Simple Summary
This review examines the current treatment landscape for uveal melanoma, highlighting effective local therapies such as enucleation and plaque brachytherapy. Despite advances in medical therapeutics, the outcome for metastatic uveal melanoma has not improved significantly in the past 50 years, with immune checkpoint inhibitors showing limited success. The review discusses the role of local therapies and examines various treatment options for uveal melanoma, ranging from conventional chemotherapy and checkpoint inhibitors to newer molecularly selective T-cell therapies. Additionally, emerging strategies such as biomarker-driven patient stratification and multimodal therapies, including inhibition of the mitogen-activated extracellular signal-regulated kinase (MEK) pathway, histone deacetylase (HDAC) inhibitors, and spliceosome-targeted treatments such as splicing factor 3b subunit 1 (SF3B1) inhibitors, are discussed as potential avenues to improve patient outcomes.
Abstract
Uveal melanoma is a rare eye malignancy with a 50% overall metastasis rate. Local control is typically successful, with low overall rates of recurrence. Survival following metastasis averages less than one year. Metastasis treatments to date have yielded poor response rates and often are seen as palliative care measures. Advanced cutaneous melanoma treatment has benefitted from the recent development of immunotherapy, in particular immune checkpoint inhibitors. Comparatively, uveal melanoma cases have not seen therapeutic benefit from these drugs. This is likely due to the difference in genetic signatures and the sun-shielded nature of uveal melanoma. The development of novel antibody modalities such as bi-specific T-cell engagers and targeted molecular therapy has benefitted outcomes for select cohorts of patients. Future avenues are currently being developed to increase the efficacy of immunotherapies for these patients. These include a combination of different therapy modalities and patient stratification based on biomarkers of sensitivity to immunotherapies and novel targeted molecular therapy.
1. Introduction
Uveal melanoma (UM) is the most common primary intraocular malignancy in adults worldwide [1]. It is characterised by melanocytic tumour growth within the uveal tract of the eye. There are an average of 9.5 cases per million of UM in Ireland and other Northern European countries each year [2]. UM represents 3–5% of all melanomas [3,4]. The average age for diagnosis is around 62 years [1].
The majority of UM patients are of Caucasian race, which is consistent with the literature on other melanoma subtypes [5,6,7]. The reason behind low rates of UM in African, Asian, and Hispanic ethnicities remains a controversial and nuanced question. Studies have theorised a link to the protective mechanisms of eumelanin against ultraviolet radiation (UVR) produced in the melanocytes of darker eye colours (more common in non-Caucasians) compared to pheomelanin, lighter eye colours (more common in Caucasians) [8,9]. However, some studies, as well as the 2018 WHO classification status [10], have refuted this claim due to the lack of support towards UVR as a factor for UM development [11].
The tumour mutational burden (TMB) is characteristically low in uveal melanoma. There are a small number of genetic mutations common to UM, including initiating mutations which do not affect prognosis, such as guanosine nucleotide-binding protein alpha-11 (GNA11), guanosine nucleotide-binding protein alpha-Q (GNAQ), cysteinyl leukotriene receptor 2 (CYSLTR2), and phospholipase C beta 4 (PLCB4) [12,13]. Mutations in BRCA1-associated protein 1 (BAP1), splicing factor 3b subunit 1 (SF3B1), initiation factor 1A X-linked (EIF1AX), and serine and arginine-rich splicing factor 2 (SRSF2) are prognostically important [12,13].
UM has overall low rates of primary tumour recurrence in patients following treatment; however, metastasis rates to distant systemic sites are as high as 50% [1]. Therapeutic approaches for advanced UM to date have been insufficient in combatting the spread of disease. New advancements in therapies are vital for metastatic uveal melanoma (mUM) patients, who have minimal effective options for treatment.
This review will focus on the pathogenesis and subsequent hallmarks of the UM disease state, the current treatments for primary and metastatic disease, as well as the recent development and future directions of immunotherapies for mUM patients.
2. Uveal Melanoma
2.1. Overview: Prognosis
UM is a malignancy of the melanocytic cells developed within the uveal tract of the eye. These melanocytes in the eye acquire mutations over time that allow the cells to uncontrollably divide and lead to tumour development. In total, 90% of UM cases develop in the choroid, 5–8% in the ciliary body, and 2–5% in the iris (see Figure 1A) [14].
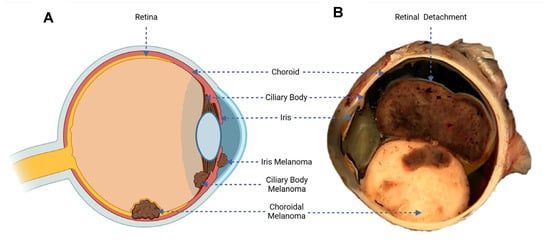
Figure 1.
(A) Diagram of the eye, illustrating the three possible sites of UM development. (B) Macroscopic cross-section of an enucleated eye, highlighting variegated UM tumour growth and a detached retinal rim [15].
The symptoms are dependent on the area of tumour growth within the eye. Metamorphopsia (painless vision loss) is the most common symptom in UMs. Larger choroidal melanomas are associated with retinal detachment, which is associated with impaired vision (see Figure 1B). Asymmetric astigmatism is common in ciliary body melanomas. Iris melanomas are mainly associated with obvious growth or change in colour of the iris [16].
Some cases can present asymptomatically, and the disease in many cases can go undetected for some time in patients, particularly choroidal and ciliary body melanomas, where the tumour is not visible subclinically [17]. In this case, the lesion is discovered as a result of routine fundus imaging or slit lamp examination of the back of the eye at optometry appointments, where a patient will be referred to ocular oncology for investigation. In cases where this may remain undetected or treatment is neglected, patients may experience a variety of symptoms, including blindness due to retinal detachment, pain resulting from glaucoma, proptosis (bulging of the eye) due to local invasion of surrounding tissues (also known as extraocular spread), and systemic spread or metastasis of tumour cells to different organs within the body [18,19].
Diagnosis of UM has improved in accuracy throughout the years with the development of diagnostic tools and with the increased experience of ocular oncologists [20]. Diagnosis can be made through various imaging techniques such as magnetic resonance imaging (MRI), computerised tomography (CT), ocular ultrasound, or fluorescein angiography [21,22]. Fine Needle Aspirate (FNA) biopsies may be needed in more challenging cases of UM, for example, to rule out a metastatic tumour. Complications that come with this procedure include possible retinal detachment and vitreous haemorrhage [23]. In choroidal tumour cases, which clinically may represent a benign naevus, clinics often adopt a “watch-and-wait” protocol, also known as active surveillance, where treatment will be deferred until further growth is observed [23]. This is mainly because the imaging of smaller uveal melanomas is hard to distinguish from a benign choroidal naevus. These nevi only develop into melanomas in about 0.0001% of cases, so distinguishing between these lesions is important for patient care [24].
Certain molecular biomarkers can impact a patient’s prognosis and likelihood of metastasis in UM. These include tumour-suppressor BAP1 expression loss (often accompanied by monosomy 3), chromosome 8 gain, and epithelioid cell type [25,26]. Loss of BAP1 expression, which often occurs alongside partial deletion of chromosome 3, is strongly associated with a high risk of metastasis and poor survival rates due to a highly aggressive tumour [25,26,27]. Similarly, the gain of chromosome 8q is associated with reduced survival outcomes and enhanced tumour proliferation. It is also linked with immune system dysfunction, which further contributes to a worse prognosis [25,28]. Other standard prognostic tests include tumour size and cell type. Tumours of a large thickness and basal diameter have been associated with a poorer prognosis. Additionally, an epithelioid cell tumour morphology is generally a more invasive phenotype, as epithelioid cells are characterized by large nuclei, which further contribute to the aggressive nature of the tumour [25,26]. The status of these biomarkers helps clinics determine how likely a patient is to metastasise, so adequate levels of surveillance can be taken following local disease control through the techniques outlined below.
2.2. Current Treatments
2.2.1. Treatment of Primary Uveal Melanoma
Upon diagnosis, patients have several different avenues of treatment, dependent on patient preference and characteristics of the tumour (e.g., approximate size and area).
Enucleation (removal of the eye) was the most common method of local treatment for UM until recent times [29]. The oncology team would surgically remove the whole eye from the body to prevent further local growth and systemic spread. The majority of UMs can be treated with this method and it is encouraged for cases of large tumour size.
More recently, targeted radiotherapy, also known as plaque brachytherapy, has emerged as a popular choice due to the eye-saving and possible sight-saving aspects of this treatment [30]. This may not be suitable for all UM cases, particularly in large UM tumours or where the placement of the tumour is unsuitable [29]. Brachytherapy isotopes include ruthenium 106 as well as iodine 125. This surgery involves the placement of a radioactive plaque into the socket of the eye, directly at the site of the tumour. This plaque will gradually kill the tumour cells, and is subsequently removed after radiotherapy treatment has finished. While this treatment is eye-saving, in many instances, treated eyes can experience vision loss and pain post-operation, due to increased risk of radiation retinopathy and secondary glaucoma [29].
Both treatments have comparable survival rates, and neither shows superiority in terms of tumour metastasis [31,32]. Other less common methods of treatment include proton beam radiotherapy, which uses protons as targeted irradiation instead of a plaque, and transpupillary thermotherapy, which uses infrared light to increase the temperature of the choroid to kill the tumour cells [33].
Interestingly, a phase 3 randomised trial is currently underway to evaluate the use of Belzupacap Sarotalocan (AU-011), a type of papillomavirus-like particle (VLP), in the treatment of patients with primary indeterminate lesions or small choroidal melanoma [34]. VLPs consist of recombinantly synthesized capsid proteins that assemble into a viral-like capsid, excluding viral genetic material. As anticancer agents, VLPs offer efficient delivery of drugs to tumours without relying on viral replication, reducing biosafety risks. Their mechanism involves binding of the VLPs to modified glycosaminoglycans (GAGs) present on cancer cells [35].
Additionally, a phase 2 clinical trial (NCT05907954) is investigating darovasertib (IDE196), a protein kinase C (PKC) inhibitor, as a neoadjuvant/adjuvant treatment prior to local therapy for patients with tumours harbouring GNA11 or GNAQ mutations [36,37]. These mutations, found in approximately 90% of UM patients, activate the PKC pathway, and promote tumorigenesis and metastasis. By inhibiting PKC’s, darovasertib can subsequently inhibit the proliferation of UM, induce tumour shrinkage, and reduce cell viability in mUM [37].
2.2.2. Treatment of Metastatic Uveal Melanoma
Metastasis in UM occurs primarily through haematogenous dissemination, in most cases to the liver (90%), followed by the lungs, bone, and brain [38]. Unlike cutaneous melanoma, UM does not travel to lymph nodes first, as the uveal tract lacks lymphatic vessels [39].
Therapies for mUM are limited, and some treatments may not be suitable for all patients. Systemic intravenous (IV) chemotherapies, primarily alkylating agents such as fotemustine and dacarbazine [40], are less commonly used methods of treatment for mUM. This is due to their overall low efficacy of <10% [41] and significantly higher toxicological profile compared to the more modern targeted therapy approaches [42].
The first use of a liver-directed therapy approach was in 1961 by Dr. Robert K. Ausman, changing the focus of metastatic treatment for liver metastasis [43]. Table 1 presents the current mainstream hepatic-based therapies used to treat metastatic disease in UM, including their efficacy rates and limitations.

Table 1.
Mainstream liver-directed therapies for mUM patients: limitations and efficacy (excl. immunotherapy).
While localised treatments for primary tumours remain quite successful for UM patients, survival rates for advanced UM have remained consistently poor over time, despite the continued development of these liver-directed therapies [7,51].
The poor overall efficiency of these therapies for mUM has led to the development of more advanced approaches to targeting metastatic tumours in these patients. At present, the main therapy developed by researchers and clinicians is immunotherapy, which has shown high efficiency and increased survival rates in metastatic melanoma cases.
3. Current Immunotherapies in mUM
Immunotherapy is commonly used for the treatment of advanced melanoma patients. These drugs target the immune system of the patients to try to improve or increase the anti-tumour immune cell response. The two main types of immunotherapies for mUM currently in use are immune checkpoint inhibitor therapies and T-cell-directed therapies.
3.1. Immune Checkpoint Inhibitor Therapy
Checkpoint proteins are a class of proteins on T-cells that are crucial players in immune regulation in the body. In healthy patients, these checkpoint proteins will attach to corresponding ligands, primarily presented by antigen-presenting cells (APC) and tissue cells in response to inflammation, to ensure the downregulation of T-cell activation and cytotoxicity when necessary. This is important in the immune tolerance of the body and dampens the immune responses, to prevent damaging healthy tissues [52].
Tumour cells can often develop mechanisms to avoid being recognised by the immune system, known as immune evasion. One of these mechanisms is the expression of checkpoint protein ligands on the surfaces of the tumour cells. These ligands can attach to the checkpoint proteins on the surface of the T-cell, subsequently deactivating such, ensuring it does not recognise the tumour and provoke an immune response [53].
Immune checkpoint inhibitors (ICIs) are a class of immunotherapy drugs derived from antibodies that use this mechanism of immune evasion to their advantage. These drugs target either the checkpoint proteins expressed on the surface of T-cells or the corresponding ligand on the tumour cell. The two main checkpoints currently targeted in melanoma are cytotoxic-T-lymphocyte-associated protein 4 (CTLA-4), programmed death-protein 1 (PD-1), and programmed death-ligand 1 (PD-L1) [54].
3.1.1. CTLA-4
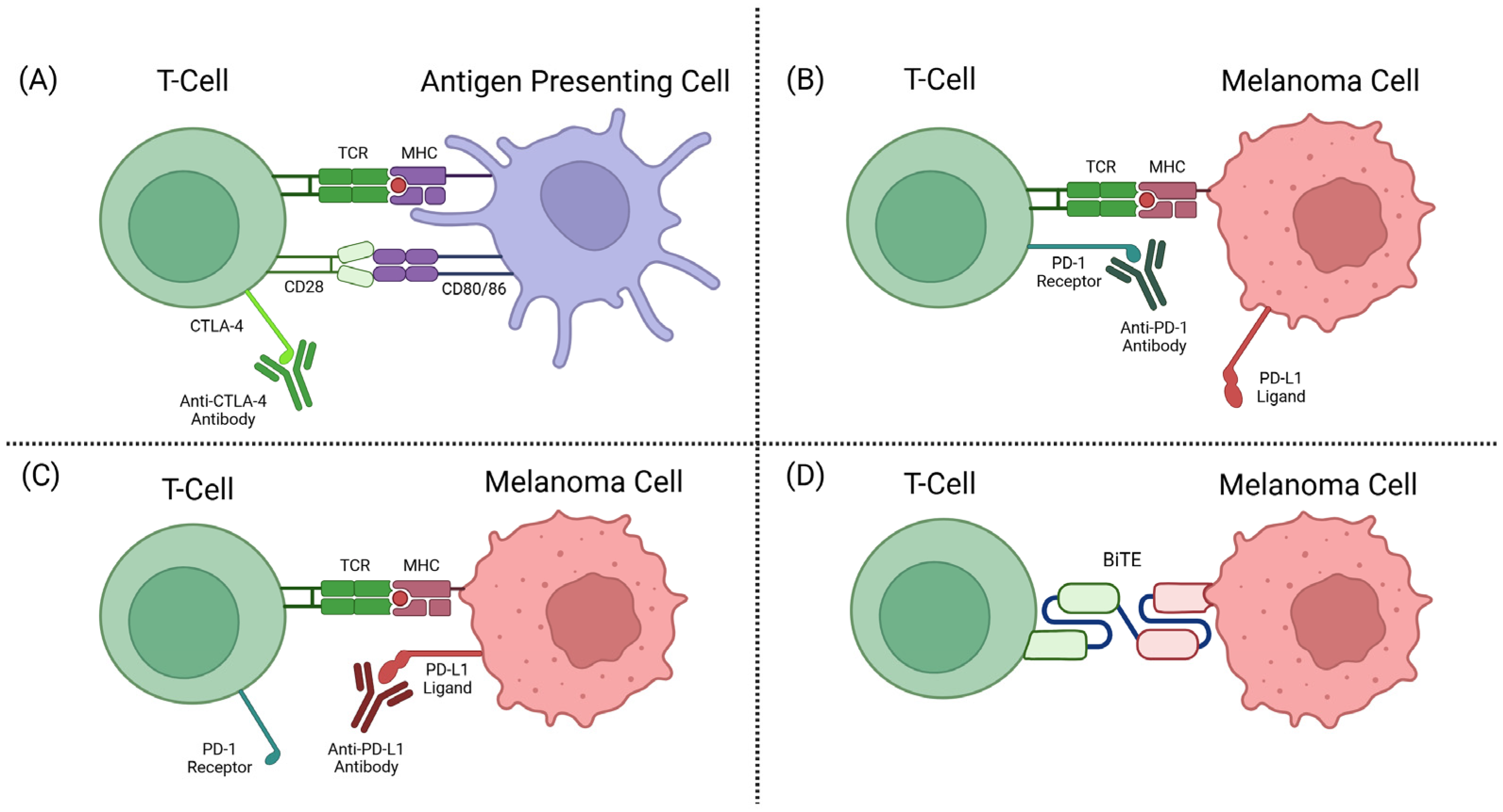
T-cell activation can occur when a T-cell receptor (TCR) recognises a peptide expressed by the major histocompatibility complex (MHC) on an APC. Then, the CD28 molecule on the T-cell surface binds to the CD80/CD86 on an APC, which acts as a costimulatory signal for full T-cell activation [55].
The CTLA-4 receptor plays a role in immune regulation by the competitive inhibition of CD28-CD80/86 binding. CTLA-4, when bound to CD80/86, also activates intracellular signalling to inhibit proliferation and activation of T-cells [56,57]. Anti-CTLA-4 ICIs target the T-cell-expressed CTLA-4 receptor that plays a role in T-cell inhibition, allowing the T-cell to remain activated and target the tumour cells (see Figure 2A).
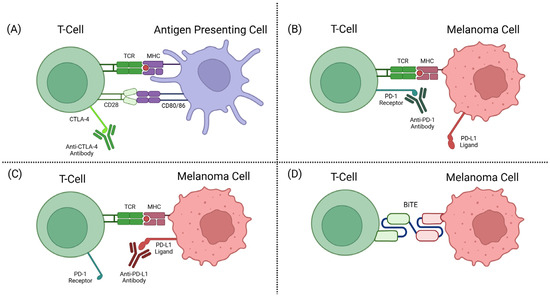
Figure 2.
(A) CTLA-4 inhibitor mechanism of action (MOA). (B) PD-1 inhibitor MOA. (C) PD-L1 inhibitor MOA. (D) BiTE MOA [58].
Ipilimumab is an anti-CTLA-4 humanised monoclonal antibody (mAb) most commonly used for melanoma patients. Ipilimumab was the first FDA-approved ICI for advanced melanoma patients, with response rates of about 10–20% [59].Despite the low response rates, this was a huge step in melanoma therapies at the time.
3.1.2. PD-1 and PDL-1 Inhibitors
In healthy individuals, PD-1 and PD-L1 interactions help to deactivate T-cells and reduce proliferation in later stages of T-cell activation. Tumours have adopted mechanisms of expressing PD-L1 to become unrecognisable as tumours by T-cells and subsequently deactivate these immune defences from killing the tumour cells [53]. Anti-PD-1 antibody drugs are a class of ICIs targeting the checkpoint protein PD-1 on a T-cell surface, or its corresponding ligand PD-L1. This prevents the interaction between PD-1 and PD-L1, as well as the subsequent deactivation mechanisms (see Figure 2B,C).
In 2014, the FDA approved nivolumab and pembrolizumab, two mAbs targeting PD-1 for unresectable or metastatic melanoma [60,61]. This therapy has been highly effective in advanced cutaneous melanoma cases, where overall response rates can reach up to ~45% [62]. However, much lower response rates are seen from UM tumours across many retrospective studies and clinical trials, averaging around 0–10% [63].
Combination therapies are now a common mode of administration for ICI therapies. This involves the administration of an anti-PD-1/anti-PD-L1 antibody combined with an anti-CTLA-4 antibody to maximise the effect, mainly a combination of ipilimumab and nivolumab [64]. This has seen even higher efficacy rates in advanced melanoma patients (58% ORR in phase 3 clinical trial) [65]. However, the increase in adverse effects compared to monotherapy makes this therapy unsuitable for immunocompromised or severely ill patients.
Despite ICI’s revolutionising the treatment of cutaneous metastatic melanoma cases, the overall responses for UM have been poor. This has been theorised to be attributed to UM’s low mutational burden average (1.1 Mut/Mb) compared to cutaneous melanomas (18 Mut/Mb) [26]. A study by Dousset et. al [66] showed a positive correlation between low TMB and poor responses to ICI therapies [67]. The characteristically low TMB rate of UM is likely related to the sun-shielded location of UM development: UM is an intraocular malignancy, partially blocked from ultraviolet radiation (UVR), unlike skin melanomas that are much more exposed to UVR [68]. High exposure to UVR results in higher rates of UV-mediated DNA mutations, and tumours are thus more likely to express immune-modulation mechanisms and neoantigen production [69].
The poor response rates seen in mUM in comparison to other forms of melanoma have gained traction within immune-oncology groups, to develop more effective immunological approaches in mUM management.
3.2. T-Cell Direction Therapy
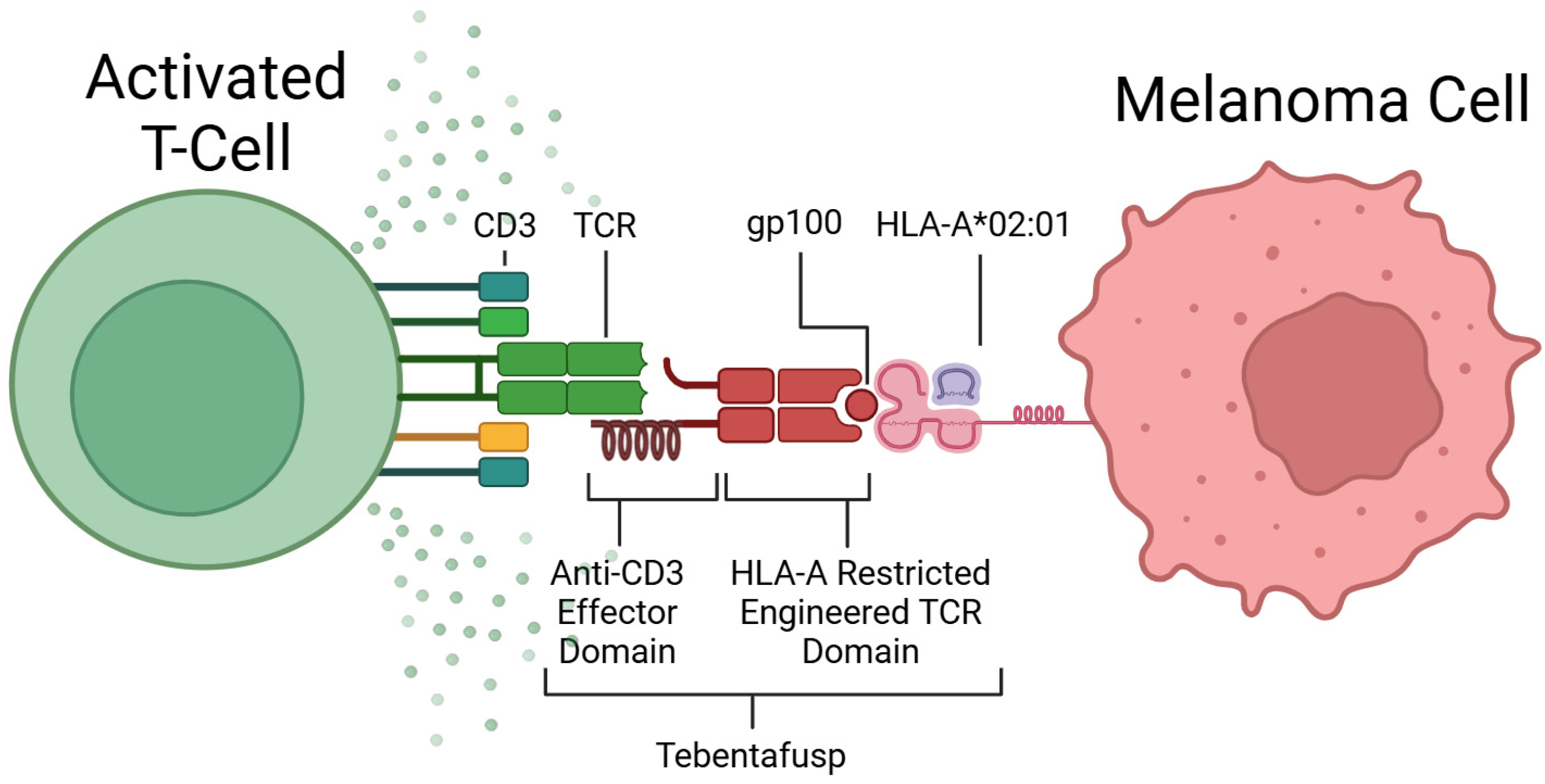
The main immunotherapy that has shown promising results in UM patients is Tebentafusp (TEBE). TEBE is the first engineered soluble T-cell receptor (TCR) that acts as a bispecific T-cell engager (BiTE) [70]. Developed by Immunocore, TEBE was approved by the FDA in January 2022, under the brand name Kimmtrak [71].
The discovery of TCR recognition of glycoprotein 100 (gp100) began the development of this form of therapy [72]. TEBE’s TCR binds to gp100, located on the surface of melanoma cells, and its anti-CD3 domain binds to CD3, located on all T-cells (see Figure 3) [68]. This bispecific binding brings T-cells into close contact with melanoma cells and creates a bridge between the two cells, where the T-cell recognises the tumour cell and induces apoptosis (see Figure 2D) [73].
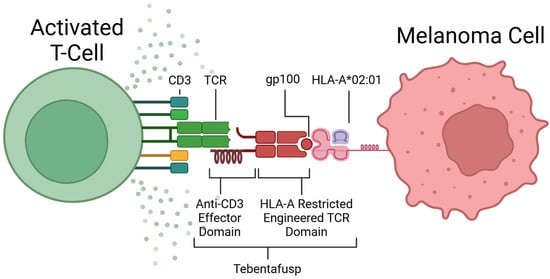
Figure 3.
Mechanism of action of Tebentafusp. Created in BioRender [74].
TEBE is used in the treatment of mUM patients who are Human Leukocyte Antigen-A*02:01 (HLA-A*02:01)-positive, which consists of around 50% of the Caucasian population [70,75]. HLA-A*02:01 is a specific variant of the human leukocyte antigen (HLA) expressed as a cellular marker on melanoma cells. Treatment is not suitable for patients without this selective biomarker, as TEBE is specific for both the HLA-A*02:01 molecule and gp100, so it will not bind if gp100 is expressed on a different variant of HLA.
TEBE’s trials to date have shown impressive results in mUM patients. In a phase 1/2 clinical trial involving eighteen mUM patients, conducted over one year, three showed partial response, eight showed stable disease, and seven showed progressive disease [76]. The study also interrogated the tumour microenvironment and immune system responses through various biomarker analyses, such as cytokine CXCL10 and CXCR3 T-cell concentration levels. They found an overall increase in T-cell markers from immunohistochemical staining of pre-treatment and post-treatment tumour biopsies.
A much larger phase 3 clinical trial including a control group showed overall survival (OS) at one year of 59% in the control vs. 73% in the TEBE-treated group, along with an increase in disease-free survival [70]. Many other clinical trials have supported the evidence that TEBE is an efficient treatment option for suitable patients.
Despite TEBE’s efficiency in response to mUM, its limitation to only HLA-A*02:01-positive patients is a valid concern for clinics. More universal treatment strategies are needed to increase the therapeutic options for mUM patients who do not qualify for TEBE.
3.3. Clinical Trials
Table 2 presents the immunotherapy and T-cell therapy treatments for mUM that are currently undergoing phase II or phase III clinical trials.
Table 2.
Current phase II/III clinical trials in metastatic uveal melanoma immunotherapy treatments [77].
4. Future Directions
To combat the efficacy issues with current treatments, researchers have been developing new strategies that can help close the gap in therapies for mUM. The three main directions focused on in this review are patient stratification, multimodal therapeutics, and emerging targeted therapeutics.
4.1. Selective Biomarkers: Patient Stratification
As previously described, across the UM population, there is a poor response rate to ICIs compared to their cutaneous counterpart [67]. This is likely down to the lower TMB rates associated with uveal melanoma, where the tumour is less likely to produce the necessary immune evasion mutations that are targeted in ICI therapies [67].
However, a subset of patients with UM with high TMB has previously been identified in the literature [79,80,81]. The patient subsets of interest described by Johansson et al. (2020) have either a unique germline mutation of Methyl-CpG Binding Domain 4 (MBD4) or UVR damage, a signature seen in iris melanoma due to tumour location [79]. These two characteristics cause the tumours to become hypermutated and increase in mutations per megabase (mut/Mb).
As previously described, high TMB is a biomarker of an increased affinity for ICI therapies, and patients with this tumour signature are potential candidates for this treatment. By analysing TMB status in patients, focusing on patients with MBD4 mutations or UVR signatures, oncologists can potentially stratify patients more likely to benefit from these treatments and, therefore, achieve better overall patient outcomes. However, further studies are needed to validate and develop these findings.
4.2. Multimodal Therapies
Another avenue currently being trialled for increasing response rates of mUM to immunotherapies is to combine them with other forms of systemic and liver-directed therapies.
In a retrospective analysis of dual-treated patients at the University Medical Centre Hamburg-Eppendorf [82], a variety of historically combined therapies, exclusive to ICI combinations, were reviewed. This included dual-ICI with TACE and surgical resection. They found that overall survival (OS) rates increased with all forms of combined ICI and other modes of therapy (22.47 months versus 11.37 months). However, they did not compare the different therapies with each other to find the combination that most significantly improves OS.
Some specific therapies are currently being evaluated for their efficacy in combination with ICIs. These include hepatic perfusion with melphalan, SIRT, TACE, and immunoembolization [83]. A Phase 1 trial of percutaneous hepatic perfusion with melphalan in a small cohort of patients showed promising results, where all patients had either stable disease, partial, or full responses [84]. Further, a retrospective study of a cohort of SIRT-treated mUM, and combined SIRT- and ICI-treated mUM showed greatly increased OS in the combined treatment group (49.6 months) versus the SIRT group (13.6 months) [85].
The combination of different modes of therapy with immunotherapy has shown promising results compared to immunotherapy alone and may be a potential avenue for increasing the efficacy of immunotherapy in mUM. However, there is a need for further studies and clinical trial results that can back up these results.
4.3. Emerging Targeted Therapies
Emerging therapies for mUM focus on novel targeted therapeutic strategies beyond current systemic and liver-directed therapies. Three targeted therapeutics of particular interest for mUM are mitogen-activated extracellular signal-regulated kinase (MEK) pathway inhibitors, histone deacetylase inhibitors, and spliceosome-targeted therapies.
GNA11/GNAQ somatic activating mutations occur in up to 90% of UM cases, which subsequently activates the MEK pathway. As a result, MEK inhibitors, such as selumetinib and trametinib, are currently being investigated as targeted therapies for mUM [86,87]. MEK inhibitors have shown success in some clinical trials but unfortunately have not been seen to improve OS due to acquired resistance [88]. To counteract this by blocking other cell signalling pathways, c-mesenchymal-epithelial transition factor (c-MET) and phosphoinositide 3-kinase (PI3K) inhibitors could be a solution to MEK inhibition resistance [88].
Roginolisib (IOA-244) is the first highly selective, allosteric modulator of Phosphoinositide 3-kinase delta (PI3Kδ) [89,90]. It inhibits PI3Kδ through non-adenosine triphosphate (ATP) competitive binding to the C-terminal region, effectively regulating its activity, while avoiding the side effects typically associated with other PI3K inhibitors [91]. Roginolisib is currently being investigated and has shown promise for overcoming resistance mechanisms in MEK-targeted therapies.
The combination of darovasertib (IDE196), a PKC inhibitor, with crizotinib, a mesenchymal-epithelial transition factor (MET) inhibitor, is another emerging strategy. This combination is being investigated as a first-line treatment in HLA-A*02:01-negative patients with mUM in a phase 2/3 multi-arm, multi-stage clinical trial (NCT05987332) [92]. The study aims to further evaluate the effectiveness of combining darovasertib and crizotinib to target GNA11 and GNAQ signalling pathways to improve clinical outcomes in patients with HLA-A*02:01-negative mUM [92].
Additionally, histone deacetylase (HDAC) inhibitors have demonstrated efficacy in reducing tumour growth in vivo. Histones are simple proteins that package DNA into structural units called nucleosomes, which organize DNA in the nucleus and play a crucial role in gene regulation [93]. By inhibiting the acetylation of histones, these agents may reverse the effects of BAP1 loss, leading to a less aggressive tumour, and potentially restoring normal gene expression [93].
The spliceosome is another promising area of research, which acts as a potential antitumoural target. The spliceosome is a large RNA–protein complex which catalyses the removal of introns from nuclear pre-mRNA. SF3B1 is a pre-mRNA splicing target that, when mutated, is a common metastatic driver in UM. Spliceostatin A and sudemycin are both examples of SF3B1 inhibitors [94]. Their mechanism of action disrupts splicing processes, leading to exon skipping or intron retention, and increasing alternative splicing patterns [94]. This SF3B1 inhibition could potentially cause an SF3B1-mutated tumour to become less aggressive in nature through defective RNA transcript production and subsequent nonsense-mediated decay activation [94].
5. Conclusions
Despite much improved local control, the rates of systemic metastasis of UM remain at 50% on average. While immunotherapies like ICIs have transformed the outcome of cutaneous melanoma cases, the same cannot be said for UM. Many studies explain this disparity by the differences in the genetic profiles of these tumours, particularly TMB. Despite ICI’s low effectiveness in UM compared to cutaneous melanoma, these therapies are currently in use for mUM cases. Protein engineering has facilitated the development of novel immunotherapies and molecular therapies for mUM, including the significant development of TEBE. More recent developments for increasing immunotherapy effectiveness include the stratification of mUM patients by genetic profiles and UVR signatures for increased ICI therapeutic benefits, multimodal therapy strategies, and emerging targeted therapies for metastatic tumours. These future directions require increased studies and development before implementation in clinical settings, but have shown early promising results. There remains a clear path for further developments and refinements of immunotherapy for mUM to continue to advance treatments and patient care worldwide.
Author Contributions
Conceptualization, K.H., K.D.R., S.K. and G.F.; methodology, K.H., G.F., K.D.R. and S.K.; software, K.H., K.D.R. and G.F.; validation, K.H., K.D.R., S.K. and G.F.; writing—original draft preparation, K.H.; writing—review and editing, K.H., G.F., S.K. and K.D.R.; visualization, K.H. and G.F; supervision, K.D.R. and S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| APC | Antigen-Presenting Cell |
| ATP | Adenosine TriPhosphate |
| BAP1 | BRCA1-associated protein 1 |
| BiTE | Bispecific T-cell Engager |
| CD | Cluster of Differentiation |
| c-MET | c-Mesenchymal-Epithelial Transition Factor |
| CT | Computerised Tomography |
| CTLA-4 | Cytotoxic T-Lymphocyte Associated Protein 4 |
| CYSLTR2 | Cysteinyl Leukotriene Receptor 2 |
| CXCL | Chemokine (C-X-C motif) Ligand |
| CXCR3 | C-X-C Chemokine Receptor 3 |
| EIF1AX | Initiation Factor 1A X-linked |
| FDA | Food and Drug Administration |
| FNA | Fine Needle Aspiration |
| GAGs | Glycosaminoglycans |
| GNA11 | Guanosine Nucleotide-Binding Protein Alpha-11 |
| GNAQ | Guanosine Nucleotide-Binding Protein Alpha-Q Gene |
| gp100 | Glycoprotein 100 |
| HDAC | Histone Deacetylase |
| HLA | Human Leukocyte Antigen |
| HR | Hazard Ratio |
| ICI | Immune Checkpoint Inhibitor |
| IV | Intravenous |
| LAG3 | Lymphocyte Activation Gene 3 |
| mAb | Monoclonal Antibody |
| MBD4 | Methyl-CpG Binding Domain Protein 4 |
| MEK | Mitogen-Activated Extracellular Signal-Regulated Kinase |
| MET | Mesenchymal-Epithelial Transition Factor |
| MHC | Major Histocompatibility Complex |
| MRI | Magnetic Resonance Imaging |
| MS | Median Survival |
| mTKI | Multitargeted Tyrosine Kinase Inhibitors |
| mUM | Metastatic Uveal Melanoma |
| OS | Overall Survival |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PHP | Percutaneous Hepatic Perfusion |
| PI3K | Phosphoinositide 3-Kinase |
| PI3Kδ | Phosphoinositide 3-kinase delta |
| PKC | Protein Kinase C |
| PLCB4 | Phospholipase C Beta 4 (PLCB4) |
| PV | Papillomaviruses |
| SF3B1 | Splicing Factor 3B Subunit 1 |
| SIRT | Selective Internal Radiotherapy |
| SRSF2 | Serine and Arginine Rich Splicing Factor 2 |
| TACE | Transarterial Chemoembolization |
| TCR | T-cell Receptor |
| TEBE | Tebentafusp |
| TMB | Tumour Mutational Burden |
| UM | Uveal Melanoma |
| UVR | Ultraviolet Radiation |
| VEGF | Vascular Endothelial Growth Factor |
| VLP | Viral-Like Particles |
References
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Baily, C.; O’Neill, V.; Dunne, M.; Cunningham, M.; Gullo, G.; Kennedy, S.; Walsh, P.M.; Deady, S.; Horgan, N. Uveal Melanoma in Ireland. Ocul. Oncol. Pathol. 2019, 5, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Gallenga, C.E.; Franco, E.; Adamo, G.G.; Violanti, S.S.; Tassinari, P.; Tognon, M.; Perri, P. Genetic Basis and Molecular Mechanisms of Uveal Melanoma Metastasis: A Focus on Prognosis. Front. Oncol. 2022, 12, 828112. [Google Scholar] [CrossRef]
- Mahendraraj, K.; Lau, C.S.; Lee, I.; Chamberlain, R.S. Trends in incidence, survival, and management of uveal melanoma: A population-based study of 7516 patients. Clin. Ophthalmol. 2016, 10, 2113–2119. [Google Scholar] [CrossRef]
- Park, S.L.; Le Marchand, L.; Wilkens, L.R.; Kolonel, L.N.; Henderson, B.E.; Zhang, Z.F.; Setiawan, V.W. Risk Factors for Malignant Melanoma in White and Non-White/Non–African American Populations. Cancer Prev. Res. 2012, 5, 423–434. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal Melanoma: Trends in Incidence, Treatment, and Survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2016, 101, 38–44. [Google Scholar] [CrossRef]
- Houtzagers, L.E.; Wierenga, A.P.A.; Ruys, A.A.M.; Luyten, G.P.M.; Jager, M.J. Iris Colour and the Risk of Developing Uveal Melanoma. Int. J. Mol. Sci. 2020, 21, 7172. [Google Scholar] [CrossRef]
- Tucker, M.A.; Shields, J.A.; Hartge, P.; Augsburger, J.; Hoover, R.N.; Fraumeni, J.F. Sunlight Exposure as Risk Factor for Intraocular Malignant Melanoma. N. Engl. J. Med. 1985, 313, 789–792. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.P.; Weis, E.; Lajous, M.; Shields, J.A.; Shields, C.L. Intermittent and Chronic Ultraviolet Light Exposure and Uveal Mela-noma: A Meta-analysis. Ophthalmology 2005, 112, 1599–1607. [Google Scholar] [CrossRef]
- Ortega, M.; Fraile Martínez, O.; García Honduvilla, N.; Coca, S.; Álvarez Mon, M.; Buján, J.; Teus, M.A. Update on uveal melanoma: Translational research from biology to clinical practice (Review). Int. J. Oncol. 2020, 57, 1262–1279. [Google Scholar] [CrossRef]
- Zohrab Beigi, Y.; Lanjanian, H.; Fayazi, R.; Salimi, M.; Haji, B.; Hoseyni, M.; Noroozizadeh, M.H.; Masoudi-Nejad, A. Heterogeneity and molecular landscape of melanoma: Implications for targeted therapy. Mol. Biomed. 2024, 5, 17. [Google Scholar]
- Damato, B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye 2012, 26, 1157–1172. [Google Scholar] [CrossRef]
- Rochfort, K. Created in BioRender. 2025. Available online: https://BioRender.com/wdemjfb (accessed on 3 April 2025).
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef]
- Damato, E.M.; Damato, B.E. Detection and time to treatment of uveal melanoma in the United Kingdom: An evaluation of 2384 patients. Ophthalmology 2012, 119, 1582–1589. [Google Scholar] [CrossRef]
- Camp, D.A.; Yadav, P.; Dalvin, L.A.; Shields, C.L. Glaucoma secondary to intraocular tumors: Mechanisms and management. Curr. Opin. Ophthalmol. 2019, 30, 71. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.E.; Coupland, S.E. Ocular melanoma. Saudi J. Ophthalmol. 2012, 26, 137–144. [Google Scholar] [CrossRef]
- Collaborative Ocular Melanoma Study Group. Accuracy of diagnosis of choroidal melanomas in the Collaborative Ocular Melanoma Study: COMS Report No. 1. Arch. Ophthalmol. 1990, 108, 1268–1273. [Google Scholar] [CrossRef]
- Patel, D.R.; Blair, K.; Patel, B.C. Ocular melanoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Solnik, M.; Paduszyńska, N.; Czarnecka, A.M.; Synoradzki, K.J.; Yousef, Y.A.; Chorągiewicz, T.; Rejdak, R.; Toro, M.D.; Zweifel, S.; Dyndor, K.; et al. Imaging of uveal melanoma—Current standard and methods in development. Cancers 2022, 14, 3147. [Google Scholar] [CrossRef]
- Kivelä, T. Diagnosis of uveal melanoma. Dev. Ophthalmol. 2012, 49, 1–15. [Google Scholar]
- Singh, A.D.; Kalyani, P.; Topham, A. Estimating the risk of malignant transformation of a choroidal nevus. Ophthalmology 2005, 112, 1784–1789. [Google Scholar] [CrossRef]
- Lamas, N.J.; Martel, A.; Nahon-Estève, S.; Goffinet, S.; Macocco, A.; Bertolotto, C.; Lassalle, S.; Hofman, P. Prognostic biomarkers in uveal melanoma: The status quo, recent advances and future directions. Cancers 2021, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, M.F.; Esmaeli, B. Molecular characteristics of uveal melanoma: Insights from the Cancer Genome Atlas (TCGA) Project. Cancers 2019, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.Q.N.; Drabarek, W.; Yavuzyigitoglu, S.; Medico Salsench, E.; Verdijk, R.M.; Naus, N.C.; de Klein, A.; Kiliç, E.; Brosens, E. Spliceosome mutations in uveal melanoma. Int. J. Mol. Sci. 2020, 21, 9546. [Google Scholar] [CrossRef]
- Ni, Y.; Zhang, Z.; Chen, G.; Long, W.; Tong, L.; Zeng, J. Integrated analyses identify potential prognostic markers for uveal melanoma. Exp. Eye Res. 2019, 187, 107780. [Google Scholar] [CrossRef]
- Shields, J.A.; Shields, C.L. Management of posterior uveal melanoma: Past, present, and future: The 2014 Charles L. Schepens lecture. Ophthalmology 2015, 122, 414–428. [Google Scholar] [CrossRef]
- Aronow, M.E.; Topham, A.K.; Singh, A.D. Uveal melanoma: 5-year update on incidence, treatment, and survival (SEER 1973–2013). Ocul. Oncol. Pathol. 2018, 4, 145–151. [Google Scholar] [CrossRef]
- Puusaari, I.; Heikkonen, J.; Summanen, P.; Tarkkanen, A.; Kivelä, T. Iodine brachytherapy as an alternative to enucleation for large uveal melanomas. Ophthalmology 2003, 110, 2223–2234. [Google Scholar] [CrossRef]
- Diener-West, M.; Earle, J.D.; Fine, S.L.; Hawkins, B.S.; Moy, C.S.; Reynolds, S.M.; Schachat, A.P.; Straatsma, B.R.; Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: Initial mortality findings. COMS Report No. 18. Arch. Ophthalmol. 2001, 119, 969–982. [Google Scholar]
- Veksler, R.; Fabian, I.D. Uveal melanoma: Diagnosis, classification and management. In Intraocular Tumours; Springer: Singapore, 2020; pp. 71–80. [Google Scholar]
- Aura Biosciences. A Phase 3 Randomized, Masked, Controlled Trial to Evaluate Efficacy and Safety of Belzupacap Sarotalocan (AU-011) Treatment Compared to Sham Control in Subjects with Primary Indeterminate Lesions or Small Choroidal Melanoma. Available online: https://clinicaltrials.gov/study/NCT06007690?id=NCT06007690&rank=1 (accessed on 2 April 2025).
- Kines, R.C.; Varsavsky, I.; Choudhary, S.; Bhattacharya, D.; Spring, S.; McLaughlin, R.; Kang, S.J.; Grossniklaus, H.E.; Vavvas, D.; Monks, S.; et al. An infrared dye–conjugated virus-like particle for the treatment of primary uveal melanoma. Mol. Cancer Ther. 2018, 17, 565–574. [Google Scholar] [CrossRef]
- IDEAYA Biosciences. (Neo)Adjuvant IDE196 (Darovasertib) in Patients With Localized Ocular Melanoma. Available online: https://clinicaltrials.gov/study/NCT05907954 (accessed on 8 April 2025).
- Cao, L.; Chen, S.; Sun, R.; Ashby, C.R., Jr.; Wei, L.; Huang, Z.; Chen, Z.S. Darovasertib, a novel treatment for metastatic uveal melanoma. Front. Pharmacol. 2023, 14, 1232787. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Ocular Melanoma Study Group. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: COMS Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [CrossRef]
- Reekie, I.R.; Sharma, S.; Foers, A.; Sherlock, J.; Coles, M.C.; Dick, A.D.; Denniston, A.K.; Buckley, C.D. The cellular composition of the uveal immune environment. Front. Med. 2021, 8, 721953. [Google Scholar] [CrossRef] [PubMed]
- Rantala, E.S.; Hernberg, M.M.; Piperno-Neumann, S.; Grossniklaus, H.E.; Kivelä, T.T. Metastatic uveal melanoma: The final frontier. Prog. Retin. Eye Res. 2022, 90, 101041. [Google Scholar] [CrossRef]
- Buder, K.; Gesierich, A.; Gelbrich, G.; Goebeler, M. Systemic treatment of metastatic uveal melanoma: Review of literature and future perspectives. Cancer Med. 2013, 2, 674. [Google Scholar] [CrossRef]
- Leyvraz, S.; Piperno-Neumann, S.; Suciu, S.; Baurain, J.F.; Zdzienicki, M.; Testori, A.; Marshall, E.; Scheulen, M.; Jouary, T.; Negrier, S.; et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): A multicentric randomized trial. Ann. Oncol. 2014, 25, 742. [Google Scholar] [CrossRef]
- Ausman, R.K. Development of a technic for isolated perfusion of the liver. N. Y. State J. Med. 1961, 61, 3993–3997. [Google Scholar]
- Agarwala, S.S.; Eggermont, A.M.M.; O’Day, S.; Zager, J.S. Metastatic melanoma to the liver: A contemporary and comprehensive review of surgical, systemic, and regional therapeutic options. Cancer 2014, 120, 781–789. [Google Scholar] [CrossRef]
- Gomez, D.; Wetherill, C.; Cheong, J.; Jones, L.; Marshall, E.; Damato, B.; Coupland, S.E.; Ghaneh, P.; Poston, G.J.; Malik, H.Z.; et al. The Liverpool uveal melanoma liver metastases pathway: Outcome following liver resection. J. Surg. Oncol. 2014, 109, 542–547. [Google Scholar] [CrossRef]
- Marquardt, S.; Kirstein, M.M.; Brüning, R.; Zeile, M.; Ferrucci, P.F.; Prevoo, W.; Radeleff, B.; Trillaud, H.; Tselikas, L.; Vicente, E.; et al. Percutaneous hepatic perfusion (chemosaturation) with melphalan in patients with intrahepatic cholangiocarcinoma: European multicentre study on safety, short-term effects and survival. Eur. Radiol. 2019, 29, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Karydis, I.; Gangi, A.; Wheater, M.J.; Choi, J.; Wilson, I.; Thomas, K.; Pearce, N.; Takhar, A.; Gupta, S.; Hardman, D.; et al. Percutaneous hepatic perfusion with melphalan in uveal melanoma: A safe and effective treatment modality in an orphan disease. J. Surg. Oncol. 2018, 117, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Valpione, S.; Aliberti, C.; Parrozzani, R.; Bazzi, M.; Pigozzo, J.; Midena, E.; Pilati, P.; Campana, L.G.; Chiarion-Sileni, V. A retrospective analysis of 141 patients with liver metastases from uveal melanoma: A two-cohort study comparing transarterial chemoembolization with CPT-11 charged microbeads and historical treatments. Melanoma Res. 2015, 25, 164–168. [Google Scholar] [CrossRef]
- Patel, K.; Sullivan, K.; Berd, D.; Mastrangelo, M.J.; Shields, C.L.; Shields, J.A.; Sato, T. Chemoembolization of the hepatic artery with BCNU for metastatic uveal melanoma: Results of a phase II study. Melanoma Res. 2005, 15, 297–304. [Google Scholar] [CrossRef]
- Alexander, H.; Wen, D.; Chu, M.; Han, C.; Hadden, P.; Thomas, R.; Bartlett, A. Selective internal radiation therapy for hepatic metastases of uveal melanoma: A systematic review. Br. J. Radiol. 2022, 95, 20210200. [Google Scholar] [CrossRef]
- Roelofsen, C.D.M.; Wierenga, A.P.A.; van Duinen, S.; Verdijk, R.M.; Bleeker, J.; Marinkovic, M.; Luyten, G.P.M.; Jager, M.J. Five decades of enucleations for uveal melanoma in one center: More tumors with high risk factors, no improvement in survival over time. Ocul. Oncol. Pathol. 2021, 7, 133–141. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7–CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, K. Created in BioRender. 2025. Available online: https://BioRender.com/8zqsz5p (accessed on 3 April 2025).
- Jain, S.; Clark, J.I. Ipilimumab for the treatment of melanoma. Melanoma Manag. 2015, 2, 33–39. [Google Scholar] [CrossRef]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA approval summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef]
- Raedler, L.A. Keytruda (pembrolizumab): First PD-1 inhibitor approved for previously treated unresectable or metastatic melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Orloff, M. Clinical trials in metastatic uveal melanoma: Immunotherapy. Ocul. Oncol. Pathol. 2021, 7, 168. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Dousset, L.D.; Poizeau, F.; Robert, C.; Mansard, S.; Mortier, L.; Caumont, C.; Routier, É.; Dupuy, A.; Rouanet, J.; Battistella, M.; et al. Positive association between location of melanoma, ultraviolet signature, tumor mutational burden, and response to anti–PD-1 therapy. JCO Precis. Oncol. 2021, 5, 1821–1829. [Google Scholar] [CrossRef]
- Hoefsmit, E.P.; Rozeman, E.A.; Van, T.M.; Dimitriadis, P.; Krijgsman, O.; Conway, J.W.; Pires da Silva, I.; van der Wal, J.E.; Ketelaars, S.L.C.; Bresser, K.; et al. Comprehensive analysis of cutaneous and uveal melanoma liver metastases. J. Immunother. Cancer 2020, 8, 001501. [Google Scholar] [CrossRef]
- Hu, D.N. Photobiology of ocular melanocytes and melanoma. Photochem. Photobiol. 2005, 81, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Mallet, J.D.; Gendron, S.P.; Drigeard Desgarnier, M.C.; Rochette, P.J. Implication of ultraviolet light in the etiology of uveal melanoma: A review. Photochem. Photobiol. 2014, 90, 15–21. [Google Scholar] [CrossRef]
- Hassel, J.C.; Piperno-Neumann, S.; Rutkowski, P.; Baurain, J.F.; Schlaak, M.; Butler, M.O.; Sullivan, R.J.; Dummer, R.; Kirkwood, J.M.; Orloff, M.; et al. Three-year overall survival with tebentafusp in metastatic uveal melanoma. N. Engl. J. Med. 2023, 389, 2256. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tebentafusp: First approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.; Schreurs, M.W.; de Boer, A.J.; Kawakami, Y.; Rosenberg, S.A.; Adema, G.J.; Figdor, C.G. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J. Exp. Med. 1994, 179, 1005–1009. [Google Scholar] [CrossRef]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef]
- Rochfort, K. Created in BioRender. 2025. Available online: https://BioRender.com/xv85lbc (accessed on 3 April 2025).
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Katovich Hurley, C. Frequencies of HLA-A2 alleles in five U.S. population groups: Predominance of A∗02011 and identification of HLA-A∗0231. Hum. Immunol. 2000, 61, 334–340. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. A TCR/anti-CD3 bispecific fusion protein targeting gp100 potently activated anti-tumor immune responses in metastatic melanoma. Clin. Cancer Res. 2020, 26, 5869. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 2 April 2025).
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Johansson, P.A.; Brooks, K.; Newell, F.; Palmer, J.M.; Wilmott, J.S.; Pritchard, A.L.; Broit, N.; Wood, S.; Carlino, M.S.; Leonard, C.; et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat. Commun. 2020, 11, 2408. [Google Scholar] [CrossRef]
- Johansson, P.A.; Stark, A.; Palmer, J.M.; Bigby, K.; Brooks, K.; Rolfe, O.; Pritchard, A.L.; Whitehead, K.; Warrier, S.; Glasson, W.; et al. Prolonged stable disease in a uveal melanoma patient with germline MBD4 nonsense mutation treated with pembrolizumab and ipilimumab. Immunogenetics 2019, 71, 433–436. [Google Scholar] [CrossRef]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Fiévet, A.; Gardrat, S.; Barnhill, R.L.; Popova, T.; Servois, V.; Rampanou, A.; Mouton, A.; et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Blomen, C.L.; Kött, J.; Hartung, T.I.; Torster, L.K.; Gebhardt, C. Combination of immune checkpoint inhibitors and liver-specific therapies in liver-metastatic uveal melanoma: Can we thus overcome its high resistance? Cancers 2021, 13, 6390. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, F.S.; De Rosa, F.; Di Terlizzi, P.; Toneatto, G.; Gabai, A.; Finocchio, L.; Salati, C.; Spadea, L.; Zeppieri, M. Uveal melanoma: Recent advances in immunotherapy. World J. Clin. Oncol. 2024, 15, 23–31. [Google Scholar] [CrossRef]
- Tong, T.M.L.; Burgmans, M.C.; Speetjens, F.M.; van Erkel, A.R.; van der Meer, R.W.; van Rijswijk, C.S.P.; Jonker-Bos, M.A.; Roozen, C.F.M.; Sporrel-Blokland, M.; Lutjeboer, J.; et al. Combining melphalan percutaneous hepatic perfusion with ipilimumab plus nivolumab in advanced uveal melanoma: First safety and efficacy data from the Phase Ib part of the CHOPIN trial. Cardiovasc. Interv. Radiol. 2023, 46, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Aedo-Lopez, V.; Gérard, C.L.; Boughdad, S.; Gautron Moura, B.; Berthod, G.; Digklia, A.; Homicsko, K.; Schaefer, N.; Duran, R.; Cuendet, M.A.; et al. Safety and efficacy of ipilimumab plus nivolumab and sequential selective internal radiation therapy in hepatic and extrahepatic metastatic uveal melanoma. Cancers 2022, 14, 1162. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Infante, J.R.; Papadopoulos, K.P.; Bendell, J.C.; Patnaik, A.; Burris, H.A.; Rasco, D.; Jones, S.F.; Smith, L.; Cox, D.S.; Durante, M.; et al. A phase 1b study of trametinib, an oral mitogen-activated protein kinase kinase (MEK) inhibitor, in combination with gemcitabine in advanced solid tumours. Eur. J. Cancer 2013, 49, 2077–2085. [Google Scholar] [CrossRef]
- Yu, D.; Zhao, W.; Vallega, K.A.; Sun, S.Y. Managing Acquired Resistance to Third-Generation EGFR Tyrosine Kinase Inhibitors Through Co-Targeting MEK/ERK Signaling. Lung Cancer Targets Ther. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Maria, A.; Simonelli, M.; Santangelo, F.; Amato, G.; Simonetti, E.; Graham, J.; Lahn, M.M.F.; Di Conza, G.; Hammett, T.; Zorrilla, R.; et al. Roginolisib, an oral, highly selective and allosteric modulator of phosphoinositide 3-kinase inhibitor delta (PI3Kδ) in patients with uveal melanoma and advanced cancers. J. Clin. Oncol. 2024, 42, 9597. [Google Scholar]
- Giacomo, A.M.D.; Simonelli, M.; Santangelo, F.; Amato, G.; Simonetti, E.; Graham, J.; Lahn, M.; Di Conza, G.; Hammett, T.; Kaur, P.; et al. 164P Roginolisib (IOA-244), the first highly selective oral allosteric modulator of phosphoinositide 3-kinase inhibitor delta (PI3Kδ), has immuno-modulatory effects associated with clinical benefits in patients with metastatic uveal melanoma. Immuno-Oncol. Technol. 2024, 24, 100793. [Google Scholar] [CrossRef]
- Carlo-Stella, C.; Lahn, M.; Hammett, T.; Kaur, P.; Zorrilla, R.; Di Conza, G.; van der Veen, L.; Pickering, C.; Santoro, A. Roginolisib a Highly Selective Allosteric Modulator of the Phosphoinositide 3-Kinase Delta (PI3Kδ) in Patients with Refractory/Relapsed Follicular Lymphoma. Blood 2023, 142, 6114. [Google Scholar] [CrossRef]
- IDEAYA Biosciences. IDE196 (Darovasertib) in Combination with Crizotinib Versus Investigator’s Choice of Treatment As First-line Therapy in HLA-A2 Negative Metastatic Uveal Melanoma (DAR-UM-2). Available online: https://clinicaltrials.gov/study/NCT05987332 (accessed on 8 April 2025).
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Wu, G.; Fan, L.; Edmonson, M.N.; Shaw, T.; Boggs, K.; Easton, J.; Rusch, M.C.; Webb, T.R.; Zhang, J.; Potter, P.M. Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA 2018, 24, 1056–1066. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).