

Unravelling Paclitaxel Resistance in Gastric Cancer: The Role of Small Extracellular Vesicles in Epithelial Mesenchymal Transition and Extracellular Matrix Remodelling

 , , , , ,
, , , , ,  ,
,  and
and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients Groups, Serum Preparation and Storage
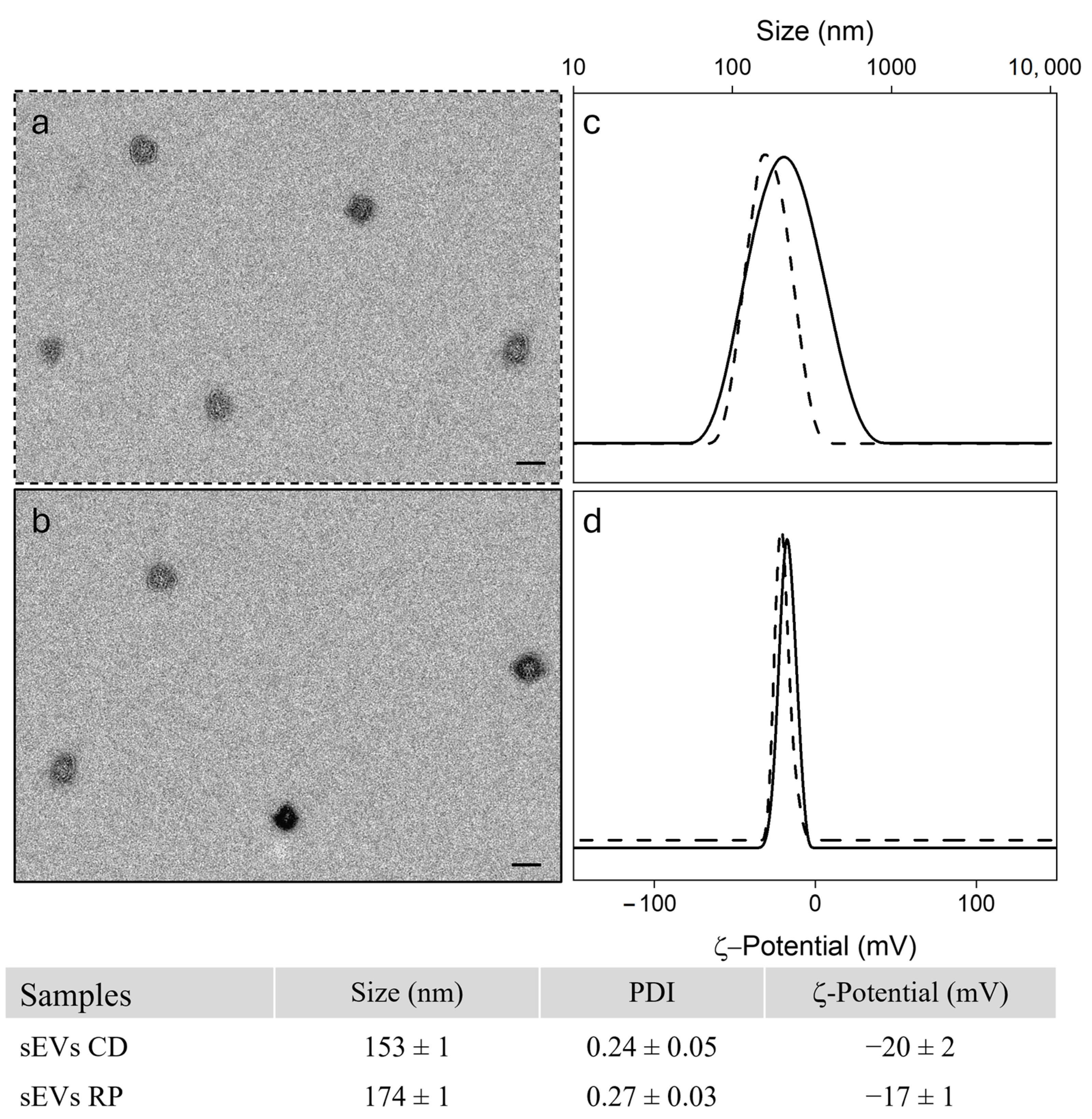
2.2. Isolation of Serum Derived Small ExtracellularVesicles
2.3. Bioinformatics Analysis
2.4. Cell Culture
2.5. RNA Extraction and Gene Expression Analysis Through ddPCR
2.6. Cell Viability of HCEC-1CT and HEPA-RG Cell Lines After Treatment with sEVs
2.7. Proteins Evaluation
2.8. Statistical Analysis
3. Results
3.1. Patients Characteristics
3.2. Bioinformatic and Experimental Analysis of Tumour-Related Proteins in Gastric Cancer sEVs
3.3. Impact of sEVs Derived from CD and RP Patients on HCEC-1CT and HEPA-RG Cell Lines Viability
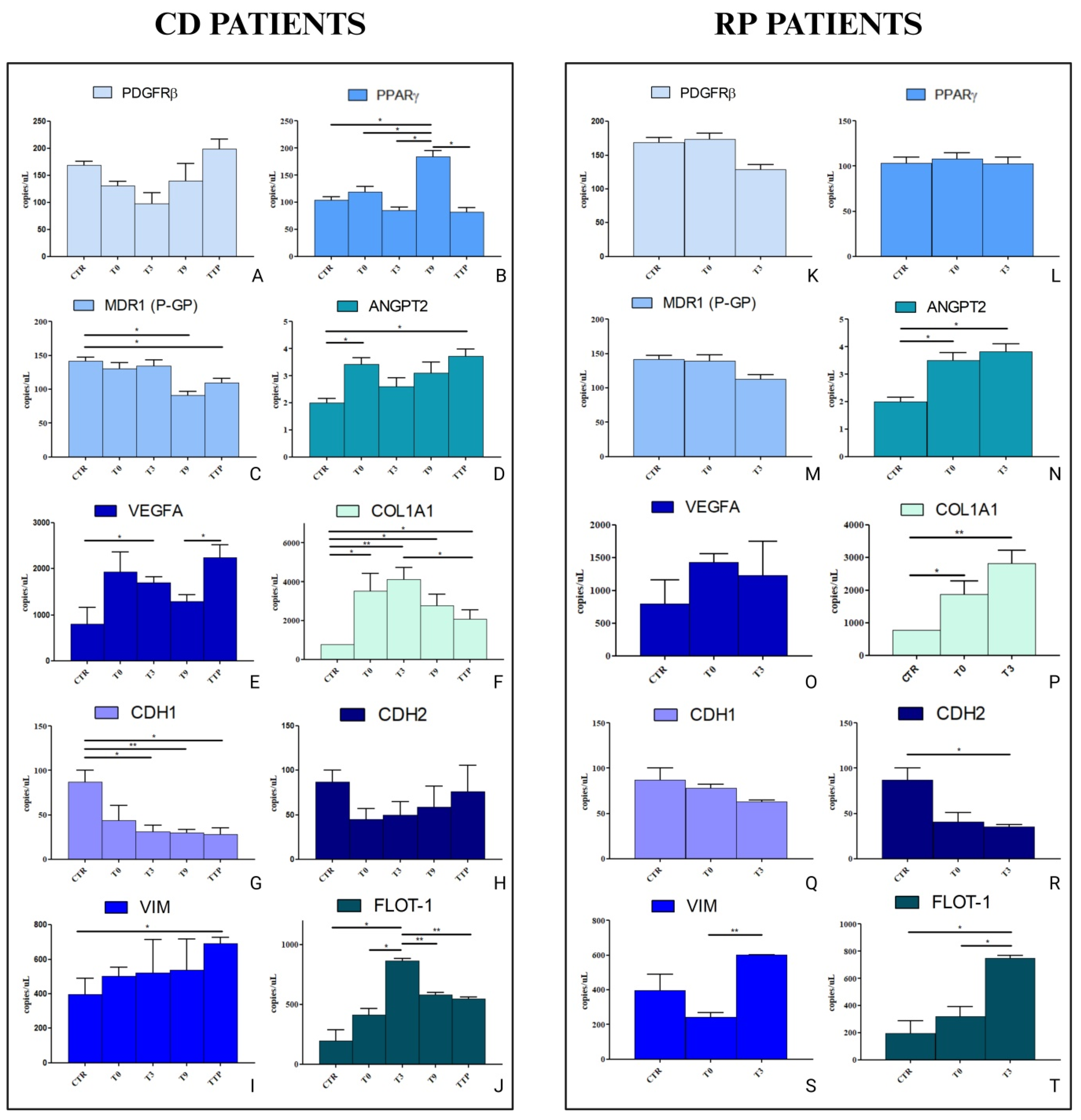
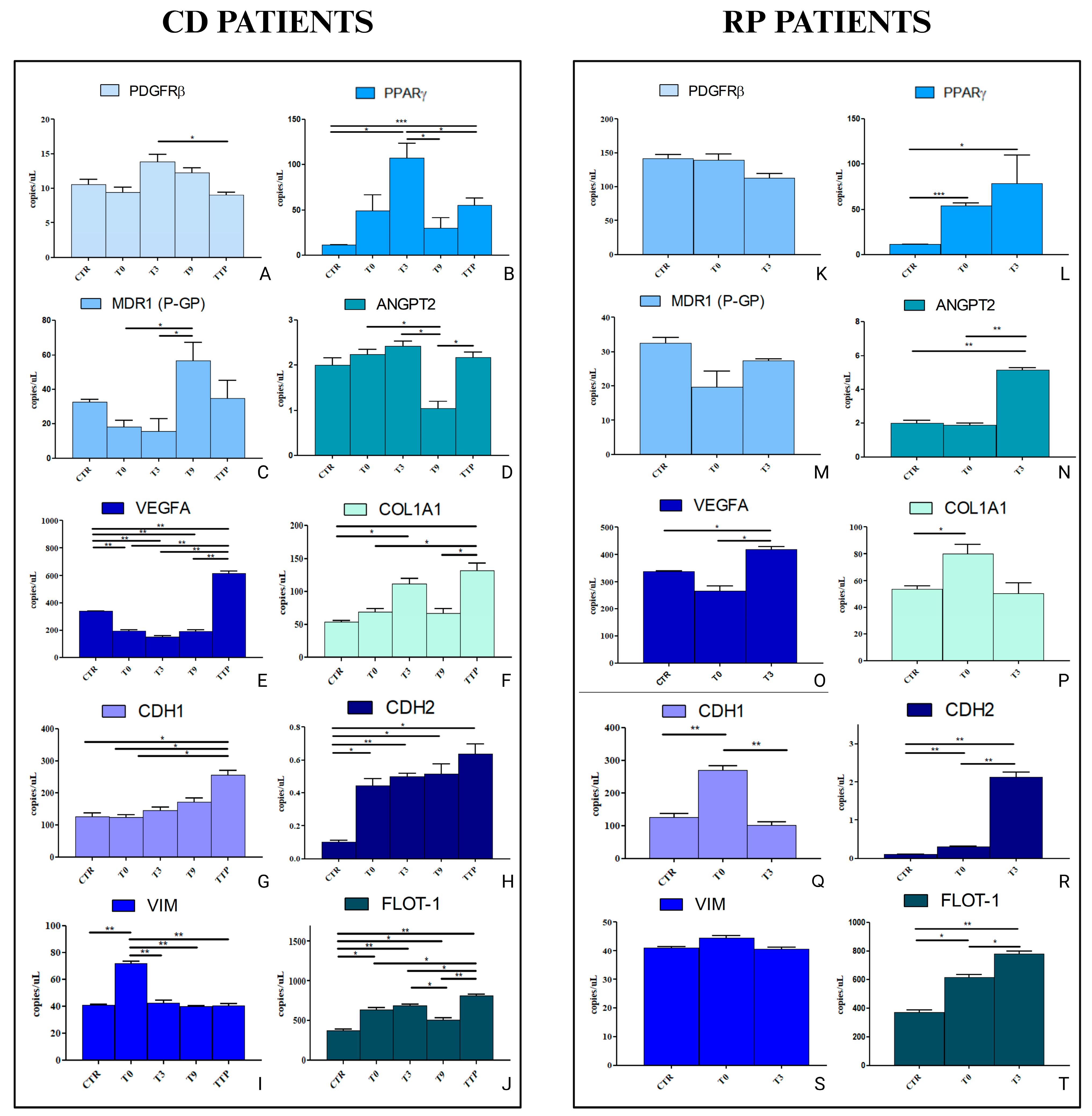
3.4. Analysis of Gene Expression in HCEC-1CT and HEPA-RG Cell Lines Treated with sEVs from CD and RP Patients Affected by Metastatic Gastric Cancer
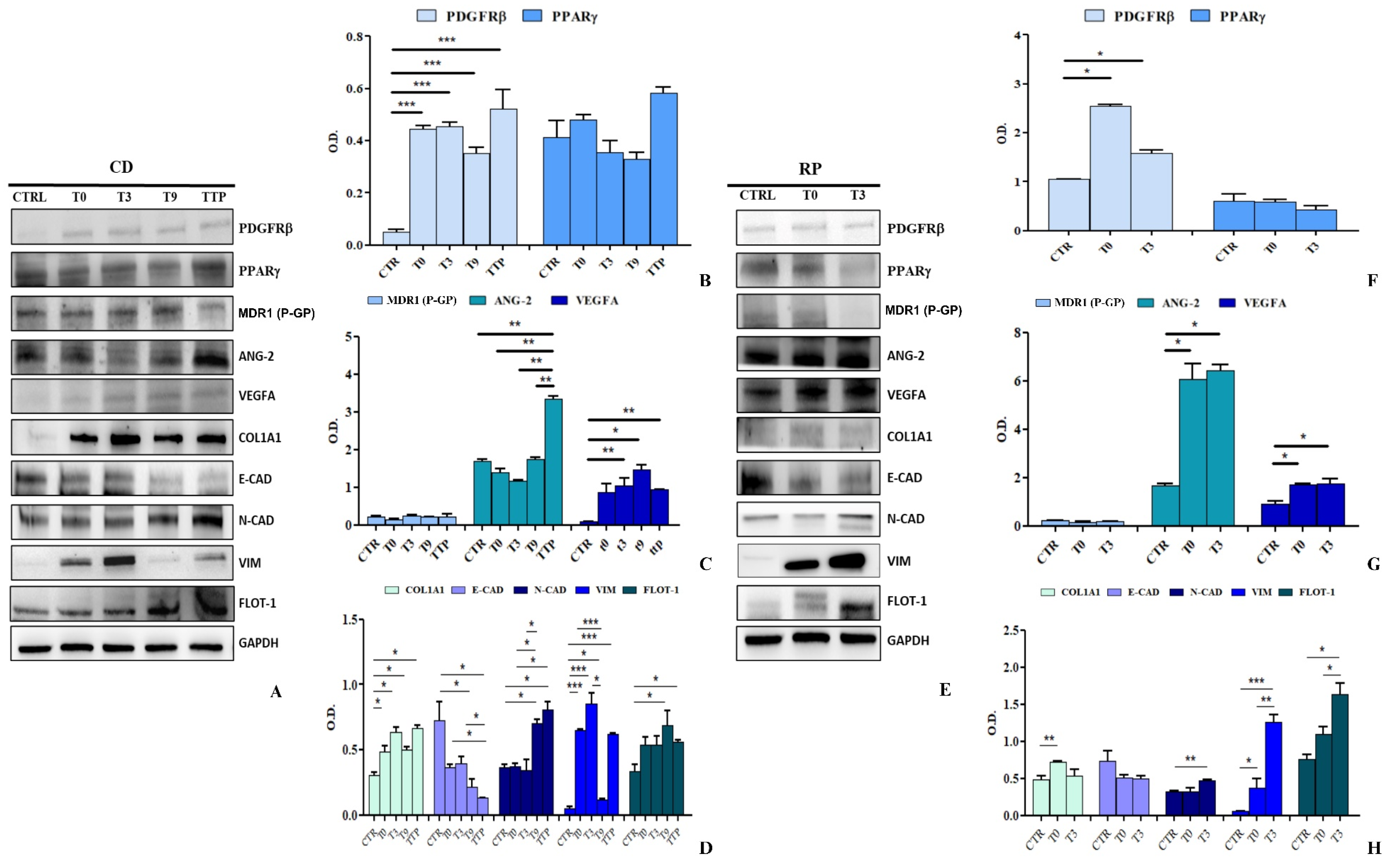
3.5. Analysis of Protein Expression in HCEC-1CT and HEPA-RG Cells Stimulated with sEVs from CD and RP Patients with Metastatic Gastric Cancer
4. Discussion
5. Conclusions and Future Direction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Sato, Y.; Okamoto, K.; Kida, Y.; Mitsui, Y.; Kawano, Y.; Sogabe, M.; Miyamoto, H.; Takayama, T. Overview of Chemotherapy for Gastric Cancer. J. Clin. Med. 2023, 12, 1336. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Anand, U.; Pandey, S.K.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.S.; Dey, A. Therapeutic Strategies to Overcome Taxane Resistance in Cancer. Drug Resist. Updat. 2021, 55, 100754. [Google Scholar] [CrossRef] [PubMed]
- Jurj, A.; Ionescu, C.; Berindan-Neagoe, I.; Braicu, C. The Extracellular Matrix Alteration, Implication in Modulation of Drug Resistance Mechanism: Friends or Foes? J. Exp. Clin. Cancer Res. 2022, 41, 276. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, J.; Gu, J.; Shi, H.; Zhang, J.; Zhang, J.; Chen, Z.S.; Fang, X.; Zhu, T.; Zhang, X. Extracellular Vesicles in Cancer Drug Resistance: Roles, Mechanisms, and Implications. Adv. Sci. 2022, 9, e2201609. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal Information for Studies of Extracellular Vesicles (MISEV2023): From Basic to Advanced Approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Scavo, M.P.; Rizzi, F.; Depalo, N.; Fanizza, E.; Ingrosso, C.; Curri, M.L.; Giannelli, G. A Possible Role of Fzd10 Delivering Exosomes Derived from Colon Cancers Cell Lines in Inducing Activation of Epithelial–Mesenchymal Transition in Normal Colon Epithelial Cell Line. Int. J. Mol. Sci. 2020, 21, 6705. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Shin, E.; Seong, K.M.; Jin, Y.W.; Youn, H.; Youn, B. The Emerging Roles of Exosomes as EMT Regulators in Cancer. Cells 2020, 9, 861. [Google Scholar] [CrossRef]
- Hraběta, J.; Belhajová, M.; Šubrtová, H.; Rodrigo, M.A.M.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 4392. [Google Scholar] [CrossRef]
- Nguyen, M.K.L.; Jose, J.; Wahba, M.; Bernaus-Esqué, M.; Hoy, A.J.; Enrich, C.; Rentero, C.; Grewal, T. Linking Late Endosomal Cholesterol with Cancer Progression and Anticancer Drug Resistance. Int. J. Mol. Sci. 2022, 23, 7206. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.-S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef]
- Schirizzi, A.; Contino, M.; Carrieri, L.; Riganti, C.; De Leonardis, G.; Scavo, M.P.; Perrone, M.G.; Miciaccia, M.; Kopecka, J.; Refolo, M.G.; et al. The Multiple Combination of Paclitaxel, Ramucirumab and Elacridar Reverses the Paclitaxel-Mediated Resistance in Gastric Cancer Cell Lines. Front. Oncol. 2023, 13, 1129832. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Cui, Q.; Zheng, H.; Ma, Y.; Kang, Y.; Tang, K. Challenges and Opportunities for Extracellular Vesicles in Clinical Oncology Therapy. Bioengineering 2023, 10, 325. [Google Scholar] [CrossRef]
- Liu, N.; Liu, M.; Fu, S.; Wang, J.; Tang, H.; Isah, A.D.; Chen, D.; Wang, X. Ang2-Targeted Combination Therapy for Cancer Treatment. Front. Immunol. 2022, 13, 949553. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus Paclitaxel versus Placebo plus Paclitaxel in Patients with Previously Treated Advanced Gastric or Gastro-Oesophageal Junction Adenocarcinoma (RAINBOW): A Double-Blind, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New Insights about the PDGF/PDGFR Signaling Pathway as a Promising Target to Develop Cancer Therapeutic Strategies. Biomed. Pharmacother. 2023, 161, 114491. [Google Scholar] [CrossRef]
- Zou, X.; Tang, X.Y.; Qu, Z.Y.; Sun, Z.W.; Ji, C.F.; Li, Y.J.; Guo, S.D. Targeting the PDGF/PDGFR Signaling Pathway for Cancer Therapy: A Review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tabernero, J.; Tomášek, J.; Chau, I.; Melichar, B.; Safran, H.; Tehfe, M.A.; Filip, D.; Topuzov, E.; Schlittler, L.; et al. Biomarker Analyses in REGARD Gastric/GEJ Carcinoma Patients Treated with VEGFR2-Targeted Antibody Ramucirumab. Br. J. Cancer 2016, 115, 974–982. [Google Scholar] [CrossRef]
- Du, S.; Yang, Z.; Lu, X.; Yousuf, S.; Zhao, M.; Li, W.; Miao, J.; Wang, X.; Yu, H.; Zhu, X.; et al. Anoikis Resistant Gastric Cancer Cells Promote Angiogenesis and Peritoneal Metastasis through C/EBPβ-Mediated PDGFB Autocrine and Paracrine Signaling. Oncogene 2021, 40, 5764–5779. [Google Scholar] [CrossRef]
- Du, S.; Wagner, N.; Wagner, K.D. The Emerging Role of PPAR Beta/Delta in Tumor Angiogenesis. PPAR Res. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, F.; Memarzia, A.; Alikhani, V.; Beigoli, S.; Boskabady, M.H. Peroxisome Proliferator-Activated Receptors: Key Regulators of Tumor Progression and Growth. Transl. Oncol. 2024, 47, 102039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ng, A.S.; Cai, S.; Li, Q.; Yang, L.; Kerr, D. Novel Therapeutic Strategies: Targeting Epithelial–Mesenchymal Transition in Colorectal Cancer. Lancet Oncol. 2021, 22, e358–e368. [Google Scholar] [CrossRef]
- de Carvalho, M.V.; Gonçalves-De-Albuquerque, C.F.; Silva, A.R. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. Int. J. Mol. Sci. 2021, 22, 805. [Google Scholar] [CrossRef]
- Bodin, S.; Planchon, D.; Morris, E.R.; Comunale, F.; Gauthier-Rouviére, C. Flotillins in Intercellular Adhesion—From Cellular Physiology to Human Diseases. J. Cell Sci. 2014, 127, 5139–5147. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Li, X.; Ai, S.; Lu, X.; Liu, S.; Guan, W. Nanotechnology-Based Strategies for Gastric Cancer Imaging and Treatment. RSC Adv. 2021, 11, 35392–35407. [Google Scholar] [CrossRef]
- Piccinno, E.; Schirizzi, A.; Scalavino, V.; De Leonardis, G.; Donghia, R.; Fantasia, A.; Ricci, A.D.; Lotesoriere, C.; Giannelli, G.; Serino, G.; et al. Circulating MiR-23b-3p, MiR-30e-3p, and MiR-205-5p as Novel Predictive Biomarkers for Ramucirumab–Paclitaxel Therapy Outcomes in Advanced Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 13498. [Google Scholar] [CrossRef] [PubMed]
- Scavo, M.P.; Cigliano, A.; Depalo, N.; Fanizza, E.; Bianco, M.G.; Denora, N.; Laquintana, V.; Curri, M.L.; Lorusso, D.; Lotesoriere, C.; et al. Frizzled-10 Extracellular Vesicles Plasma Concentration Is Associated with Tumoral Progression in Patients with Colorectal and Gastric Cancer. J. Oncol. 2019, 2019, 2715968, Erratum in J. Oncol. 2020, 2020, 6153432. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. clinicians 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Jeon, Y.; Lim, S.H.; Lee, J.; Kang, W.K.; Jang, J.Y.; Jeong, S.Y.; Choi, D.; Kim, S.T. The Role of Ramucirumab plus Paclitaxel as Second-Line Therapy after Failure of Nivolumab plus Doublet Chemotherapy in Patients with Advanced Gastric Cancer. J. Gastrointest. Oncol. 2023, 14, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Muriithi, W.; Macharia, L.W.; Heming, C.P.; Echevarria, J.L.; Nyachieo, A.; Filho, P.N.; Neto, V.M. ABC Transporters and the Hallmarks of Cancer: Roles in Cancer Aggressiveness beyond Multidrug Resistance. Cancer Biol. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Gao, Z.; Jin, L.; Geng, H.; Liang, Z. Novel Role of CircRNAs in the Drug Resistance of Gastric Cancer: Regulatory Mechanisms and Future for Cancer Therapy. Front. Pharmacol. 2024, 15, 1435264. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lei, Y.; Wang, Y.; Lai, J.; Wang, J.; Xia, F. Mechanism of Multidrug Resistance to Chemotherapy Mediated by P-glycoprotein (Review). Int. J. Oncol. 2023, 63, 1–19. [Google Scholar] [CrossRef]
- Wu, Q.; Sharma, D. Autophagy and Breast Cancer: Connected in Growth, Progression, and Therapy. Cells 2023, 12, 1156. [Google Scholar] [CrossRef]
- Li, Y.; Lu, L.; Wu, X.; Li, Q.; Zhao, Y.; Du, F.; Chen, Y.; Shen, J.; Xiao, Z.; Wu, Z.; et al. The Multifaceted Role of Long Non-Coding RNA in Gastric Cancer: Current Status and Future Perspectives. Int. J. Biol. Sci. 2021, 17, 2737–2755. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.-X.; Wu, H.-T.; Li, X.-L.; Wen, X.-F.; Du, C.-W.; Zhang, G.-J. Collagen 1A1 (COL1A1) Promotes Metastasis of Breast Cancer and Is a Potential Therapeutic Target. Discov. Med. 2019, 25, 211–223. [Google Scholar]
- Wang, M.; Qiu, R.; Yu, S.; Xu, X.; Li, G.; Gu, R.; Tan, C.; Zhu, W.; Shen, B. Paclitaxel-Resistant Gastric Cancer MGC-803 Cells Promote Epithelial-to-Mesenchymal Transition and Chemoresistance in Paclitaxel-Sensitive Cells via Exosomal Delivery of MiR-155-5p. Int. J. Oncol. 2018, 54, 326–338. [Google Scholar] [CrossRef]
- Lopez, K.; Lai, S.W.T.; Lopez Gonzalez, E.D.J.; Dávila, R.G.; Shuck, S.C. Extracellular Vesicles: A Dive into Their Role in the Tumor Microenvironment and Cancer Progression. Front. Cell Dev. Biol. 2023, 11, 1154576. [Google Scholar] [CrossRef]
- Xu, T.; Wang, M.; Jiang, L.; Ma, L.; Wan, L.; Chen, Q.; Wei, C.; Wang, Z. CircRNAs in Anticancer Drug Resistance: Recent Advances and Future Potential. Mol. Cancer 2020, 19, 127. [Google Scholar] [CrossRef]
- Kubo, T.; Piperdi, S.; Rosenblum, J.; Antonescu, C.R.; Chen, W.; Kim, H.S.; Huvos, A.G.; Sowers, R.; Meyers, P.A.; Healey, J.H.; et al. Platelet-Derived Growth Factor Receptor as a Prognostic Marker and a Therapeutic Target for Imatinib Mesylate Therapy in Osteosarcoma. Cancer 2008, 112, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, X.; Guo, J.; Yang, B.; Li, Y. Challenges and Future of HER2-Positive Gastric Cancer Therapy. Front. Oncol. 2023, 13, 1080990. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Siddhartha, R.; Garg, M. Interplay Between Extracellular Matrix Remodeling and Angiogenesis in Tumor Ecosystem. Mol. Cancer Ther. 2023, 22, 291–305. [Google Scholar] [CrossRef]
- Taeger, J.; Moser, C.; Hellerbrand, C.; Mycielska, M.E.; Glockzin, G.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O.; Lang, S.A. Targeting FGFR/PDGFR/VEGFR Impairs Tumor Growth, Angiogenesis, and Metastasis by Effects on Tumor Cells, Endothelial Cells, and Pericytes in Pancreatic Cancer. Mol. Cancer Ther. 2011, 10, 2157–2167. [Google Scholar] [CrossRef]
- Cao, Z.; Quazi, S.; Arora, S.; Osellame, L.D.; Burvenich, I.J.; Janes, P.W.; Scott, A.M. Cancer-Associated Fibroblasts as Therapeutic Targets for Cancer: Advances, Challenges, and Future Prospects. J. Biomed. Sci. 2025, 32, 7. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Assay ID (Bio-Rad) |
|---|---|---|
| PDGFRB | Platelet Derived Growth Factor Receptor Beta | qHsaCID0013272 |
| PPARG | Peroxisome Proliferator-activated Receptor Gamma | qHsaCID0011718 |
| PGP | PhosphoGlycoProtein | qHsaCED0002291 |
| ANGPT2 | Angiopoietin 2 | qHsaCID0017615 |
| VEGFA | Vascular Endothelial Growth Factor A | qHsaCED0043454 |
| COL1A1 | Collagen type I alpha 1 chain | qHsaCED0043248 |
| CDH1 | E-cadherin | qHsaCID0015365 |
| CDH2 | N-cadherin | qHsaCID0015189 |
| VIM | Vimentin | qHsaCID0012604 |
| FLOT1 | Flotillin 1 | qHsaCED0037092 |
| Group | Progression-Free Survival (PFS), Months | Overall Survival (OS), Months | Key Features | |
|---|---|---|---|---|
| 1 | RP (n = 16) | ≤3 months (median: 2.68 months) | Median: 6.30 months | Progression at first evaluation |
| 2 | CD (n = 25) | >3 months (median: 10.38 months) | Median: 12.47 months | Stable disease or partial response at initial evaluation continued therapy until progression or toxicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panzetta, G.; Schirizzi, A.; Balestra, F.; De Luca, M.; Depalo, N.; Rizzi, F.; Ricci, A.D.; De Leonardis, G.; Lotesoriere, C.; Giannelli, G.; et al. Unravelling Paclitaxel Resistance in Gastric Cancer: The Role of Small Extracellular Vesicles in Epithelial Mesenchymal Transition and Extracellular Matrix Remodelling. Cancers 2025, 17, 1360. https://doi.org/10.3390/cancers17081360
Panzetta G, Schirizzi A, Balestra F, De Luca M, Depalo N, Rizzi F, Ricci AD, De Leonardis G, Lotesoriere C, Giannelli G, et al. Unravelling Paclitaxel Resistance in Gastric Cancer: The Role of Small Extracellular Vesicles in Epithelial Mesenchymal Transition and Extracellular Matrix Remodelling. Cancers. 2025; 17(8):1360. https://doi.org/10.3390/cancers17081360
Chicago/Turabian StylePanzetta, Giorgia, Annalisa Schirizzi, Francesco Balestra, Maria De Luca, Nicoletta Depalo, Federica Rizzi, Angela Dalia Ricci, Giampiero De Leonardis, Claudio Lotesoriere, Gianluigi Giannelli, and et al. 2025. "Unravelling Paclitaxel Resistance in Gastric Cancer: The Role of Small Extracellular Vesicles in Epithelial Mesenchymal Transition and Extracellular Matrix Remodelling" Cancers 17, no. 8: 1360. https://doi.org/10.3390/cancers17081360
APA StylePanzetta, G., Schirizzi, A., Balestra, F., De Luca, M., Depalo, N., Rizzi, F., Ricci, A. D., De Leonardis, G., Lotesoriere, C., Giannelli, G., D’Alessandro, R., & Scavo, M. P. (2025). Unravelling Paclitaxel Resistance in Gastric Cancer: The Role of Small Extracellular Vesicles in Epithelial Mesenchymal Transition and Extracellular Matrix Remodelling. Cancers, 17(8), 1360. https://doi.org/10.3390/cancers17081360