
Targeting Autophagy for Pituitary Tumors

Simple Summary
Abstract
1. Introduction
2. Autophagy and Cancer
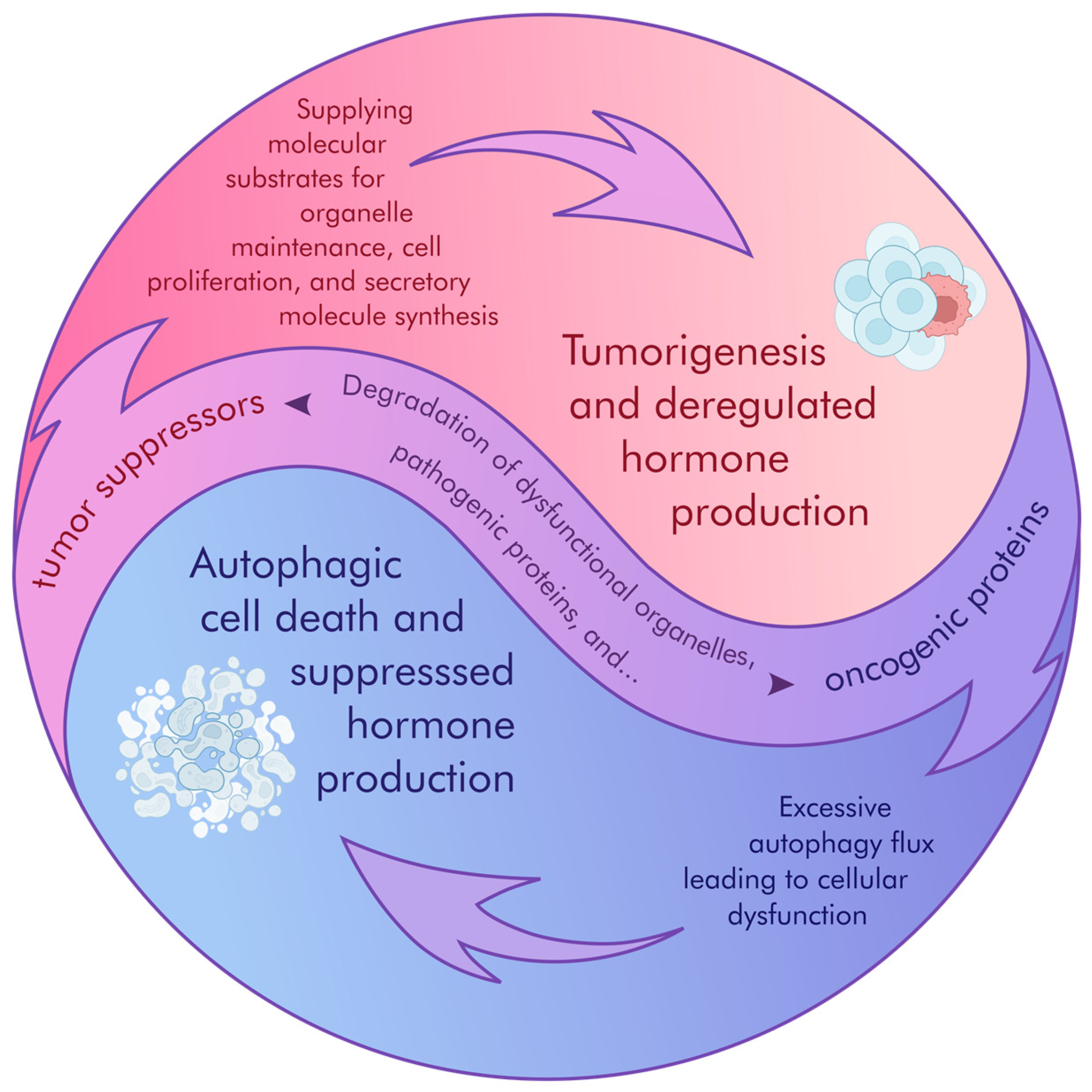
2.1. Crosstalk Between Autophagy and Cancer
2.2. Autophagy as a Cancer Inhibitor
2.3. Mitophagy’s Dual Role in Cancer Growth over Time
3. Autophagy and Hormone Sorting
3.1. Degradation of Secretory Granules
3.2. Unconventional Hormone Secretion
3.3. Dysfunction of SV Degradation
4. Targeting Autophagy as a Therapeutic Approach for Pituitary Tumors
4.1. Enhanced Autophagic Activity
4.2. Inhibited Autophagic Activity in Pituitary Tumors
5. Interpretation of Autophagy Activity
6. Limitations of Current Models
7. Clinical Perspective
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daly, A.F.; Rixhon, M.; Adam, C.; Dempegioti, A.; Tichomirowa, M.A.; Beckers, A. High prevalence of pituitary adenomas: A cross-sectional study in the province of Liege, Belgium. J. Clin. Endocrinol. Metab. 2006, 91, 4769–4775. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Karavitaki, N.; Wass, J.A. Prevalence of pituitary adenomas: A community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin. Endocrinol. 2010, 72, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The prevalence of pituitary adenomas: A systematic review. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Law, F.; Seo, J.H.; Wang, Z.; DeLeon, J.L.; Bolis, Y.; Brown, A.; Zong, W.X.; Du, G.; Rocheleau, C.E. The VPS34 PI3K negatively regulates RAB-5 during endosome maturation. J. Cell Sci. 2017, 130, 2007–2017. [Google Scholar] [CrossRef]
- Lee, H.N.; Zarza, X.; Kim, J.H.; Yoon, M.J.; Kim, S.H.; Lee, J.H.; Paris, N.; Munnik, T.; Otegui, M.S.; Chung, T. Vacuolar Trafficking Protein VPS38 Is Dispensable for Autophagy. Plant Physiol. 2018, 176, 1559–1572. [Google Scholar] [CrossRef]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Chen, H.Y.; White, E. Role of autophagy in cancer prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, B.; Wu, Y.; Jin, F.; Xia, Y.; Liu, X. Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer 2010, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.M.; Wang, G.L.; Chen, L.; Xu, Y.Y.; He, S.; Cao, X.L.; Qin, J.; Zhou, J.M.; Zhang, Y.X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef]
- Vega-Rubín-de-Celis, S.; Zou, Z.; Fernández, Á.F.; Ci, B.; Kim, M.; Xiao, G.; Xie, Y.; Levine, B. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4176–4181. [Google Scholar] [CrossRef]
- Peng, Y.; Miao, H.; Wu, S.; Yang, W.; Zhang, Y.; Xie, G.; Xie, X.; Li, J.; Shi, C.; Ye, L.; et al. ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 2016, 12, 2167–2182. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Vernucci, E.; Tomino, C.; Molinari, F.; Limongi, D.; Aventaggiato, M.; Sansone, L.; Tafani, M.; Russo, M.A. Mitophagy and Oxidative Stress in Cancer and Aging: Focus on Sirtuins and Nanomaterials. Oxidative Med. Cell. Longev. 2019, 2019, 6387357. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Marusawa, H.; Wang, H.Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef]
- Gustafsson, A.B. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 2011, 32, 267–274. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Hecht, L.; Podber, E.; Ryan, J. Phosphatases of rat liver. I. The dephosphorylation of adenosinetriphosphate. J. Biol. Chem. 1952, 194, 153–170. [Google Scholar] [CrossRef]
- Pearson, B.; Novikoff, A.B.; Morrione, T.G. The histochemical localization of alkaline phosphatase during carcinogenesis in rats fed p-dimethylaminoazobenzene. Cancer Res. 1950, 10, 557–564. [Google Scholar]
- Novikoff, A.B.; Ryan, J.; Podber, E. The effects of ribonuclease on the ribonucleic acid and enzyme activities of microsomes isolated from rat liver homogenates. J. Histochem. Cytochem. 1954, 2, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B.; Holt, S.J. Esterase-rich bodies in osmium-fixed cells of rat kidney and liver. J. Biophys. Biochem. Cytol. 1957, 3, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Boudier, J.A.; Picard, D. Granulolysis in neurosecretory neurons of the rat supraoptico-posthypophyseal system. Cell Tissue Res. 1976, 172, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Vomund, A.N.; Peterson, O.J.; Srivastava, N.; Li, T.; Kain, L.; Beatty, W.L.; Zhang, B.; Hsieh, C.S.; Teyton, L.; et al. Crinophagic granules in pancreatic β cells contribute to mouse autoimmune diabetes by diversifying pathogenic epitope repertoire. Nat. Commun. 2024, 15, 8318. [Google Scholar] [CrossRef]
- Sirek, A.M.; Horvath, E.; Ezrin, C.; Kovacs, K. Effect of starvation on pituitary growth hormone cells and blood growth hormone and prolactin levels in the rat. Nutr. Metab. 1976, 20, 67–75. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Tulipano, G.; Giustina, A. Autophagy in normal pituitary and pituitary tumor cells and its potential role in the actions of somatostatin receptor ligands in acromegaly. Rev. Endocr. Metab. Disord. 2021, 22, 147–160. [Google Scholar] [CrossRef]
- Meda, P. Lysosomes in normal pancreatic beta cells. Diabetologia 1978, 14, 305–310. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef]
- Davis, S.; Wang, J.; Ferro-Novick, S. Crosstalk between the Secretory and Autophagy Pathways Regulates Autophagosome Formation. Dev. Cell 2017, 41, 23–32. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.D.; Resnik, R.; Vaccaro, M.I. Secretory Autophagy and Its Relevance in Metabolic and Degenerative Disease. Front. Endocrinol. 2020, 11, 266. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Wu, H.T.; Wang, Y.C.; Chang, C.J.; Shan, Y.S.; Wu, S.R.; Chiu, Y.C.; Hsu, C.L.; Juan, H.F.; Lan, K.Y.; et al. Secretory autophagy promotes RAB37-mediated insulin secretion under glucose stimulation both in vitro and in vivo. Autophagy 2023, 19, 1239–1257. [Google Scholar] [CrossRef]
- Leidal, A.M.; Huang, H.H.; Marsh, T.; Solvik, T.; Zhang, D.; Ye, J.; Kai, F.; Goldsmith, J.; Liu, J.Y.; Huang, Y.H.; et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 2020, 22, 187–199. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion—New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, 213686. [Google Scholar] [CrossRef]
- Colletti, M.; Ceglie, D.; Di Giannatale, A.; Nazio, F. Autophagy and Exosomes Relationship in Cancer: Friends or Foes? Front. Cell Dev. Biol. 2020, 8, 614178. [Google Scholar] [CrossRef]
- Satou, M.; Wang, J.; Nakano-Tateno, T.; Teramachi, M.; Aoki, S.; Sugimoto, H.; Chik, C.; Tateno, T. Autophagy inhibition suppresses hormone production and cell growth in pituitary tumor cells: A potential approach to pituitary tumors. Mol. Cell. Endocrinol. 2024, 586, 112196. [Google Scholar] [CrossRef]
- Wang, C.; Tan, C.; Wen, Y.; Zhang, D.; Li, G.; Chang, L.; Su, J.; Wang, X. FOXP1-induced lncRNA CLRN1-AS1 acts as a tumor suppressor in pituitary prolactinoma by repressing the autophagy via inactivating Wnt/β-catenin signaling pathway. Cell Death Dis. 2019, 10, 499. [Google Scholar] [CrossRef]
- Lin, S.J.; Wu, Z.R.; Cao, L.; Zhang, Y.; Leng, Z.G.; Guo, Y.H.; Shang, H.B.; Zhao, W.G.; Zhang, X.; Wu, Z.B. Pituitary Tumor Suppression by Combination of Cabergoline and Chloroquine. J. Clin. Endocrinol. Metab. 2017, 102, 3692–3703. [Google Scholar] [CrossRef]
- Coker-Gurkan, A.; Ayhan-Sahin, B.; Keceloglu, G.; Obakan-Yerlikaya, P.; Arisan, E.D.; Palavan-Unsal, N. Atiprimod induce apoptosis in pituitary adenoma: Endoplasmic reticulum stress and autophagy pathways. J. Cell Biochem. 2019, 120, 19749–19763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yang, Y.; Wang, D.; Xie, B.; Li, X.; Xu, B. HIF-1α Inhibition Sensitized Pituitary Adenoma Cells to Temozolomide by Regulating Presenilin 1 Expression and Autophagy. Technol. Cancer Res. Treat. 2016, 15, Np95–Np104. [Google Scholar] [CrossRef]
- Cecenarro, L.A.; Moyano Crespo, G.D.; Guido, C.B.; Pérez, P.A.; Faure, E.E.; Picech, F.; Rossetto, S.V.; De Battista, J.C.; Gutiérrez, S.; Torres, A.I.; et al. Ultrastructural and Molecular Evidence of Macroautophagy in Functioning PitNETs and Experimental Pituitary Tumors. Neuroendocrinology 2023, 113, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Ding, X.; Zang, Z.; Zhang, Y.; Tang, X.; Pei, X.; Chen, Q.; Yin, H.; Zheng, X.; Chen, Y.; et al. Drp1-Mediated Mitochondrial Metabolic Dysfunction Inhibits the Tumor Growth of Pituitary Adenomas. Oxidative Med. Cell. Longev. 2022, 2022, 5652586. [Google Scholar] [CrossRef]
- Xu, L.; Ning, R.; Du, X.; Zhang, Y.; Gu, C.; Wang, B.; Bian, L.; Sun, Q.; Sun, Y.; Ren, J. Bone Morphogenetic Protein Signaling Agonist SB4 (BMPSB4) Inhibits Corticotroph Pituitary Neuroendocrine Tumors by Activation of Autophagy via a BMP4/SMADs-Dependent Pathway. ACS Pharmacol. Transl. Sci. 2024, 7, 1951–1970. [Google Scholar] [CrossRef]
- Xi, X.; Liu, N.; Wang, Q.; Chu, Y.; Yin, Z.; Ding, Y.; Lu, Y. ACT001, a novel PAI-1 inhibitor, exerts synergistic effects in combination with cisplatin by inhibiting PI3K/AKT pathway in glioma. Cell Death Dis. 2019, 10, 757. [Google Scholar] [CrossRef]
- Cai, L.; Wu, Z.R.; Cao, L.; Xu, J.D.; Lu, J.L.; Wang, C.D.; Jin, J.H.; Wu, Z.B.; Su, Z.P. ACT001 inhibits pituitary tumor growth by inducing autophagic cell death via MEK4/MAPK pathway. Acta Pharmacol. Sin. 2022, 43, 2386–2396. [Google Scholar] [CrossRef]
- Zhu, J.; Tang, C.; Cong, Z.; Yuan, F.; Cai, X.; Yang, J.; Ma, C. ACT001 reverses resistance of prolactinomas via AMPK-mediated EGR1 and mTOR pathways. Endocr.-Relat. Cancer 2021, 29, 33–46. [Google Scholar] [CrossRef]
- Wang, C.; Dai, S.; Zhao, X.; Zhang, Y.; Gong, L.; Fu, K.; Ma, C.; Peng, C.; Li, Y. Celastrol as an emerging anticancer agent: Current status, challenges and therapeutic strategies. Biomed. Pharmacother. 2023, 163, 114882. [Google Scholar] [CrossRef]
- Cai, Z.; Qian, B.; Pang, J.; Tan, Z.B.; Zhao, K.; Lei, T. Celastrol Induces Apoptosis and Autophagy via the AKT/mTOR Signaling Pathway in the Pituitary ACTH-secreting Adenoma Cells. Curr. Med. Sci. 2022, 42, 387–396. [Google Scholar] [CrossRef]
- Sun, H.; Hu, B.; Wu, C.; Jiang, T. Targeting the SPHK1/S1P/S1PR2 axis ameliorates GH-secreted pituitary adenoma progression. Eur. J. Clin. Investig. 2024, 54, e14117. [Google Scholar] [CrossRef] [PubMed]
- Leng, Z.G.; Lin, S.J.; Wu, Z.R.; Guo, Y.H.; Cai, L.; Shang, H.B.; Tang, H.; Xue, Y.J.; Lou, M.Q.; Zhao, W.; et al. Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy 2017, 13, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, M.; Ji, C.; Chen, Z.; Yang, H.; Wang, L.; Yu, Y.; Qiao, N.; Ma, Z.; Ye, Z.; et al. Treatment of acromegaly by rosiglitazone via upregulating 15-PGDH in both pituitary adenoma and liver. iScience 2021, 24, 102983. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Choi, S.I.; Jeung, E.B.; Yoo, Y.M. Cyclosporine A induces apoptotic and autophagic cell death in rat pituitary GH3 cells. PLoS ONE 2014, 9, e108981. [Google Scholar] [CrossRef]
- Pelicci, G.; Pagliacci, M.C.; Lanfrancone, L.; Pelicci, P.G.; Grignani, F.; Nicoletti, I. Inhibitory effect of the somatostatin analog octreotide on rat pituitary tumor cell (GH3) proliferation in vitro. J. Endocrinol. Investig. 1990, 13, 657–662. [Google Scholar] [CrossRef]
- Tulipano, G.; Giustina, A. Effects of octreotide on autophagy markers and cell viability markers related to metabolic activity in rat pituitary tumor cells. Pituitary 2020, 23, 223–231. [Google Scholar] [CrossRef]
- Dagistanli, F.K.; Ozkaya, H.M.; Kucukyoruk, B.; Biceroglu, H.; Metin, D.; Gazioglu, N.; Oz, B.; Kadioglu, P.; Ozturk, M. Preoperative Somatostatin Analogue Treatment Might Trigger Apoptosis and Autophagy in Tumor Tissues of Patients with Acromegaly: A Pilot Study. Exp. Clin. Endocrinol. Diabetes 2018, 126, 168–175. [Google Scholar] [CrossRef]
- Changotra, H.; Kaur, S.; Yadav, S.S.; Gupta, G.L.; Parkash, J.; Duseja, A. ATG5: A central autophagy regulator implicated in various human diseases. Cell Biochem. Funct. 2022, 40, 650–667. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med. 2009, 15, 217–224. [Google Scholar] [CrossRef]
- Geng, X.; Ma, L.; Li, Z.; Li, Z.; Li, J.; Li, M.; Wang, Q.; Chen, Z.; Sun, Q. Bromocriptine Induces Autophagy-Dependent Cell Death in Pituitary Adenomas. World Neurosurg. 2017, 100, 407–416. [Google Scholar] [CrossRef]
- Lin, S.J.; Leng, Z.G.; Guo, Y.H.; Cai, L.; Cai, Y.; Li, N.; Shang, H.B.; Le, W.D.; Zhao, W.G.; Wu, Z.B. Suppression of mTOR pathway and induction of autophagy-dependent cell death by cabergoline. Oncotarget 2015, 6, 39329–39341. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Tang, H.; Zhang, Y.; Zhang, Q.F.; Liu, X.Y.; Liu, Y.T.; Gu, W.T.; Zheng, Y.Z.; Shang, H.B.; Wang, Y.; et al. DEPTOR inhibits cell proliferation and confers sensitivity to dopamine agonist in pituitary adenoma. Cancer Lett. 2019, 459, 135–144. [Google Scholar] [CrossRef]
- Lin, S.; Han, C.; Lou, X.; Wu, Z.B. Hydroxychloroquine overcomes cabergoline resistance in a patient with Lactotroph Pituitary neuroendocrine tumor: A case report. Front. Endocrinol. 2022, 13, 955100. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, L.; Zhang, D.; Huang, X.; Yang, J.; Li, W.; Wang, C.; Meng, X.; Huang, G. Intranasal 15d-PGJ2 inhibits the growth of rat lactotroph pituitary neuroendocrine tumors by inducing PPARγ-dependent apoptotic and autophagic cell death. Front. Neurosci. 2023, 17, 1109675. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cao, X.; Zhu, Q. p62/SQSTM1 in cancer: Phenomena, mechanisms, and regulation in DNA damage repair. Cancer Metastasis Rev. 2025, 44, 33. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, Y.; Sun, T.; Zhu, P.; Li, J.; Zhang, Q.; Wang, X.; Jiang, J.; Chen, G.; Zhao, X. p62/SQSTM1 accumulation due to degradation inhibition and transcriptional activation plays a critical role in silica nanoparticle-induced airway inflammation via NF-κB activation. J. Nanobiotechnol. 2020, 18, 77. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef]
- Vats, S.; Manjithaya, R. A reversible autophagy inhibitor blocks autophagosome-lysosome fusion by preventing Stx17 loading onto autophagosomes. Mol. Biol. Cell 2019, 30, 2283–2295. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef]
- Mizushima, N.; Murphy, L.O. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends Biochem. Sci. 2020, 45, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Habanjar, O.; Diab-Assaf, M.; Caldefie-Chezet, F.; Delort, L. 3D Cell Culture Systems: Tumor Application, Advantages, and Disadvantages. Int. J. Mol. Sci. 2021, 22, 12200. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Enhanced Autophagic Activity | ||||
|---|---|---|---|---|
| Drug (Concentration/Dose) | Target (Species) | Effect on Cell Proliferation | Effect on Hormone Production/Secretion | Reference Number |
| BMPSB4 (39.05 μM a) | AtT20 (m) | Suppressed | NA | [55] |
| ACT001 (9.56 μM a, 22.65 μM a, 20 μM b) | GH3 (r), MMQ (r), NFPT (h) | Suppressed | NA | [57,58] |
| Celastrol (1 μM a) | AtT20 (m) | Suppressed | NA | [60] |
| JTE-013 (41.17 μM a) | GH3 (r) | Suppressed | GH suppressed | [61] |
| SKF83959 (5 μM b) | GH3 (r), MMQ (r) | Suppressed | NA | [62] |
| Rosiglitazone (50 μM b) | GH3 (r) | Suppressed | GH suppressed | [63] |
| CsA (0.1 μM b) | GH3 (r) | Suppressed | NA | [64] |
| Octreotide (100 nM c) | GH3 (r) | NA | NA | [66] |
| BRC (110 μM a, 60 μM a) | GH3 (r), MMQ (r) | Suppressed | PRL suppressed | [70] |
| CAB (100 μM a, 50 μM a) | GH3 (r), MMQ (r) | Suppressed | NA | [71] |
| Inhibited Autophagic Activity | ||||
|---|---|---|---|---|
| Drug (Concentration/Dose) | Target (Species) | Effect on Cell Proliferation | Effect on Hormone Production/Secretion | Reference Number |
| CQ (5 μM b, 10 μM b) | AtT20 (m), GH4 (r) | Suppressed | ACTH and GH suppressed | [48] |
| bafilomycin A1 (1 nM b) | AtT20 (m) | Suppressed | ACTH suppressed | [48] |
| Monensin (2.5 μM b) | AtT20 (m) | Suppressed | ACTH suppressed | [48] |
| CQ + TMZ (5 μM b + 20 μM b, 10 μM b + 400 μM b) | AtT20 (m), GH4 (r) | Suppressed | ACTH and GH suppressed | [48] |
| CQ + CAB (20 μM c + 100 μM c, 20 μM c + 50 μM c, 20 μM c + 50 or 100 μM c) | GH3 (r), MMQ (r), Prolactinoma (h) | Suppressed | PRL suppressed | [50] |
| HCQ (200 mg/day c) + CAB (3 mg/week c) | Prolactinoma (h) | No recurrence | PRL suppressed | [73] |
| 15d-PGJ2 (4 μg/day c) | Prolactinoma (r) | Suppressed | PRL suppressed | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, E.; Satou, M.; Tateno, T. Targeting Autophagy for Pituitary Tumors. Cancers 2025, 17, 1402. https://doi.org/10.3390/cancers17091402
Yin E, Satou M, Tateno T. Targeting Autophagy for Pituitary Tumors. Cancers. 2025; 17(9):1402. https://doi.org/10.3390/cancers17091402
Chicago/Turabian StyleYin, Evan, Motoyasu Satou, and Toru Tateno. 2025. "Targeting Autophagy for Pituitary Tumors" Cancers 17, no. 9: 1402. https://doi.org/10.3390/cancers17091402
APA StyleYin, E., Satou, M., & Tateno, T. (2025). Targeting Autophagy for Pituitary Tumors. Cancers, 17(9), 1402. https://doi.org/10.3390/cancers17091402