenges. J. Thorac. Oncol. 2023, 18, 419–435. [Google Scholar] [CrossRef]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef]
- Digumarthy, S.R.; Mendoza, D.P.; Zhang, E.W.; Lennerz, J.K.; Heist, R.S. Clinicopathologic and Imaging Features of Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. Cancers 2019, 11, 2033. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients With Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Peled, N.; Greer, J.; Wu, W.; Choi, P.; Berger, A.H.; Wong, S.; Jen, K.Y.; Seo, Y.; Hann, B.; et al. MET Exon 14 Mutation Encodes an Actionable Therapeutic Target in Lung Adenocarcinoma. Cancer Res. 2017, 77, 4498–4505. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Schiering, N.; Knapp, S.; Marconi, M.; Flocco, M.M.; Cui, J.; Perego, R.; Rusconi, L.; Cristiani, C. Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc. Natl. Acad. Sci. USA 2003, 100, 12654–12659. [Google Scholar] [CrossRef]
- Abella, J.V.; Peschard, P.; Naujokas, M.A.; Lin, T.; Saucier, C.; Urbé, S.; Park, M. Met/Hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol. Cell. Biol. 2005, 25, 9632–9645. [Google Scholar] [CrossRef]
- Lee, G.D.; Lee, S.E.; Oh, D.Y.; Yu, D.B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.Y.; et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef]
- Lee, J.H.; Gao, C.F.; Lee, C.C.; Kim, M.D.; Vande Woude, G.F. An alternatively spliced form of Met receptor is tumorigenic. Exp. Mol. Med. 2006, 38, 565–573. [Google Scholar] [CrossRef]
- Park, S.; Koh, J.; Kim, D.W.; Kim, M.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Chung, D.H.; Heo, D.S. MET amplification, protein expression, and mutations in pulmonary adenocarcinoma. Lung Cancer 2015, 90, 381–387. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Oxnard, G.R.; Elkin, S.; Sullivan, E.K.; Carter, J.L.; Barbie, D.A. Response to Crizotinib in a Patient With Lung Adenocarcinoma Harboring a MET Splice Site Mutation. Clin. Lung Cancer 2015, 16, e101–e104. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.G.; Mambetsariev, I.; Salgia, R. Prolonged survival and response to tepotinib in a non-small-cell lung cancer patient with brain metastases harboring MET exon 14 mutation: A research report. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005785. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, X.; Xu, Y.; Zhao, J.; Zhong, W.; Gao, X.; Chen, M.; Wang, M. Remarkable pathological response to neoadjuvant tepotinib in lung adenocarcinoma with MET exon 14 skipping mutation: A case report. Thorac. Cancer 2024, 15, 2339–2343. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Sun, X.; Li, C.W.; Wang, W.J.; Chen, M.K.; Li, H.; Lai, Y.J.; Hsu, J.L.; Koller, P.B.; Chan, L.C.; Lee, P.C.; et al. Inhibition of c-MET upregulates PD-L1 expression in lung adenocarcinoma. Am. J. Cancer Res. 2020, 10, 564–571. [Google Scholar] [PubMed]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Adesina, A.; Ahmad, S.; Bowler-Barnett, E.H.; Bye-A-Jee, H.; Carpentier, D.; Denny, P.; et al. UniProt: The Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2024, 52, D609–D617. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell. Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Hassn Mesrati, M.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11, 1850. [Google Scholar] [CrossRef]
- Weng, X.; Maxwell-Warburton, S.; Hasib, A.; Ma, L.; Kang, L. The membrane receptor CD44: Novel insights into metabolism. Trends Endocrinol. Metab. 2022, 33, 318–332. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Zeilstra, J.; Joosten, S.P.; van Andel, H.; Tolg, C.; Berns, A.; Snoek, M.; van de Wetering, M.; Spaargaren, M.; Clevers, H.; Pals, S.T. Stem cell CD44v isoforms promote intestinal cancer formation in Apc(min) mice downstream of Wnt signaling. Oncogene 2014, 33, 665–670. [Google Scholar] [CrossRef]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef]
- Clarke, M.R.; Landreneau, R.J.; Resnick, N.M.; Crowley, R.; Dougherty, G.J.; Cooper, D.L.; Yousem, S.A. Prognostic significance of CD44 expression in adenocarcinoma of the lung. Clin. Mol. Pathol. 1995, 48, M200–M204. [Google Scholar] [CrossRef]
- Ramasami, S.; Kerr, K.M.; Chapman, A.D.; King, G.; Cockburn, J.S.; Jeffrey, R.R. Expression of CD44v6 but not E-cadherin or beta-catenin influences prognosis in primary pulmonary adenocarcinoma. J. Pathol. 2000, 192, 427–432. [Google Scholar] [CrossRef]
- Lee, L.N.; Kuo, S.H.; Lee, Y.C.; Chang, Y.L.; Chang, H.C.; Jan, I.S.; Yang, P.C. CD44 splicing pattern is associated with disease progression in pulmonary adenocarcinoma. J. Formos. Med. Assoc. 2005, 104, 541–548. [Google Scholar] [PubMed]
- Suzuki, H.; Yamashiro, K. Reduced expression of CD44 v3 and v6 is related to invasion in lung adenocarcinoma. Lung Cancer 2002, 38, 137–141. [Google Scholar] [CrossRef]
- Ciulean, I.S.; Fischer, J.; Quaiser, A.; Bach, C.; Abken, H.; Tretbar, U.S.; Fricke, S.; Koehl, U.; Schmiedel, D.; Grunwald, T. CD44v6 specific CAR-NK cells for targeted immunotherapy of head and neck squamous cell carcinoma. Front. Immunol. 2023, 14, 1290488. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Remon, J.; Hellmann, M.D. First-Line Immunotherapy for Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 586–597. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, W.; Xuan, L.; Yu, Y.; Zheng, W.; Tao, F.; Nemechek, J.; He, C.; Ma, W.; Han, X.; et al. PD-1 signalling defines and protects leukaemic stem cells from T cell receptor-induced cell death in T cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2023, 25, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wartewig, T.; Daniels, J.; Schulz, M.; Hameister, E.; Joshi, A.; Park, J.; Morrish, E.; Venkatasubramani, A.V.; Cernilogar, F.M.; van Heijster, F.H.A.; et al. PD-1 instructs a tumor-suppressive metabolic program that restricts glycolysis and restrains AP-1 activity in T cell lymphoma. Nat. Cancer 2023, 4, 1508–1525. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nowakowski, G.S.; Wang, M.L.; Ansell, S.M. Advances in CD30- and PD-1-targeted therapies for classical Hodgkin lymphoma. J. Hematol. Oncol. 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ren, S.; Chen, Y.; Liu, A.; Wu, Q.; Jiang, T.; Lv, P.; Song, D.; Hu, F.; Lan, J.; et al. PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance. Sci. Adv. 2023, 9, eade4186. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Abu Hejleh, T.; Furqan, M.; Ballas, Z.; Clamon, G. The clinical significance of soluble PD-1 and PD-L1 in lung cancer. Crit. Rev. Oncol. Hematol. 2019, 143, 148–152. [Google Scholar] [CrossRef]
- Kruger, S.; Legenstein, M.L.; Rösgen, V.; Haas, M.; Modest, D.P.; Westphalen, C.B.; Ormanns, S.; Kirchner, T.; Heinemann, V.; Holdenrieder, S.; et al. Serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death ligand 1 (sPD-L1) in advanced pancreatic cancer. Oncoimmunology 2017, 6, e1310358. [Google Scholar] [CrossRef]
- Niu, M.; Liu, Y.; Yi, M.; Jiao, D.; Wu, K. Biological Characteristics and Clinical Significance of Soluble PD-1/PD-L1 and Exosomal PD-L1 in Cancer. Front. Immunol. 2022, 13, 827921. [Google Scholar] [CrossRef]
- Sorensen, S.F.; Demuth, C.; Weber, B.; Sorensen, B.S.; Meldgaard, P. Increase in soluble PD-1 is associated with prolonged survival in patients with advanced EGFR-mutated non-small cell lung cancer treated with erlotinib. Lung Cancer 2016, 100, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Zhao, Z.; Arooj, S.; Fu, Y.; Liao, G. Soluble PD-1: Predictive, Prognostic, and Therapeutic Value for Cancer Immunotherapy. Front. Immunol. 2020, 11, 587460. [Google Scholar] [CrossRef] [PubMed]
- Széles, Á.; Fazekas, T.; Váncsa, S.; Váradi, M.; Kovács, P.T.; Krafft, U.; Grünwald, V.; Hadaschik, B.; Csizmarik, A.; Hegyi, P.; et al. Pre-treatment soluble PD-L1 as a predictor of overall survival for immune checkpoint inhibitor therapy: A systematic review and meta-analysis. Cancer Immunol. Immunother. 2023, 72, 1061–1073. [Google Scholar] [CrossRef]
- Himuro, H.; Nakahara, Y.; Igarashi, Y.; Kouro, T.; Higashijima, N.; Matsuo, N.; Murakami, S.; Wei, F.; Horaguchi, S.; Tsuji, K.; et al. Clinical roles of soluble PD-1 and PD-L1 in plasma of NSCLC patients treated with immune checkpoint inhibitors. Cancer Immunol. Immunother. 2023, 72, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Jiao, Z.; Lu, G.; Yao, B.; Wang, T.; Rong, W.; Xu, J.; Fan, T.; Sun, X.; Yang, R.; et al. PD-L1 lncRNA splice isoform promotes lung adenocarcinoma progression via enhancing c-Myc activity. Genome Biol. 2021, 22, 104. [Google Scholar] [CrossRef]
- Nielsen, C.; Ohm-Laursen, L.; Barington, T.; Husby, S.; Lillevang, S.T. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005, 235, 109–116. [Google Scholar] [CrossRef]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef]
- Silvestri, I.; Manigrasso, J.; Andreani, A.; Brindani, N.; Mas, C.; Reiser, J.B.; Vidossich, P.; Martino, G.; McCarthy, A.A.; De Vivo, M.; et al. Targeting the conserved active site of splicing machines with specific and selective small molecule modulators. Nat. Commun. 2024, 15, 4980. [Google Scholar] [CrossRef]
- Alsafadi, S.; Dayot, S.; Tarin, M.; Houy, A.; Bellanger, D.; Cornella, M.; Wassef, M.; Waterfall, J.J.; Lehnert, E.; Roman-Roman, S.; et al. Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene 2021, 40, 85–96. [Google Scholar] [CrossRef]
- Esfahani, M.S.; Lee, L.J.; Jeon, Y.J.; Flynn, R.A.; Stehr, H.; Hui, A.B.; Ishisoko, N.; Kildebeck, E.; Newman, A.M.; Bratman, S.V.; et al. Functional significance of U2AF1 S34F mutations in lung adenocarcinomas. Nat. Commun. 2019, 10, 5712. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef]
- Spinello, A.; Borišek, J.; Malcovati, L.; Magistrato, A. Investigating the Molecular Mechanism of H3B-8800: A Splicing Modulator Inducing Preferential Lethality in Spliceosome-Mutant Cancers. Int. J. Mol. Sci. 2021, 22, 11222. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, A.B.; Tang, Y. Therapeutic approaches to treat human spliceosomal diseases. Curr. Opin. Biotechnol. 2019, 60, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Tsai, J.H.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B.; et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017, 8, 15522. [Google Scholar] [CrossRef]
- Folco, E.G.; Coil, K.E.; Reed, R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes. Dev. 2011, 25, 440–444. [Google Scholar] [CrossRef]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes. Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Aptullahoglu, E.; Ciardullo, C.; Wallis, J.P.; Marr, H.; Marshall, S.; Bown, N.; Willmore, E.; Lunec, J. Splicing Modulation Results in Aberrant Isoforms and Protein Products of p53 Pathway Genes and the Sensitization of B Cells to Non-Genotoxic MDM2 Inhibition. Int. J. Mol. Sci. 2023, 24, 2410. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Teng, T.; Huang, C.L.; Pazolli, E.; Banka, D.; Cheung-Ong, K.; Eifert, C.; Furman, C.; Wu, Z.J.; Seiler, M.; et al. Sensitivity to splicing modulation of BCL2 family genes defines cancer therapeutic strategies for splicing modulators. Nat. Commun. 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M. E7107, a new 7-urethan derivative of pladienplide D, displays curative effect against several human tumor xenografts. Proc. Am. Assoc. Cancer Res. 2004, 45, 691-a. [Google Scholar]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Corrionero, A.; Miñana, B.; Valcárcel, J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011, 25, 445–459. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; El Hassouni, B.; Funel, N.; Gandellini, P.; Lagerweij, T.; Buonamici, S.; Blijlevens, M.; Zeeuw van der Laan, E.A.; Zaffaroni, N.; et al. Splicing modulation as novel therapeutic strategy against diffuse malignant peritoneal mesothelioma. EBioMedicine 2019, 39, 215–225. [Google Scholar] [CrossRef]
- Sette, C.; Paronetto, M.P. Somatic Mutations in Core Spliceosome Components Promote Tumorigenesis and Generate an Exploitable Vulnerability in Human Cancer. Cancers 2022, 14, 1827. [Google Scholar] [CrossRef]
- Wu, T.; Millar, H.; Gaffney, D.; Beke, L.; Mannens, G.; Vinken, P.; Sommers, I.; Thuring, J.-W.; Sun, W.; Moy, C.; et al. Abstract 4859: JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Cancer Res. 2018, 78, 4859. [Google Scholar] [CrossRef]
- Vieito, M.; Moreno, V.; Spreafico, A.; Brana, I.; Wang, J.S.; Preis, M.; Hernández, T.; Genta, S.; Hansen, A.R.; Doger, B.; et al. Phase 1 Study of JNJ-64619178, a Protein Arginine Methyltransferase 5 Inhibitor, in Advanced Solid Tumors. Clin. Cancer Res. 2023, 29, 3592–3602. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef]
- Yamanaka, S.; Horiuchi, Y.; Matsuoka, S.; Kido, K.; Nishino, K.; Maeno, M.; Shibata, N.; Kosako, H.; Sawasaki, T. A proximity biotinylation-based approach to identify protein-E3 ligase interactions induced by PROTACs and molecular glues. Nat. Commun. 2022, 13, 183. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Balcells, C.; Ma, Y.; Poon, E.; Eckold, C.; Valbuena, G.N.; Xu, Y.; et al. Indisulam targets RNA splicing and metabolism to serve as a therapeutic strategy for high-risk neuroblastoma. Nat. Commun. 2022, 13, 1380. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Fu, Z.; Zhang, X.; Zhou, X.; Ur-Rehman, U.; Yu, M.; Liang, H.; Guo, H.; Guo, X.; Kong, Y.; Su, Y.; et al. In vivo self-assembled small RNAs as a new generation of RNAi therapeutics. Cell Res. 2021, 31, 631–648. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, G.; Tian, C.; Jiang, W.; Jin, L.; Zhang, C.; Chen, X. Exosome-mediated small RNA delivery for gene therapy. Wiley Interdiscip. Rev. RNA 2016, 7, 758–771. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Madanayake, T.W.; Welsh, E.A.; Darville, L.N.F.; Koomen, J.M.; Chalfant, C.E.; Haura, E.B.; Robinson, T.J. Inhibition of Epidermal Growth Factor Receptor Signaling by Antisense Oligonucleotides as a Novel Approach to Epidermal Growth Factor Receptor Inhibition. Nucleic Acid Ther. 2022, 32, 391–400. [Google Scholar] [CrossRef]
- Shi, B.; An, K.; Wang, Y.; Fei, Y.; Guo, C.; Cliff Zhang, Q.; Yang, Y.G.; Tian, X.; Kan, Q. RNA Structural Dynamics Modulate EGFR-TKI Resistance Through Controlling YRDC Translation in NSCLC Cells. Genom. Proteom. Bioinform. 2023, 21, 850–865. [Google Scholar] [CrossRef]
- Wang, L.; Fu, H.; Song, L.; Wu, Z.; Yu, J.; Guo, Q.; Chen, C.; Yang, X.; Zhang, J.; Wang, Q.; et al. Overcoming AZD9291 Resistance and Metastasis of NSCLC via Ferroptosis and Multitarget Interference by Nanocatalytic Sensitizer Plus AHP-DRI-12. Small 2023, 19, e2204133. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, S.; de Gusmao, C.M.; Zhao, B.; Chin, D.H.; DiDonato, R.L.; Nguyen, M.A.; Nakayama, T.; Hu, C.A.; Soucy, A.; et al. A framework for individualized splice-switching oligonucleotide therapy. Nature 2023, 619, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Kral, A.J.; Voss, D.; Schäfer, B.; Sudheendran, K.; Danielsen, M.; Caruthers, M.H.; Krainer, A.R. Screening Splice-Switching Antisense Oligonucleotides in Pancreas-Cancer Organoids. Nucleic Acid Ther. 2024, 34, 188–198. [Google Scholar] [CrossRef]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mangala, L.S.; Rodriguez-Aguayo, C.; Kong, X.; Lopez-Berestein, G.; Sood, A.K. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018, 37, 107–124. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, J.S.; Kim, S.W.; Yun, C.O. Evolution of oncolytic adenovirus for cancer treatment. Adv. Drug Deliv. Rev. 2012, 64, 720–729. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Deng, H.; Liu, H.; de Silva, T.; Xue, Y.; Mohamud, Y.; Ng, C.S.; Qu, J.; Zhang, J.; Jia, W.W.G.; Lockwood, W.W.; et al. Coxsackievirus Type B3 Is a Potent Oncolytic Virus against KRAS-Mutant Lung Adenocarcinoma. Mol. Ther. Oncolytics 2019, 14, 266–278. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef]
- Yan, Y.; Su, C.; Hang, M.; Huang, H.; Zhao, Y.; Shao, X.; Bu, X. Recombinant Newcastle disease virus rL-RVG enhances the apoptosis and inhibits the migration of A549 lung adenocarcinoma cells via regulating alpha 7 nicotinic acetylcholine receptors in vitro. Virol. J. 2017, 14, 190. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.H.; Kwon, Y.G.; Kim, E.C.; Kim, N.K.; Choi, H.J.; Yun, C.O. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Hall, G.; Pulido, J.; Diaz, R.M.; Thompson, J.; Chong, H.; Selby, P.; Coffey, M.; Pandha, H.; Chester, J.; et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J. Clin. Investig. 2010, 120, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Liu, J.B.; Li, W.; Ma, Y.S.; Fu, D. The power and the promise of CRISPR/Cas9 genome editing for clinical application with gene therapy. J. Adv. Res. 2022, 40, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef]
- Fan, W.; Huang, J.; Tian, F.; Hong, X.; Zhu, K.; Zhan, Y.; Li, X.; Wang, X.; Wang, X.; Cai, L.; et al. m(6)A-Modified SNRPA Controls Alternative Splicing of ERCC1 Exon 8 to Induce Cisplatin Resistance in Lung Adenocarcinoma. Adv. Sci. 2024, 11, e2404609. [Google Scholar] [CrossRef]
- Thomas, J.D.; Polaski, J.T.; Feng, Q.; De Neef, E.J.; Hoppe, E.R.; McSharry, M.V.; Pangallo, J.; Gabel, A.M.; Belleville, A.E.; Watson, J.; et al. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat. Genet. 2020, 52, 84–94. [Google Scholar] [CrossRef]
- Li, K.; Peng, Z.Y.; Wang, R.; Li, X.; Du, N.; Liu, D.P.; Zhang, J.; Zhang, Y.F.; Ma, L.; Sun, Y.; et al. Enhancement of TKI sensitivity in lung adenocarcinoma through m6A-dependent translational repression of Wnt signaling by circ-FBXW7. Mol. Cancer 2023, 22, 103. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, S.; Chen, T.; Fitzgerald, M.; Liu, S.; Peng, C.; Tang, K.H.; Cao, S.; Chouitar, J.; Wu, J.; et al. Targeting HER2 Exon 20 Insertion-Mutant Lung Adenocarcinoma with a Novel Tyrosine Kinase Inhibitor Mobocertinib. Cancer Res. 2021, 81, 5311–5324. [Google Scholar] [CrossRef]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; de Marinis, F. Recent Advances on the Role of EGFR Tyrosine Kinase Inhibitors in the Management of NSCLC With Uncommon, Non Exon 20 Insertions, EGFR Mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Liao, B.C.; Liao, W.Y.; Markovets, A.; Stetson, D.; Thress, K.; Yang, J.C. Exon 16-Skipping HER2 as a Novel Mechanism of Osimertinib Resistance in EGFR L858R/T790M-Positive Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2020, 15, 50–61. [Google Scholar] [CrossRef]
- Yamaoka, T.; Tsurutani, J.; Sagara, H.; Ohmori, T. HER2-D16 oncogenic driver mutation confers osimertinib resistance in EGFR mutation-positive non-small cell lung cancer. Transl. Lung Cancer Res. 2020, 9, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef]
- Yu, M.; Nah, G.S.S.; Krishnan, V.; Sulaimi, F.N.B.; Ng, K.P.; Wang, C.; Bhatt, S.; Chuah, C.; Bergstrom, D.E.; Ong, S.T. The BIM deletion polymorphism potentiates the survival of leukemia stem and progenitor cells and impairs response to targeted therapies. Leukemia 2024, 39, 134–143. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Tanimoto, A.; Takeuchi, S.; Arai, S.; Fukuda, K.; Yamada, T.; Roca, X.; Ong, S.T.; Yano, S. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2017, 23, 3139–3149. [Google Scholar] [CrossRef]
- Takeuchi, S.; Hase, T.; Shimizu, S.; Ando, M.; Hata, A.; Murakami, H.; Kawakami, T.; Nagase, K.; Yoshimura, K.; Fujiwara, T.; et al. Phase I study of vorinostat with gefitinib in BIM deletion polymorphism/epidermal growth factor receptor mutation double-positive lung cancer. Cancer Sci. 2020, 111, 561–570. [Google Scholar] [CrossRef]
- Bao, Y.; Zhang, S.; Zhang, X.; Pan, Y.; Yan, Y.; Wang, N.; Ren, Y.; Zuo, J.; Zong, W.X.; Wang, Z.; et al. RBM10 Loss Promotes EGFR-Driven Lung Cancer and Confers Sensitivity to Spliceosome Inhibition. Cancer Res. 2023, 83, 1490–1502. [Google Scholar] [CrossRef]
- Blakely, C.M.; Urisman, A.; Gubens, M.A.; Mulvey, C.K.; Allen, G.M.; Shiboski, S.C.; Rotow, J.K.; Chakrabarti, T.; Kerr, D.L.; Aredo, J.V.; et al. Neoadjuvant Osimertinib for the Treatment of Stage I-IIIA Epidermal Growth Factor Receptor-Mutated Non-Small Cell Lung Cancer: A Phase II Multicenter Study. J. Clin. Oncol. 2024, 42, 3105–3114. [Google Scholar] [CrossRef]
- Nanjo, S.; Wu, W.; Karachaliou, N.; Blakely, C.M.; Suzuki, J.; Chou, Y.T.; Ali, S.M.; Kerr, D.L.; Olivas, V.R.; Shue, J.; et al. Deficiency of the splicing factor RBM10 limits EGFR inhibitor response in EGFR-mutant lung cancer. J. Clin. Investig. 2022, 132, e145099. [Google Scholar] [CrossRef]
- Yang, K.; Halima, A.; Chan, T.A. Antigen presentation in cancer—Mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 604–623. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Douillard, J.Y.; Blumenschein, G.R., Jr. Immunotherapy for non-small cell lung cancer: Novel approaches to improve patient outcome. J. Thorac. Oncol. 2011, 6, 1763–1773. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, Y.; Oyang, L.; Wu, N.; Tang, Y.; Su, M.; Luo, X.; Wang, Y.; Sheng, X.; Ma, J.; et al. Impacts and mechanisms of alternative mRNA splicing in cancer metabolism, immune response, and therapeutics. Mol. Ther. 2022, 30, 1018–1035. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, C.; Kadota, K.; Yamada, K.; Fujimoto, S.; Ibuki, E.; Ishikawa, R.; Haba, R.; Yajima, T. CD44v6 downregulation as a prognostic factor for distant recurrence in resected stage I lung adenocarcinomas. Clin. Exp. Med. 2023, 23, 5191–5200. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EGFR AS Isoform | AS Type | Number of a.a.s | Protein Size (kDa) | Predicted Subcellular Location |
|---|---|---|---|---|
| Isoform a | Canonical isoform | 1210 | 134.3 | Cell surface protein |
| Isoform b | Alternative last exon | 628 | 69.2 | Secreted protein |
| Isoform c | Alternative last exon | 405 | 44.7 | Secreted protein |
| Isoform d | Alternative last exon | 705 | 77.3 | Secreted protein |
| Isoform e | Exon skipping and alternative last exon | 1091 | 120.7 | Cell surface protein |
| Isoform f | Alternative last exon | 1136 | 125.8 | Cell surface protein |
| Isoform g | Exon skipping | 1165 | 129.2 | Cell surface protein |
| Isoform h | Alternative first exon | 1157 | 128.7 | Cell surface protein |
| Isoform I (EGFRvIII) | Alternative first exon | 943 | 104.3 | Cell surface protein |
| Gene | AS Event | Biological or Clinical Implications | AS-Related Therapy | Reference(s) |
|---|---|---|---|---|
| KRAS | Inclusion of either exon 4A (KRAS4A) or exon 4B (KRAS4B) | KRAS4A induces metastatic LUAD in vivo | ASO | [51,52] |
| EGFR | Deletion of exons 2–7 (EGFRvIII) | EGFRvIII contributes to resistance against TKIs | ASO | [53,54] |
| c-Met | Deletion of exon 14 (MET-ΔEx14) | MET-ΔEx14 confers clinical sensitivity to MET inhibitors | Small-molecule inhibitors | [55,56] |
| CD44 | Inclusion of variant exon 6 (CD44v6) | CD44v6 is implicated in the process of lung tumorigenesis | CAR T cell therapy | [57,58] |
| PD-1 | Deletion of exon 3 (sPD-1) | sPD-1 enhances anti-tumor immunity | Anti-PD-1/PD-L1 therapy | [59,60] |
| Bcl-x | Alternative last exon (Bcl-xL) | Bcl-xL inhibits tumor cell apoptosis | ASO | [61,62] |
| CDC25C | Exon 3 skipping (CDC25C-ΔEx3) | CDC25C-ΔEx3 inhibits cell proliferation | / | [63] |
| KAT2A | Alternative 5′ splicing | The splicing variant promotes LUAD progression | / | [64] |
| EIF4H | Exon 5 skipping (EIF4H-ΔEx5) | EIF4H-ΔEx5 promotes LUAD progression | / | [65] |
| IMPAD1 | Alternative last exon | The splicing variant promotes LUAD proliferation | / | [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, W.; Xu, M.; Wong, N.; Ng, C.S.-H. Alternative Splicing in Lung Adenocarcinoma: From Bench to Bedside. Cancers 2025, 17, 1329. https://doi.org/10.3390/cancers17081329
Luo W, Xu M, Wong N, Ng CS-H. Alternative Splicing in Lung Adenocarcinoma: From Bench to Bedside. Cancers. 2025; 17():1329. https://doi.org/10.3390/cancers17081329
Chicago/Turabian StyleLuo, Wenjie, Mingjing Xu, Nathalie Wong, and Calvin Sze-Hang Ng. 2025. "Alternative Splicing in Lung Adenocarcinoma: From Bench to Bedside" Cancers 17, no. : 1329. https://doi.org/10.3390/cancers17081329
APA StyleLuo, W., Xu, M., Wong, N., & Ng, C. S.-H. (2025). Alternative Splicing in Lung Adenocarcinoma: From Bench to Bedside. Cancers, 17(), 1329. https://doi.org/10.3390/cancers17081329