
Identification of Driver Mutations and Risk Stratification in Lung Adenocarcinoma via Liquid Biopsy

 , ,
, ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Participant and Clinicopathological Data
2.2. Sample Collection and Processing
2.3. Sequencing and Analysis
2.4. Data Visualization and Statistical Analysis
3. Results
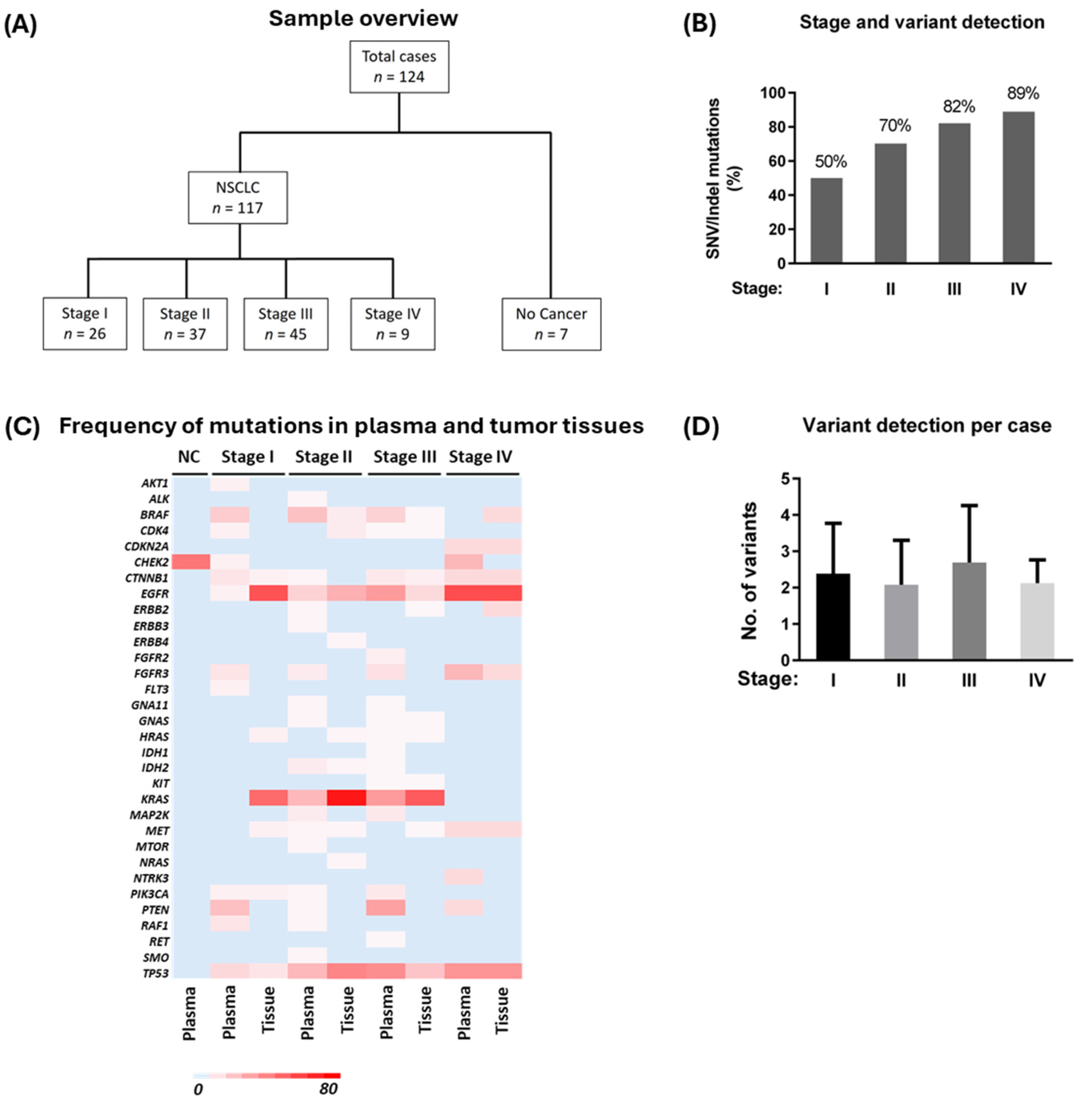
3.1. Participants and Sample Overview
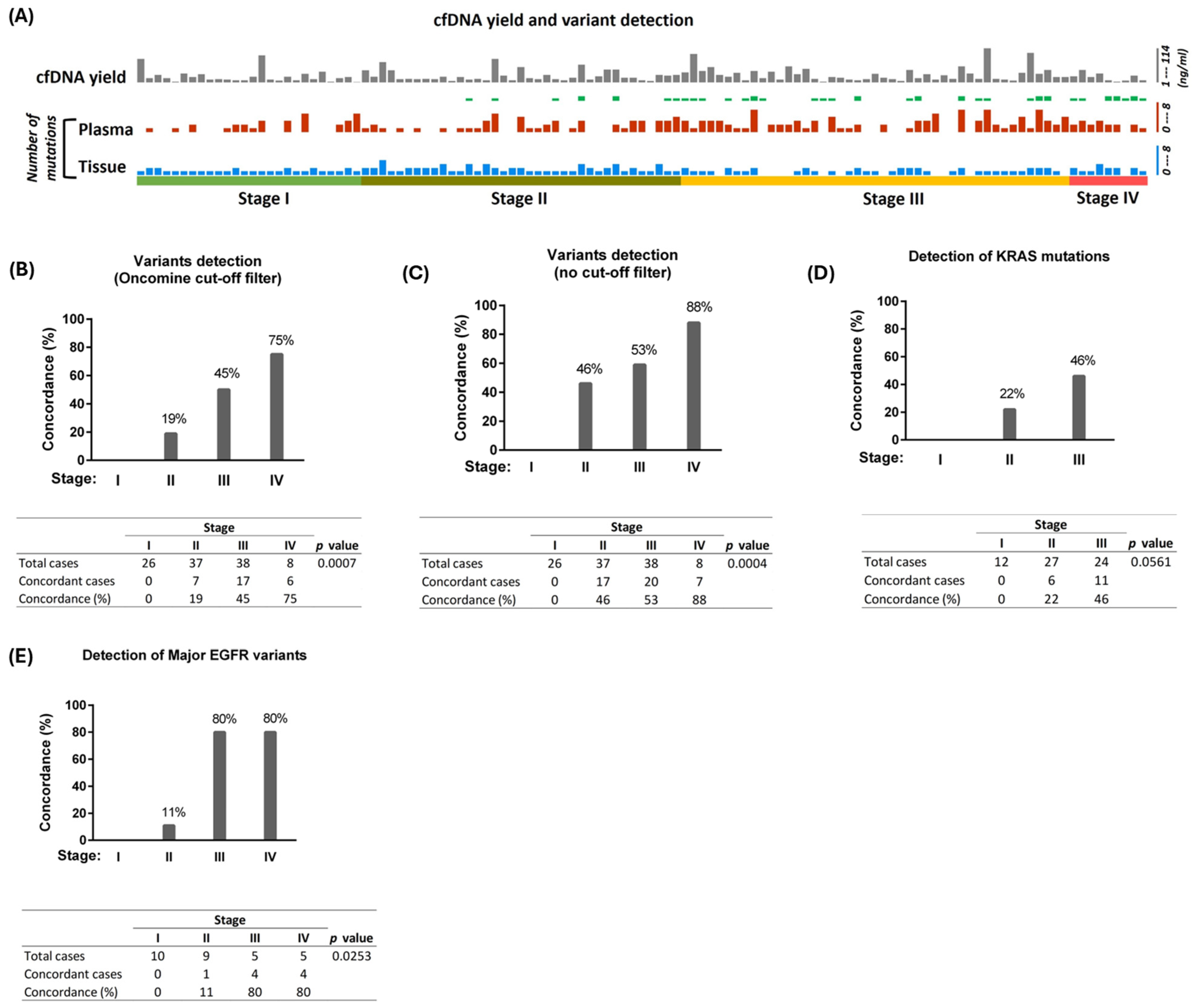
3.2. Variant Detection in Plasma cfDNA and Matched Tumor Tissues from Stage I to IV Patients
3.3. Concordance of Detection of Variants Across All Four Stages of NSCLC
3.4. Detection of Major Driver Mutations
3.5. Presence of Actionable Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cfDNA | Cell-free DNA |
| cfRNA | Cell-free RNA |
| CNV | Copy number variation |
| ctDNA | Circulating tumor DNA |
| FFPE | Formalin-fixed paraffin-embedded |
| Indel | Insertion and deletion |
| NSCLC | Non-small cell lung cancer |
| SD | Standard deviation |
| SNV | Single nucleotide variant |
| TNA | Total nucleic acid |
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics 2023; Canadian Cancer Society: Toronto, ON, Canada, 2023; Available online: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2023-statistics/2023_PDF_EN.pdf (accessed on 1 November 2024).
- Bach, P.B.; Mirkin, J.N.; Oliver, T.K.; Azzoli, C.G.; Berry, D.A.; Brawley, O.W.; Byers, T.; Colditz, G.A.; Gould, M.K.; Jett, J.R.; et al. Benefits and Harms of CT Screening for Lung Cancer: A Systematic Review. JAMA 2012, 307, 2418–2429. [Google Scholar] [CrossRef] [PubMed]
- Pathak, G.P.; Shah, R.; Castonguay, M.; Cheng, A.; Fris, J.; Murphy, R.; Darling, G.; Ednie, A.; French, D.; Henteleff, H.; et al. Temporal Effect on PD-L1 Detection and Novel Insights Into Its Clinical Implications in Non–Small Cell Lung Cancer. Cancer Med. 2024, 13, e70262. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Zhang, J.; IJzerman, M.J.; Oberoi, J.; Karnchanachari, N.; Bergin, R.J.; Franchini, F.; Druce, P.; Wang, X.; Emery, J.D. Time to diagnosis and treatment of lung cancer: A systematic overview of risk factors, interventions and impact on patient outcomes. Lung Cancer 2022, 166, 27–39. [Google Scholar] [CrossRef]
- Frangioni, J.V. New Technologies for Human Cancer Imaging. J. Clin. Oncol. 2008, 26, 4012–4021. [Google Scholar] [CrossRef]
- Alwithenani, A.; Bethune, D.; Castonguay, M.; Drucker, A.; Flowerdew, G.; Forsythe, M.; French, D.; Fris, J.; Greer, W.; Henteleff, H.; et al. Profiling non-small cell lung cancer reveals that PD-L1 is associated with wild type EGFR and vascular invasion, and immunohistochemistry quantification of PD-L1 correlates weakly with RT-qPCR. PLoS ONE 2021, 16, e0251080. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Remon, J.; Soria, J.C.; Peters, S. Early and locally advanced non-small-cell lung cancer: An update of the ESMO Clinical Practice Guidelines focusing on diagnosis, staging, systemic and local therapy. Ann. Oncol. 2021, 32, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Batool, S.M.; Yekula, A.; Khanna, P.; Hsia, T.; Gamblin, A.S.; Ekanayake, E.; Escobedo, A.K.; You, D.G.; Castro, C.M.; Im, H.; et al. The Liquid Biopsy Consortium: Challenges and opportunities for early cancer detection and monitoring. Cell Rep. Med. 2023, 4, 101198. [Google Scholar] [CrossRef]
- Russo, A.; Lee, J.K.; Pasquina, L.W.; Del Re, M.; Dilks, H.H.; Murugesan, K.; Madison, R.W.; Lee, Y.; Schrock, A.B.; Comment, L.; et al. Liquid Biopsy of Lung Cancer Before Pathological Diagnosis Is Associated With Shorter Time to Treatment. JCO Precis. Oncol. 2024, 8, e2300535. [Google Scholar] [CrossRef]
- Garcia-Pardo, M.; Czarnecka, K.; Law, J.H.; Salvarrey, A.; Fernandes, R.; Fan, J.; Corke, L.; Waddell, T.K.; Yasufuku, K.; Donahoe, L.L.; et al. Plasma-first: Accelerating lung cancer diagnosis and molecular profiling through liquid biopsy. Ther. Adv. Med. Oncol. 2022, 14, 17588359221126151. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Souza, V.G.P.; Forder, A.; Brockley, L.J.; Pewarchuk, M.E.; Telkar, N.; de Araújo, R.P.; Trejo, J.; Benard, K.; Seneda, A.L.; Minutentag, I.W.; et al. Liquid Biopsy in Lung Cancer: Biomarkers for the Management of Recurrence and Metastasis. Int. J. Mol. Sci. 2023, 24, 8894. [Google Scholar] [CrossRef]
- Bonanno, L.; Dal Maso, A.; Pavan, A.; Zulato, E.; Calvetti, L.; Pasello, G.; Guarneri, V.; Conte, P.; Indraccolo, S. Liquid biopsy and non-small cell lung cancer: Are we looking at the tip of the iceberg? Br. J. Cancer 2022, 127, 383–393. [Google Scholar] [CrossRef]
- Angeles, A.K.; Christopoulos, P.; Yuan, Z.; Bauer, S.; Janke, F.; Ogrodnik, S.J.; Reck, M.; Schlesner, M.; Meister, M.; Schneider, M.A.; et al. Early identification of disease progression in ALK-rearranged lung cancer using circulating tumor DNA analysis. Npj Precis. Oncol. 2021, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Breadner, D.; Hwang, D.M.; Husereau, D.; Cheema, P.; Doucette, S.; Ellis, P.M.; Kassam, S.; Leighl, N.; Maziak, D.E.; Selvarajah, S.; et al. Implementation of Liquid Biopsy in Non-Small-Cell Lung Cancer: An Ontario Perspective. Curr. Oncol. 2024, 31, 6017–6031. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.E.; Reuland, D.S.; Reddy, S.M.; Nagle, M.; Clark, S.D.; Weber, R.P.; Enyioha, C.; Malo, T.L.; Brenner, A.T.; Armstrong, C.; et al. Screening for Lung Cancer With Low-Dose Computed Tomography: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2021, 325, 971–987. [Google Scholar] [CrossRef]
- American Lung Association. State of Lung Cancer; American Lung Association: Chicago, IL, USA, 2022. [Google Scholar]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Low, S.K.; Ariyasu, R.; Uchibori, K.; Hayashi, R.; Chan, H.T.; Chin, Y.M.; Akita, T.; Harutani, Y.; Kiritani, A.; Tsugitomi, R.; et al. Rapid genomic profiling of circulating tumor DNA in non-small cell lung cancer using Oncomine Precision Assay with GenexusTM integrated sequencer. Transl. Lung Cancer Res. 2022, 11, 711–721. [Google Scholar] [CrossRef]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1-16PO.17.00011. [Google Scholar] [CrossRef]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef]
- Sunaga, N.; Miura, Y.; Kasahara, N.; Sakurai, R. Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer. Cancers 2021, 13, 5956. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef] [PubMed]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef]
- Sugimoto, A.; Matsumoto, S.; Udagawa, H.; Itotani, R.; Usui, Y.; Umemura, S.; Nishino, K.; Nakachi, I.; Kuyama, S.; Daga, H.; et al. A Large-Scale Prospective Concordance Study of Plasma- and Tissue-Based Next-Generation Targeted Sequencing for Advanced Non–Small Cell Lung Cancer (LC-SCRUM-Liquid). Clin. Cancer Res. 2023, 29, 1506–1514. [Google Scholar] [CrossRef]
- Nicholas, C.; Beharry, A.; Bendzsak, A.M.; Bisson, K.R.; Dadson, K.; Dudani, S.; Iafolla, M.; Irshad, K.; Perdrizet, K.; Raskin, W.; et al. Point of Care Liquid Biopsy for Cancer Treatment—Early Experience from a Community Center. Cancers 2024, 16, 2505. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef]
- Park, S.; Olsen, S.; Ku, B.M.; Lee, M.S.; Jung, H.A.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.L.; et al. High concordance of actionable genomic alterations identified between circulating tumor DNA–based and tissue-based next-generation sequencing testing in advanced non–small cell lung cancer: The Korean Lung Liquid Versus Invasive Biopsy Program. Cancer 2021, 127, 3019–3028. [Google Scholar] [CrossRef]
- Itotani, R.; Matsumoto, S.; Udagawa, H.; Nishino, K.; Nakachi, I.; Miyamoto, S.; Hara, S.; Kuyama, S.; Ebi, N.; Tsubata, Y.; et al. A large scale prospective concordance study of oncogene driver detection between plasma- and tissue-based NGS analysis in advanced non-small cell lung cancer (NSCLC). Ann. Oncol. 2019, 30, v641-2. [Google Scholar] [CrossRef]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef]
- Iams, W.T.; Mackay, M.; Ben-Shachar, R.; Drews, J.; Manghnani, K.; Hockenberry, A.J.; Cristofanilli, M.; Nimeiri, H.; Guinney, J.; Benson, A.B. Concurrent Tissue and Circulating Tumor DNA Molecular Profiling to Detect Guideline-Based Targeted Mutations in a Multicancer Cohort. JAMA Netw. Open 2024, 7, e2351700. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Velculescu, V.E. Pushing the Boundaries of Liquid Biopsies for Early Precision Intervention. Cancer Discov. 2024, 14, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Clarke, M.A. Liquid Biopsy for Cancer Detection: Clinical and Epidemiologic Considerations. Clin. Cancer Res. 2021, 27, 5733–5735. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.D.; Madison, R.W.; Pasquina, L.W.; Brown, D.W.; Huang, Y.; Hughes, J.D.; Graf, R.P.; Oxnard, G.R.; Husain, H. Measurement of ctDNA Tumor Fraction Identifies Informative Negative Liquid Biopsy Results and Informs Value of Tissue Confirmation. Clin. Cancer Res. 2024, 30, 2452–2460. [Google Scholar] [CrossRef]
- Forsythe, M.L.; Alwithenani, A.; Bethune, D.; Castonguay, M.; Drucker, A.; Flowerdew, G.; French, D.; Fris, J.; Greer, W.; Henteleff, H.; et al. Molecular profiling of non-small cell lung cancer. PLoS ONE 2020, 15, e0236580. [Google Scholar] [CrossRef]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS Pathway in Non-Small Cell Lung Cancer. Oncologist 2016, 21, 1450–1460. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients With Oncogenic Driver Molecular Alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Ancevski Hunter, K.; Friedland, D.M.; Villaruz, L.C.; Burns, T.F. First-Line Osimertinib in Patients with Treatment-Naive Somatic or Germline EGFR T790M–Mutant Metastatic NSCLC. J. Thorac. Oncol. 2018, 13, e3–e5. [Google Scholar] [CrossRef]
- Chang, W.J.; Sung, J.S.; Lee, S.Y.; Kang, E.J.; Kwon, N.J.; Kim, H.M.; Shin, S.W.; Choi, J.Y.; Choi, Y.J.; Kim, J.W.; et al. The Clinical Significance of RAS, PIK3CA, and PTEN Mutations in Non-Small Cell Lung Cancer Using Cell-Free DNA. J. Clin. Med. 2020, 9, 2642. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Ghini, V.; Laera, L.; Fantechi, B.; del Monte, F.; Benelli, M.; McCartney, A.; Tenori, L.; Luchinat, C.; Pozzessere, D. Metabolomics to Assess Response to Immune Checkpoint Inhibitors in Patients with Non-Small-Cell Lung Cancer. Cancers 2020, 12, 3574. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, T.; Kisluk, J.; Pietrowska, K.; Godzien, J.; Kozlowski, M.; Reszeć, J.; Sierko, E.; Naumnik, W.; Mróz, R.; Moniuszko, M.; et al. The Ability of Metabolomics to Discriminate Non-Small-Cell Lung Cancer Subtypes Depends on the Stage of the Disease and the Type of Material Studied. Cancers 2021, 13, 3314. [Google Scholar] [CrossRef] [PubMed]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef]
- Silveira, C.; Sousa, A.C.; Janeiro, A.; Malveiro, S.; Teixeira, E.; Brysch, E.; Pantarotto, M.; Felizardo, M.; Madureira, R.; Nogueira, F.; et al. Detection and quantification of EGFR T790M mutation in liquid biopsies by droplet digital PCR. Transl. Lung Cancer Res. 2021, 10, 1200–1208. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Characteristics | Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|---|
| Total cases (n = 117) | ||||
| No. of cases at each stage | 26 | 37 | 45 | 9 |
| Average primary tumor size (cm) | 2.55 | 3.42 | 5.66 | 3.55 |
| Median age (SD) | 69 (8) | 67 (7) | 66 (8) | 67 (7) |
| Sex | ||||
| Female | 18 (69%) | 25 (68%) | 21 (47%) | 4 (44%) |
| Male | 8 (31%) | 12 (32%) | 24 (53%) | 5 (56%) |
| Characteristics | Number of Cases (%) | |||
|---|---|---|---|---|
| SNV/Indel variants | Stage I | Stage II | Stage III | Stage IV |
| Plasma+, Tissue+ | 13 (50) | 26 (70) | 30 (67) | 8 (89) |
| Plasma+, Tissue− | 0 (0) | 0 (0) | 7 (15) * | 0 (0) |
| Plasma−, Tissue+ | 13 (50) | 11 (30) | 4 (9) | 0 (0) |
| Plasma−, Tissue− | 0 (0) | 0 (0) | 4 (9) ** | 1 (11) |
| CNV/fusion/LoF | Stage I | Stage II | Stage III | Stage IV |
| Plasma+, Tissue+ | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Plasma+, Tissue− | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Plasma−, Tissue+ | 0 | 0 | 4 (9) | 0 (0) |
| Plasma−, Tissue− | 26 (100) | 37 (100) | 41 (91) | 9 (100) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, G.P.; Shah, R.; Reiman, T.; Wallace, A.; Carter, M.D.; Snow, S.; Fris, J.; Xu, Z. Identification of Driver Mutations and Risk Stratification in Lung Adenocarcinoma via Liquid Biopsy. Cancers 2025, 17, 1340. https://doi.org/10.3390/cancers17081340
Pathak GP, Shah R, Reiman T, Wallace A, Carter MD, Snow S, Fris J, Xu Z. Identification of Driver Mutations and Risk Stratification in Lung Adenocarcinoma via Liquid Biopsy. Cancers. 2025; 17(8):1340. https://doi.org/10.3390/cancers17081340
Chicago/Turabian StylePathak, Gopal P., Rashmi Shah, Tony Reiman, Alison Wallace, Michael D. Carter, Stephanie Snow, John Fris, and Zhaolin Xu. 2025. "Identification of Driver Mutations and Risk Stratification in Lung Adenocarcinoma via Liquid Biopsy" Cancers 17, no. 8: 1340. https://doi.org/10.3390/cancers17081340
APA StylePathak, G. P., Shah, R., Reiman, T., Wallace, A., Carter, M. D., Snow, S., Fris, J., & Xu, Z. (2025). Identification of Driver Mutations and Risk Stratification in Lung Adenocarcinoma via Liquid Biopsy. Cancers, 17(8), 1340. https://doi.org/10.3390/cancers17081340