Interplay Between the Cytoskeleton and DNA Damage Response in Cancer Progression


 and
and
Simple Summary
Abstract
1. Introduction
2. The Cytoskeleton and Cancer
2.1. Microfilaments
2.2. Microtubules
2.3. Intermediate Filaments
3. DNA Damage Response and Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR Pathway | DNA Damage Type | Key Players |
|---|---|---|
| BER | SSB due to ROS, alkylation, depurination, deamination | DNA glycosylases, APE1, DNA Pol β, XRCC1 [64] |
| MMR | Wrong base pairing due to DNA replication mistakes | PCNA, MSH2, MSH3, MSH6, MLH1, PMS2, Exo1, DNA Pol δ [69,70] |
| TLS | Interstrand cross-links | PCNA, DNA Pol η, κ, ζ, θ, λ, Rev1 [77,78,79] |
| NER | DNA damage caused by UV radiation and bulky adducts due to chemicals | RAD23B, XPA, XPB, XPC, XPD, XPF, XPG, TFIIH complex, RPA, ERCC1 [75,76] |
| HR | DSB during S/G2 phases of the cell cycle | BRCA1, BRCA2, RAD51, RAD52, RAD54, WRN, BLM [94] |
| NHEJ | DSB during G0/G1 phases of the cell cycle | γH2A.X, 53BP1, Ku70/80, DNA-PKc, XRCC4, DNA Ligase IV [93,95] |
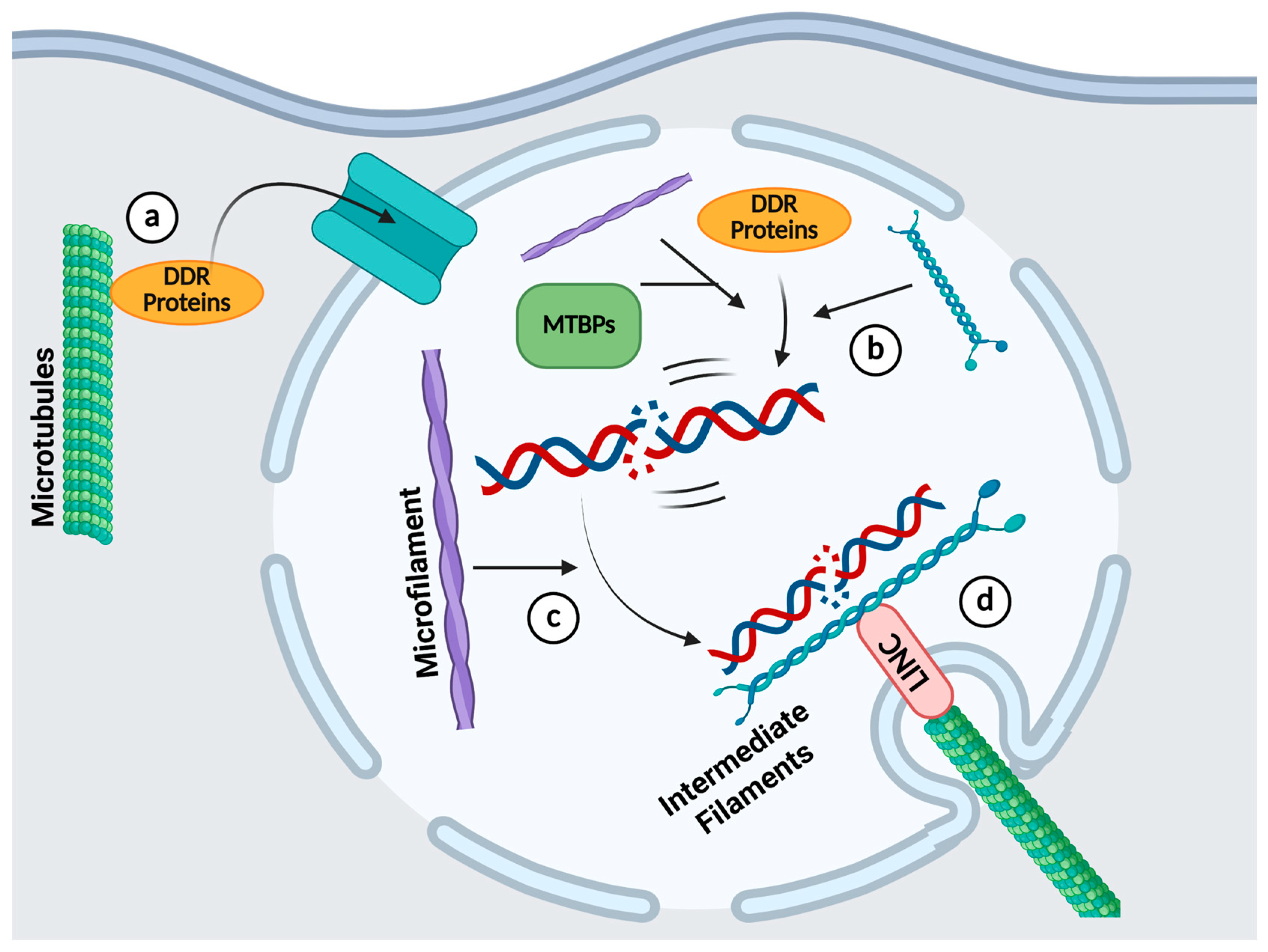
4. The Roles of the Cytoskeleton in DDR
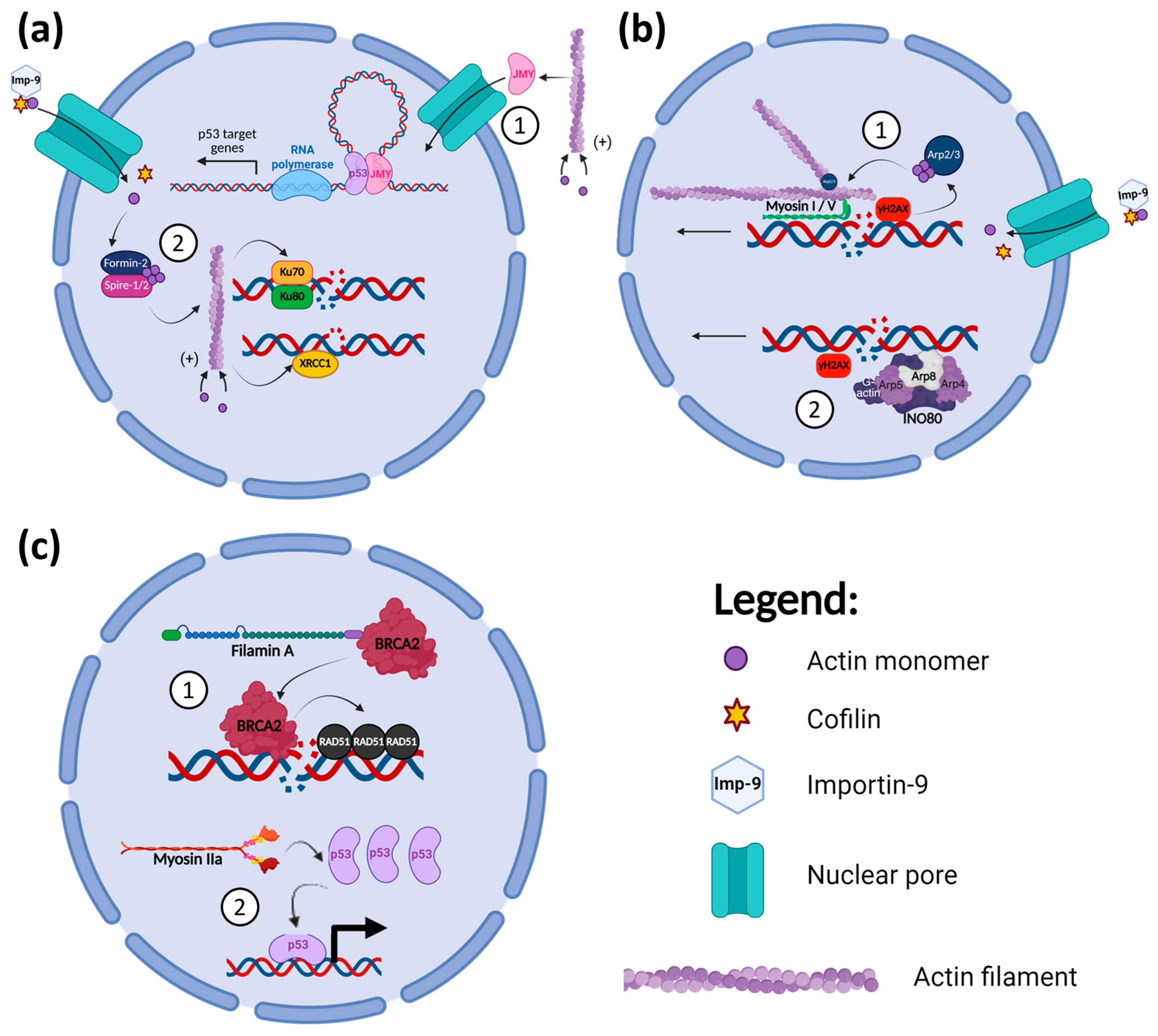
4.1. Microfilaments and DDR
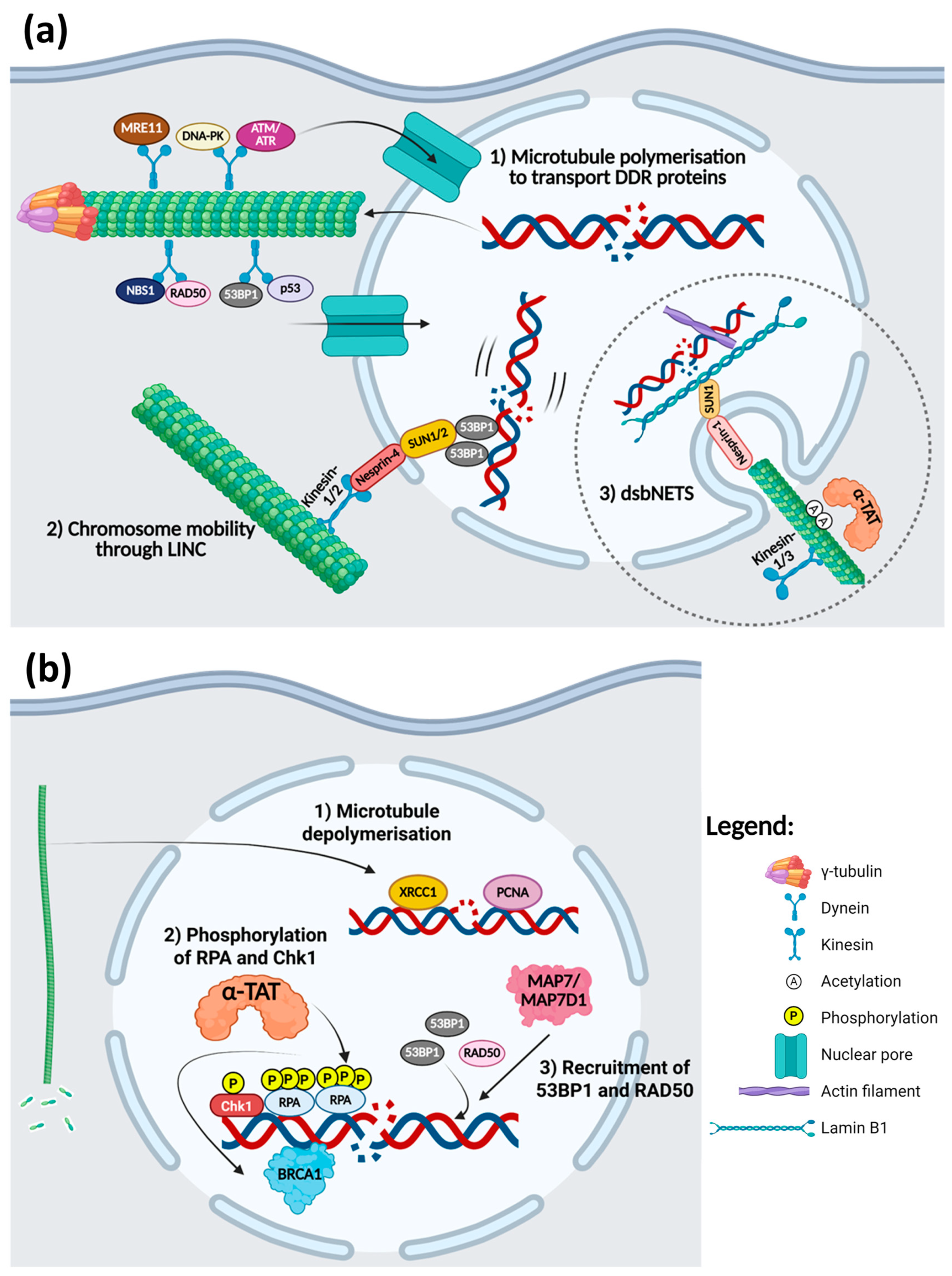
4.2. Microtubules and DDR
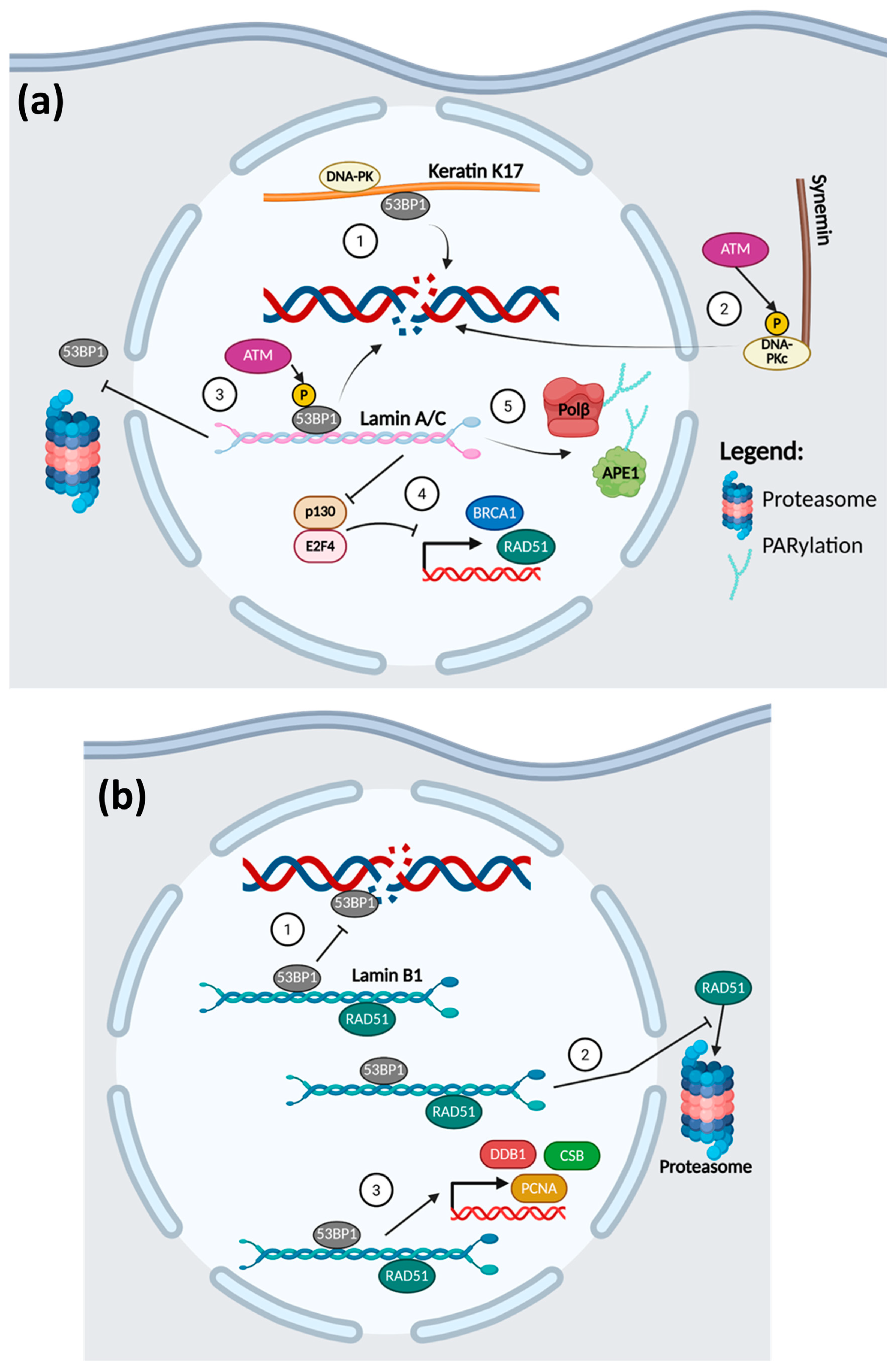
4.3. Intermediate Filaments and DDR
5. Targeting the Cytoskeleton in Conjunction with DNA Damage Induction in Cancer
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-TAT | α-tubulin acetyltranferase |
| ABP | Actin binding protein |
| ADC | Antibody-drug conjugate |
| AML | Acute myeloid leukemia |
| ATM | Ataxia Telangiectasia Mutated |
| BER | Base excision repair |
| CSC | Cancer stem cells |
| CTL | Cytotoxic T-cells |
| DDR | DNA damage response |
| DSB | Double stranded break |
| dsbNETs | DSB-capturing nuclear envelope tubules |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| HNPCC | Hereditary non-polyposis colorectal cancer |
| HNSCC | Head and neck squamous cell carcinoma |
| HR | Homologous recombination |
| ICL | Interstrand cross-links |
| IF | Intermediate filaments |
| JMY | Junction-mediating and regulatory protein |
| LINC | Linker of nucleoskeleton and cytoskeleton |
| MAP | Microtubule-associated proteins |
| MET | Mesenchymal-epithelial transition |
| MMR | Mismatch repair |
| MRN | MRE11/RAD50/NBS1 complex |
| MTA | Microtubule targeting agent |
| MTBP | Microtubules binding protein |
| MTOC | Microtubule organising centre |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end joining |
| NK cell | Natural killer cell |
| NSCLC | Non-small cell lung cancer |
| OGG1 | Oxoguanine glycosylase 1 |
| PCNA | Proliferating cell nuclear antigen |
| PTM | Post-translational modification |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| SSB | Single strand break |
| TLS | Translesion synthesis |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response—A double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jurkovicova, D.; Neophytou, C.M.; Gasparovic, A.C.; Goncalves, A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int. J. Mol. Sci. 2022, 23, 14672. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef]
- Moujaber, O.; Stochaj, U. The Cytoskeleton as Regulator of Cell Signaling Pathways. Trends Biochem. Sci. 2020, 45, 96–107. [Google Scholar] [CrossRef]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed]
- dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef]
- Jiang, X.; Qin, Y.; Kun, L.; Zhou, Y. The Significant Role of the Microfilament System in Tumors. Front. Oncol. 2021, 11, 620390. [Google Scholar] [CrossRef]
- Suresh, R.; Diaz, R.J. The remodelling of actin composition as a hallmark of cancer. Transl. Oncol. 2021, 14, 101051. [Google Scholar] [CrossRef]
- Gibieža, P.; Petrikaite, V. The regulation of actin dynamics during cell division and malignancy. Am. J. Cancer Res. 2021, 11, 4050–4069. [Google Scholar]
- An, J.H.; Kim, J.W.; Jang, S.M.; Kim, C.H.; Kang, E.J.; Choi, K.H. Gelsolin negatively regulates the activity of tumor suppressor p53 through their physical interaction in hepatocarcinoma HepG2 cells. Biochem. Biophys. Res. Commun. 2011, 412, 44–49. [Google Scholar] [CrossRef]
- Huang, B.; Deng, S.; Loo, S.Y.; Datta, A.; Yap, Y.L.; Yan, B.; Ooi, C.H.; Dinh, T.D.; Zhuo, J.; Tochhawng, L.; et al. Gelsolin-mediated activation of PI3K/Akt pathway is crucial for hepatocyte growth factor-induced cell scattering in gastric carcinoma. Oncotarget 2016, 7, 25391–25407. [Google Scholar] [CrossRef]
- Deng, R.; Hao, J.; Han, W.; Ni, Y.; Huang, X.; Hu, Q. Gelsolin regulates proliferation, apoptosis, migration and invasion in human oral carcinoma cells. Oncol. Lett. 2015, 9, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.J.; Wu, T.I.; Huang, Y.H.; Chang, T.C.; Wang, C.S.; Tsai, M.M.; Hsu, C.Y.; Tsai, M.H.; Lai, C.H.; Lin, K.H. Overexpression of gelsolin in human cervical carcinoma and its clinicopathological significance. Gynecol. Oncol. 2011, 120, 135–144. [Google Scholar] [CrossRef]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Kumar, A.P.; Lim, K.H.; Tan, T.Z.; Yang, H.; Hooi, S.C.; Goh, Y.C.; Maciver, S.K.; et al. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832–52848. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.; Tan, E.H.; Yan, B.; Tochhawng, L.; Jayapal, M.; Koh, S.; Tay, H.K.; Maciver, S.K.; Hooi, S.C.; Salto-Tellez, M.; et al. Gelsolin induces colorectal tumor cell invasion via modulation of the urokinase-type plasminogen activator cascade. PLoS ONE 2012, 7, e43594. [Google Scholar] [CrossRef]
- Wang, P.W.; Abedini, M.R.; Yang, L.X.; Ding, A.A.; Figeys, D.; Chang, J.Y.; Tsang, B.K.; Shieh, D.B. Gelsolin regulates cisplatin sensitivity in human head-and-neck cancer. Int. J. Cancer 2014, 135, 2760–2769. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Huang, Y.; Zhao, J.; Wu, L.; Qi, Q.; Liu, Y.; Li, G.; Li, J.; Liu, H.; Wu, H. Cofilin: A Promising Protein Implicated in Cancer Metastasis and Apoptosis. Front. Cell Dev. Biol. 2021, 9, 599065. [Google Scholar] [CrossRef]
- Sidani, M.; Wessels, D.; Mouneimne, G.; Ghosh, M.; Goswami, S.; Sarmiento, C.; Wang, W.; Kuhl, S.; El-Sibai, M.; Backer, J.M.; et al. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J. Cell Biol. 2007, 179, 777–791. [Google Scholar] [CrossRef]
- Prendergast, G.C. Actin’ up: RhoB in cancer and apoptosis. Nat. Rev. Cancer 2001, 1, 162–168. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef]
- Amos, L.A.; Schlieper, D. Microtubules and maps. Adv. Protein. Chem. 2005, 71, 257–298. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Ludueña, R.F.; Banerjee, A. The Isotypes of Tubulin. In The Role of Microtubules in Cell Biology, Neurobiology, and Oncology; Fojo, T., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 123–175. [Google Scholar]
- Wade, R.H. On and around microtubules: An overview. Mol. Biotechnol. 2009, 43, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, T.; Mirigian, M.; Selcuk Yasar, M.; Ross, J.L. Mechanics of microtubules. J. Biomech. 2010, 43, 23–30. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Hasegawa, S.; Miyoshi, Y.; Egawa, C.; Ishitobi, M.; Taguchi, T.; Tamaki, Y.; Monden, M.; Noguchi, S. Prediction of Response to Docetaxel by Quantitative Analysis of Class I and III β-Tubulin Isotype mRNA Expression in Human Breast Cancers. Clin. Cancer Res. 2003, 9, 2992–2997. [Google Scholar] [PubMed]
- Noguchi, S. Predictive factors for response to docetaxel in human breast cancers. Cancer Sci. 2006, 97, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Mialhe, A.; Lafanechèere, L.; Treilleux, I.; Peloux, N.; Dumontet, C.; Brémond, A.; Panh, M.-H.; Payan, R.; Wehland, J.; Margolis, R.-L.; et al. Tubulin Detyrosination Is a Frequent Occurrence in Breast Cancers of Poor Prognosis. Cancer Res. 2001, 61, 5024–5027. [Google Scholar]
- Dharmapal, D.; Jyothy, A.; Mohan, A.; Balagopal, P.G.; George, N.A.; Sebastian, P.; Maliekal, T.T.; Sengupta, S. beta-Tubulin Isotype, TUBB4B, Regulates The Maintenance of Cancer Stem Cells. Front. Oncol. 2021, 11, 788024. [Google Scholar] [CrossRef]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III β-Tubulin Overexpression Is a Prominent Mechanism of Paclitaxel Resistance in Ovarian Cancer Patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar] [CrossRef]
- Ferrandina, G.; Zannoni, G.F.; Martinelli, E.; Paglia, A.; Gallotta, V.; Mozzetti, S.; Scambia, G.; Ferlini, C. Class III beta-tubulin overexpression is a marker of poor clinical outcome in advanced ovarian cancer patients. Clin. Cancer Res. 2006, 12, 2774–2779. [Google Scholar] [CrossRef]
- Tsourlakis, M.C.; Weigand, P.; Grupp, K.; Kluth, M.; Steurer, S.; Schlomm, T.; Graefen, M.; Huland, H.; Salomon, G.; Steuber, T.; et al. betaIII-tubulin overexpression is an independent predictor of prostate cancer progression tightly linked to ERG fusion status and PTEN deletion. Am. J. Pathol. 2014, 184, 609–617. [Google Scholar] [CrossRef]
- Zhang, H.L.; Ruan, L.; Zheng, L.M.; Whyte, D.; Tzeng, C.M.; Zhou, X.W. Association between class III beta-tubulin expression and response to paclitaxel/vinorebine-based chemotherapy for non-small cell lung cancer: A meta-analysis. Lung Cancer 2012, 77, 9–15. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Mariani, M.; Petrella, L.; Martinelli, E.; Zannoni, G.F.; Vellone, V.; Ferrandina, G.; Shahabi, S.; Scambia, G.; Ferlini, C. Class III beta-tubulin and the cytoskeletal gateway for drug resistance in ovarian cancer. J. Cell. Physiol. 2012, 227, 1034–1041. [Google Scholar] [CrossRef]
- Hwang, J.E.; Hong, J.Y.; Kim, K.; Kim, S.H.; Choi, W.Y.; Kim, M.J.; Jung, S.H.; Shim, H.J.; Bae, W.K.; Hwang, E.C.; et al. Class III beta-tubulin is a predictive marker for taxane-based chemotherapy in recurrent and metastatic gastric cancer. BMC Cancer 2013, 13, 431. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; Terry, S.; Maille, P.; Allory, Y.; Sirab, N.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Coppolani, E.; Paule, B.; et al. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010, 70, 9253–9264. [Google Scholar] [CrossRef]
- Wattanathamsan, O.; Thararattanobon, R.; Rodsiri, R.; Chanvorachote, P.; Vinayanuwattikun, C.; Pongrakhananon, V. Tubulin acetylation enhances lung cancer resistance to paclitaxel-induced cell death through Mcl-1 stabilization. Cell Death Discov. 2021, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Smoter, M.; Bodnar, L.; Duchnowska, R.; Stec, R.; Grala, B.; Szczylik, C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother. Pharmacol. 2011, 68, 553–557. [Google Scholar] [CrossRef]
- Bhat, K.M.; Setaluri, V. Microtubule-associated proteins as targets in cancer chemotherapy. Clin. Cancer Res. 2007, 13, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. Intermediate Filaments. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect. Biol. 2016, 8, a018242. [Google Scholar] [CrossRef]
- Fuchs, E.; Weber, K. Intermediate filaments: Structure, dynamics, function, and disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef]
- Daly, N.; Meleady, P.; Walsh, D.; Clynes, M. Regulation of keratin and integrin gene expression in cancer and drug resistance. Cytotechnology 1998, 27, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, S.; Inokuchi, M.; Enjoji, M.; Ishikawa, T.; Takagi, Y.; Kato, K.; Yamada, H.; Kojima, K.; Sugihara, K. Vimentin expression is associated with decreased survival in gastric cancer. Oncol. Rep. 2011, 25, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Sutoh Yoneyama, M.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Naito, Z. Nestin in gastrointestinal and other cancers: Effects on cells and tumor angiogenesis. World J. Gastroenterol. 2011, 17, 409–418. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef]
- Baraks, G.; Tseng, R.; Pan, C.H.; Kasliwal, S.; Leiton, C.V.; Shroyer, K.R.; Escobar-Hoyos, L.F. Dissecting the Oncogenic Roles of Keratin 17 in the Hallmarks of Cancer. Cancer Res. 2022, 82, 1159–1166. [Google Scholar] [CrossRef]
- Elazezy, M.; Schwentesius, S.; Stegat, L.; Wikman, H.; Werner, S.; Mansour, W.Y.; Failla, A.V.; Peine, S.; Muller, V.; Thiery, J.P.; et al. Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer. Cancers 2021, 13, 3869. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, A.R.; Nyaga, S.G.; Jaruga, P.; Lohani, A.; Dizdaroglu, M.; Evans, M.K. Cellular repair of oxidatively induced DNA base lesions is defective in prostate cancer cell lines, PC-3 and DU-145. Carcinogenesis 2004, 25, 1359–1370. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef]
- Mambo, E.; Nyaga, S.G.; Bohr, V.A.; Evans, M.K. Defective repair of 8-hydroxyguanine in mitochondria of MCF-7 and MDA-MB-468 human breast cancer cell lines. Cancer Res. 2002, 62, 1349–1355. [Google Scholar] [PubMed]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef]
- Peltomaki, P. Deficient DNA mismatch repair: A common etiologic factor for colon cancer. Hum. Mol. Genet. 2001, 10, 735–740. [Google Scholar] [CrossRef]
- Umar, A.; Boyer, J.C.; Thomas, D.C.; Nguyen, D.C.; Risinger, J.I.; Boyd, J.; Ionov, Y.; Perucho, M.; Kunkel, T.A. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem. 1994, 269, 14367–14370. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Hayashi, S.; O’Shea, A.M.; Burgart, L.J.; Smyrk, T.C.; Shimizu, D.; Waring, P.M.; Ruszkiewicz, A.R.; Pollett, A.F.; Redston, M.; et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology 2007, 133, 48–56. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- de Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005, 579, 873–876. [Google Scholar] [CrossRef]
- Zafar, M.K.; Eoff, R.L. Translesion DNA Synthesis in Cancer: Molecular Mechanisms and Therapeutic Opportunities. Chem. Res. Toxicol. 2017, 30, 1942–1955. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef]
- Anand, J.; Chiou, L.; Sciandra, C.; Zhang, X.; Hong, J.; Wu, D.; Zhou, P.; Vaziri, C. Roles of trans-lesion synthesis (TLS) DNA polymerases in tumorigenesis and cancer therapy. NAR Cancer 2023, 5, zcad005. [Google Scholar] [CrossRef]
- Lin, X.; Okuda, T.; Trang, J.; Howell, S.B. Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2006, 69, 1748–1754. [Google Scholar] [CrossRef]
- Bostian, A.C.; Maddukuri, L.; Reed, M.R.; Savenka, T.; Hartman, J.H.; Davis, L.; Pouncey, D.L.; Miller, G.P.; Eoff, R.L. Kynurenine Signaling Increases DNA Polymerase Kappa Expression and Promotes Genomic Instability in Glioblastoma Cells. Chem. Res. Toxicol. 2016, 29, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Chen, Z.; Wang, S.; Wang, H.W.; Qiu, W.; Zhao, L.; Xu, R.; Luo, H.; Chen, Y.; Chen, D.; et al. The Error-Prone DNA Polymerase kappa Promotes Temozolomide Resistance in Glioblastoma through Rad17-Dependent Activation of ATR-Chk1 Signaling. Cancer Res. 2016, 76, 2340–2353. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, W.; Wang, H.W.; Wang, S.; Chen, Y.; Zhang, X.; Yang, J.; Zhao, S.; Ding, H.F.; Lu, D. Analysis of specialized DNA polymerases expression in human gliomas: Association with prognostic significance. Neuro-Oncology 2010, 12, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, Y.W.; Liu, X.; Chu, P.; Loria, S.; Wang, Y.; Yen, Y.; Chou, K.M. Expression of DNA translesion synthesis polymerase eta in head and neck squamous cell cancer predicts resistance to gemcitabine and cisplatin-based chemotherapy. PLoS ONE 2013, 8, e83978. [Google Scholar] [CrossRef]
- Ceppi, P.; Novello, S.; Cambieri, A.; Longo, M.; Monica, V.; Lo Iacono, M.; Giaj-Levra, M.; Saviozzi, S.; Volante, M.; Papotti, M.; et al. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2009, 15, 1039–1045. [Google Scholar] [CrossRef]
- Teng, K.Y.; Qiu, M.Z.; Li, Z.H.; Luo, H.Y.; Zeng, Z.L.; Luo, R.Z.; Zhang, H.Z.; Wang, Z.Q.; Li, Y.H.; Xu, R.H. DNA polymerase eta protein expression predicts treatment response and survival of metastatic gastric adenocarcinoma patients treated with oxaliplatin-based chemotherapy. J. Transl. Med. 2010, 8, 126. [Google Scholar] [CrossRef]
- Xie, K.; Doles, J.; Hemann, M.T.; Walker, G.C. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20792–20797. [Google Scholar] [CrossRef]
- Lemee, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.L.; Leduc, C.; et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair 2005, 4, 583–593. [Google Scholar] [CrossRef]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Shi, L.; Yu, X.; Gu, Y.; Sun, X. LINP1 facilitates DNA damage repair through non-homologous end joining (NHEJ) pathway and subsequently decreases the sensitivity of cervical cancer cells to ionizing radiation. Cell Cycle 2018, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, D.; Xie, X.; He, Y.; Lv, M.; Jiang, X. miR-129-3p inhibits NHEJ pathway by targeting SAE1 and represses gastric cancer progression. Int. J. Clin. Exp. Pathol. 2019, 12, 1539–1547. [Google Scholar]
- Yang, Y.; Yang, C.; Li, T.; Yu, S.; Gan, T.; Hu, J.; Cui, J.; Zheng, X. The Deubiquitinase USP38 Promotes NHEJ Repair through Regulation of HDAC1 Activity and Regulates Cancer Cell Response to Genotoxic Insults. Cancer Res. 2020, 80, 719–731. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Glass, J. The phenotypic radiation resistance of CD44+/CD24(-or low) breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Valentino, T.; D’Angelo, D.; De Martino, I.; Postiglione, I.; Pacelli, R.; Croce, C.M.; Fedele, M.; Fusco, A. HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene 2011, 30, 3024–3035. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef]
- Li, Y.; Yang, D.Q. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt in cancer cells with overactivated Akt. Mol. Cancer Ther. 2010, 9, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hurst, V.; Shimada, K.; Gasser, S.M. Nuclear Actin and Actin-Binding Proteins in DNA Repair. Trends Cell Biol. 2019, 29, 462–476. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Belin, B.; Mullins, R.D. Actin binding to WH2 domains regulates nuclear import of the multifunctional actin regulator JMY. Mol. Biol. Cell 2012, 23, 853–863. [Google Scholar] [CrossRef]
- Belin, B.J.; Lee, T.; Mullins, R.D. DNA damage induces nuclear actin filament assembly by Formin-2 and Spire-(1/2) that promotes efficient DNA repair. eLife 2015, 4, e07735. [Google Scholar] [CrossRef]
- Coutts, A.S.; Weston, L.; La Thangue, N.B. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc. Natl. Acad. Sci. USA 2009, 106, 19872–19877. [Google Scholar] [CrossRef]
- Rodriguez-Pastrana, I.; Birli, E.; Coutts, A.S. p53-dependent DNA repair during the DNA damage response requires actin nucleation by JMY. Cell Death Differ. 2023, 30, 1636–1647. [Google Scholar] [CrossRef]
- Andrin, C.; McDonald, D.; Attwood, K.M.; Rodrigue, A.; Ghosh, S.; Mirzayans, R.; Masson, J.Y.; Dellaire, G.; Hendzel, M.J. A requirement for polymerized actin in DNA double-strand break repair. Nucleus 2012, 3, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Hurst, V.; Challa, K.; Shimada, K.; Gasser, S.M. Cytoskeleton integrity influences XRCC1 and PCNA dynamics at DNA damage. Mol. Biol. Cell 2021, 32, br6. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Kulashreshtha, M.; Mehta, I.S.; Kumar, P.; Rao, B.J. Chromosome territory relocation during DNA repair requires nuclear myosin 1 recruitment to chromatin mediated by Upsilon-H2AX signaling. Nucleic Acids Res. 2016, 44, 8272–8291. [Google Scholar] [CrossRef]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef]
- Cook, A.W.; Toseland, C.P. The roles of nuclear myosin in the DNA damage response. J. Biochem. 2021, 169, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zagelbaum, J.; Schooley, A.; Zhao, J.; Schrank, B.R.; Callen, E.; Zha, S.; Gottesman, M.E.; Nussenzweig, A.; Rabadan, R.; Dekker, J.; et al. Multiscale reorganization of the genome following DNA damage facilitates chromosome translocations via nuclear actin polymerization. Nat. Struct. Mol. Biol. 2023, 30, 99–106. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef]
- Shen, X.; Ranallo, R.; Choi, E.; Wu, C. Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 2003, 12, 147–155. [Google Scholar] [CrossRef]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar] [CrossRef]
- Osakabe, A.; Takahashi, Y.; Murakami, H.; Otawa, K.; Tachiwana, H.; Oma, Y.; Nishijima, H.; Shibahara, K.I.; Kurumizaka, H.; Harata, M. DNA binding properties of the actin-related protein Arp8 and its role in DNA repair. PLoS ONE 2014, 9, e108354. [Google Scholar] [CrossRef]
- Kapoor, P.; Chen, M.; Winkler, D.D.; Luger, K.; Shen, X. Evidence for monomeric actin function in INO80 chromatin remodeling. Nat. Struct. Mol. Biol. 2013, 20, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, X.; Bao, S.; Guo, R.; Johnson, D.G.; Shen, X.; Li, L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 17274–17279. [Google Scholar] [CrossRef]
- Sokolova, V.; Lee, G.; Mullins, A.; Mody, P.; Watanabe, S.; Tan, D. DNA-translocation-independent role of INO80 remodeler in DNA damage repairs. J. Biol. Chem. 2023, 299, 105245. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shen, Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J. Biol. Chem. 2001, 276, 48318–48324. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Wang, Q.; Lu, H.; Brenneman, M.; Fan, F.; Shen, Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009, 69, 7978–7985. [Google Scholar] [CrossRef]
- Schramek, D.; Sendoel, A.; Segal, J.P.; Beronja, S.; Heller, E.; Oristian, D.; Reva, B.; Fuchs, E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 2014, 343, 309–313. [Google Scholar] [CrossRef]
- Tuan, N.M.; Lee, C.H. Role of Anillin in Tumour: From a Prognostic Biomarker to a Novel Target. Cancers 2020, 12, 1600. [Google Scholar] [CrossRef]
- Wei, S.; Teng, S.; Yao, J.; Gao, W.; Zang, J.; Wang, G.; Hu, Z. Develop a circular RNA-related regulatory network associated with prognosis of gastric cancer. Cancer Med. 2020, 9, 8589–8599. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Jia, X.; Song, W.; Wu, H.; Zhu, H.; Xuan, Z.; Du, Y.; Zhu, X.; Song, G.; et al. Targeting anillin inhibits tumorigenesis and tumor growth in hepatocellular carcinoma via impairing cytokinesis fidelity. Oncogene 2022, 41, 3118–3130. [Google Scholar] [CrossRef]
- Ma, J.; Liu, X.; Liu, P.; Lu, W.; Shen, X.; Ma, R.; Zong, H. Identification of a new p53 responsive element in the promoter region of anillin. Int. J. Mol. Med. 2020, 45, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hemmerich, P.; Grosse, F. Centrosomal localization of DNA damage checkpoint proteins. J. Cell. Biochem. 2007, 101, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Rong, Z.; Liu, C.; Qin, X.; Zhang, X.; Chen, Q. DNA damage promotes microtubule dynamics through a DNA-PK-AKT axis for enhanced repair. J. Cell Biol. 2021, 220, e201911025. [Google Scholar] [CrossRef]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef]
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. OncoTargets Ther. 2017, 10, 5633–5642. [Google Scholar] [CrossRef]
- Lawrimore, J.; Barry, T.M.; Barry, R.M.; York, A.C.; Friedman, B.; Cook, D.M.; Akialis, K.; Tyler, J.; Vasquez, P.; Yeh, E.; et al. Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol. Biol. Cell 2017, 28, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Lottersberger, F.; Karssemeijer, R.A.; Dimitrova, N.; de Lange, T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 2015, 163, 880–893. [Google Scholar] [CrossRef]
- Shokrollahi, M.; Stanic, M.; Hundal, A.; Chan, J.N.Y.; Urman, D.; Jordan, C.A.; Hakem, A.; Espin, R.; Hao, J.; Krishnan, R.; et al. DNA double-strand break-capturing nuclear envelope tubules drive DNA repair. Nat. Struct. Mol. Biol. 2024, 31, 1319–1330. [Google Scholar] [CrossRef]
- Ryu, N.M.; Kim, J.M. The role of the alpha-tubulin acetyltransferase alphaTAT1 in the DNA damage response. J. Cell Sci. 2020, 133, jcs246702. [Google Scholar] [CrossRef]
- Zhao, M.; Geng, R.; Guo, X.; Yuan, R.; Zhou, X.; Zhong, Y.; Huo, Y.; Zhou, M.; Shen, Q.; Li, Y.; et al. PCAF/GCN5-Mediated Acetylation of RPA1 Promotes Nucleotide Excision Repair. Cell Rep. 2017, 20, 1997–2009. [Google Scholar] [CrossRef]
- Dullovi, A.; Ozgencil, M.; Rajvee, V.; Tse, W.Y.; Cutillas, P.R.; Martin, S.A.; Horejsi, Z. Microtubule-associated proteins MAP7 and MAP7D1 promote DNA double-strand break repair in the G1 cell cycle phase. iScience 2023, 26, 106107. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Seymour, I.; Markiewicz, E.; Bekker-Jensen, S.; Mailand, N.; Hutchison, C.J. Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 2015, 14, 162–169. [Google Scholar] [CrossRef]
- Etourneaud, L.; Moussa, A.; Rass, E.; Genet, D.; Willaume, S.; Chabance-Okumura, C.; Wanschoor, P.; Picotto, J.; Theze, B.; Depagne, J.; et al. Lamin B1 sequesters 53BP1 to control its recruitment to DNA damage. Sci. Adv. 2021, 7, eabb3799. [Google Scholar] [CrossRef] [PubMed]
- Redwood, A.B.; Perkins, S.M.; Vanderwaal, R.P.; Feng, Z.; Biehl, K.J.; Gonzalez-Suarez, I.; Morgado-Palacin, L.; Shi, W.; Sage, J.; Roti-Roti, J.L.; et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle 2011, 10, 2549–2560. [Google Scholar] [CrossRef]
- Liu, N.A.; Sun, J.; Kono, K.; Horikoshi, Y.; Ikura, T.; Tong, X.; Haraguchi, T.; Tashiro, S. Regulation of homologous recombinational repair by lamin B1 in radiation-induced DNA damage. FASEB J. 2015, 29, 2514–2525. [Google Scholar] [CrossRef]
- Maynard, S.; Keijzers, G.; Akbari, M.; Ezra, M.B.; Hall, A.; Morevati, M.; Scheibye-Knudsen, M.; Gonzalo, S.; Bartek, J.; Bohr, V.A. Lamin A/C promotes DNA base excision repair. Nucleic Acids Res. 2019, 47, 11709–11728. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Regulation of nucleotide excision repair by nuclear lamin b1. PLoS ONE 2013, 8, e69169. [Google Scholar] [CrossRef]
- Nair, R.R.; Hsu, J.; Jacob, J.T.; Pineda, C.M.; Hobbs, R.P.; Coulombe, P.A. A role for keratin 17 during DNA damage response and tumor initiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2020150118. [Google Scholar] [CrossRef]
- Russell, M.A. Synemin Redefined: Multiple Binding Partners Results in Multifunctionality. Front. Cell Dev. Biol. 2020, 8, 159. [Google Scholar] [CrossRef]
- Deville, S.S.; Vehlow, A.; Forster, S.; Dickreuter, E.; Borgmann, K.; Cordes, N. The Intermediate Filament Synemin Regulates Non-Homologous End Joining in an ATM-Dependent Manner. Cancers 2020, 12, 1717. [Google Scholar] [CrossRef]
- Ma, J.; Sun, F.; Li, C.; Zhang, Y.; Xiao, W.; Li, Z.; Pan, Q.; Zeng, H.; Xiao, G.; Yao, K.; et al. Depletion of intermediate filament protein Nestin, a target of microRNA-940, suppresses tumorigenesis by inducing spontaneous DNA damage accumulation in human nasopharyngeal carcinoma. Cell Death Dis. 2014, 5, e1377. [Google Scholar] [CrossRef]
- Cancer Research UK. Treatment Breakdown by Demographics, 2013–2021. Available online: https://nhsd-ndrs.shinyapps.io/treatment_by_demographic_factors/ (accessed on 1 August 2024).
- Sadoughi, F.; Mirsafaei, L.; Dana, P.M.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Montazer, M.; Hosseinpour, M.; Yousefi, B. The role of DNA damage response in chemo- and radio-resistance of cancer cells: Can DDR inhibitors sole the problem? DNA Repair 2021, 101, 103074. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.M.; Zhou, S.; Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics 2022, 12, 656. [Google Scholar] [CrossRef]
- Gatti, L.; Zunino, F. Overview of tumor cell chemoresistance mechanisms. Methods Mol. Med. 2005, 111, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Taha, R.; Omri, H.E. Current Opinions on Chemoresistance: An Overview. Bioinformation 2018, 14, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Curtin, N.J. Targeting the DNA damage response for cancer therapy. Biochem. Soc. Trans. 2023, 51, 207–221. [Google Scholar] [CrossRef]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: Progress made and future hopes. npj Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef]
- Hsu, W.H.; Zhao, X.; Zhu, J.; Kim, I.K.; Rao, G.; McCutcheon, J.; Hsu, S.T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef]
- Li, Q.; Qian, W.; Zhang, Y.; Hu, L.; Chen, S.; Xia, Y. A new wave of innovations within the DNA damage response. Signal Transduct. Target. Ther. 2023, 8, 338. [Google Scholar] [CrossRef]
- Jiang, S.; Pan, A.W.; Lin, T.Y.; Zhang, H.; Malfatti, M.; Turteltaub, K.; Henderson, P.T.; Pan, C.X. Paclitaxel Enhances Carboplatin-DNA Adduct Formation and Cytotoxicity. Chem. Res. Toxicol. 2015, 28, 2250–2252. [Google Scholar] [CrossRef] [PubMed]
- Arun, B.K.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bell-McGuinn, K.M.; Bach, B.A.; Kundu, M.G.; et al. Efficacy and safety of first-line veliparib and carboplatin-paclitaxel in patients with HER2- advanced germline BRCA+ breast cancer: Subgroup analysis of a randomised clinical trial. Eur. J. Cancer 2021, 154, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembe, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef]
- Chao, O.S.; Goodman, O.B., Jr. DNA-PKc inhibition overcomes taxane resistance by promoting taxane-induced DNA damage in prostate cancer cells. Prostate 2021, 81, 1032–1048. [Google Scholar] [CrossRef]
- Zhang, H.; Kreis, J.; Schelhorn, S.E.; Dahmen, H.; Grombacher, T.; Zuhlsdorf, M.; Zenke, F.T.; Guan, Y. Mapping combinatorial drug effects to DNA damage response kinase inhibitors. Nat. Commun. 2023, 14, 8310. [Google Scholar] [CrossRef]
- Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. [Google Scholar] [CrossRef]
- Wang, X.; Gigant, B.; Zheng, X.; Chen, Q. Microtubule-targeting agents for cancer treatment: Seven binding sites and three strategies. MedComm–Oncology 2023, 2, e46. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Leong, H.C.; Datta, A.; Gopal, V.; Kumar, A.P.; Yap, C.T. PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression. Cancers 2022, 14, 1652. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Pianowski, Z. Photopharmacology of Antimitotic Agents. Int. J. Mol. Sci. 2022, 23, 5657. [Google Scholar] [CrossRef]
- Gao, J.; Nakamura, F. Actin-Associated Proteins and Small Molecules Targeting the Actin Cytoskeleton. Int. J. Mol. Sci. 2022, 23, 2118. [Google Scholar] [CrossRef]
| Cytoskeletal Class | Key Proteins | DDR Pathway | Impact on the DDR | Ref. |
|---|---|---|---|---|
| Microfilaments | Nuclear actin, Formin-2 and Spire-1/2 | NHEJ | Nuclear actin polymerisation by Formin-2 and Spire-1/2:
| [110,113,114] |
| BER | ||||
| Filamin-A, Arp2/3, Nuclear actin, Myosin I and V, INO80 complex | HR |
| [115,116,117,118,119,122,123,124,128] | |
| Cytoplasmic actin, JMY, Myosin IIa | p53 | Myosin IIa increases p53 retention and accumulation in the nucleus, while JMY acts as its transcription co-activator, augmenting its target gene expression. | [109,111,112,129] | |
| INO80 complex | NER | INO80 complex helps to recruit XPA and XPC to the DNA to initiate NER. | [125] | |
| Microtubules | αβ-Tubulin, γ-TubulinDynein | NHEJ and HR | Microtubule dynamics and dynein help in the transport of DSB repair proteins, such as ATM, MRN11, NBS1, etc., into the nucleus upon DNA damage induction. | [134,136,137] |
| αβ-Tubulin, Kinesin-1/2, MAP7/MAP7D1 | NHEJ |
| [135,138,139,143] | |
| α-TAT, αβ-Tubulin, Kinesin-1/3 | NHEJ and HR | Promote the creation of dsbNETs with the help of the LINC complex, lamin B1, and actin filament. | [140] | |
| α-TAT | HR | Promotes hyperphosphorylation of RPA and phosphorylation of Chk1, increasing BRCA1 recruitment to DSB sites. | [141,143] | |
| αβ-Tubulin | BER | Microtubule depolymerisation increases XRCC1 and PCNA accumulation on damaged DNA for BER to take place. | [114] | |
| Intermediate Filaments | Lamin A/C, Synemin, K17 | NHEJ |
| [144,150,152] |
| Lamin B1 | NHEJ | Inhibits NHEJ as overexpression sequesters 53BP1 from being recruited to DSBs. | [145] | |
| Lamin A/C, Lamin B1 | HR |
| [146,147] | |
| Lamin A/C | BER | Fosters the PARylation of APE1 and Polβ to increase their activities. | [148] | |
| Lamin B1 | NER | Expression of NER key proteins, DDB1, CSB, and PCNA, is dependent upon the presence of lamin B1. | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halim, C.E.; Deng, S.; Crasta, K.C.; Yap, C.T. Interplay Between the Cytoskeleton and DNA Damage Response in Cancer Progression. Cancers 2025, 17, 1378. https://doi.org/10.3390/cancers17081378
Halim CE, Deng S, Crasta KC, Yap CT. Interplay Between the Cytoskeleton and DNA Damage Response in Cancer Progression. Cancers. 2025; 17(8):1378. https://doi.org/10.3390/cancers17081378
Chicago/Turabian StyleHalim, Clarissa Esmeralda, Shuo Deng, Karen Carmelina Crasta, and Celestial T. Yap. 2025. "Interplay Between the Cytoskeleton and DNA Damage Response in Cancer Progression" Cancers 17, no. 8: 1378. https://doi.org/10.3390/cancers17081378
APA StyleHalim, C. E., Deng, S., Crasta, K. C., & Yap, C. T. (2025). Interplay Between the Cytoskeleton and DNA Damage Response in Cancer Progression. Cancers, 17(8), 1378. https://doi.org/10.3390/cancers17081378