Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy

{kind=link}
Abstract
: Estrogen receptor (ERα) signaling plays a key role in hormonal cancer progression. ERα is a ligand-dependent transcription factor that modulates gene transcription via recruitment to the target gene chromatin. Emerging evidence suggests that ERα signaling has the potential to contribute to epigenetic changes. Estrogen stimulation is shown to induce several histone modifications at the ERα target gene promoters including acetylation, phosphorylation and methylation via dynamic interactions with histone modifying enzymes. Deregulation of enzymes involved in the ERα -mediated epigenetic pathway could play a vital role in ERα driven neoplastic processes. Unlike genetic alterations, epigenetic changes are reversible, and hence offer novel therapeutic opportunities to reverse ERα driven epigenetic changes. In this review, we summarize current knowledge on mechanisms by which ERα signaling potentiates epigenetic changes in cancer cells via histone modifications.1. Introduction
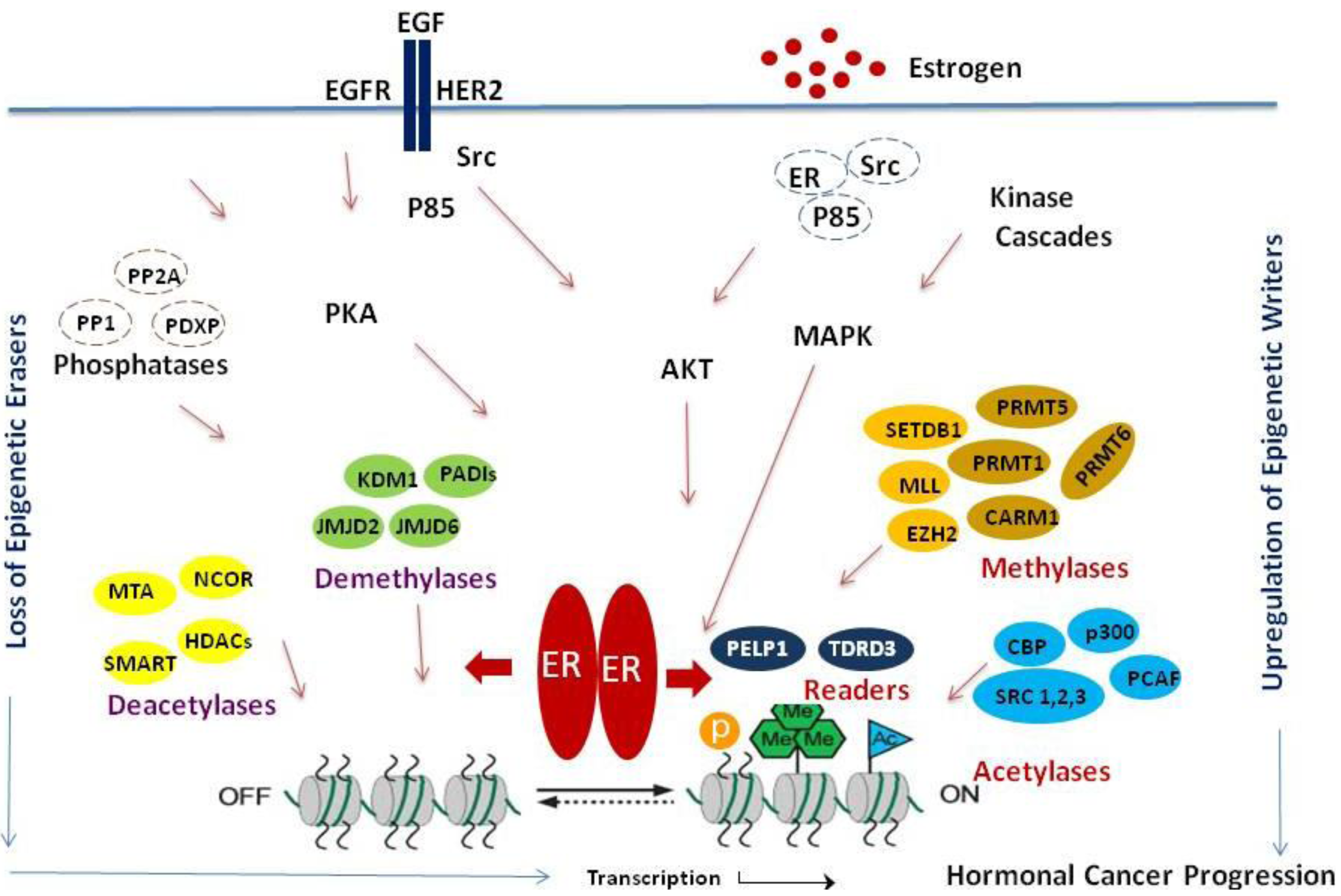
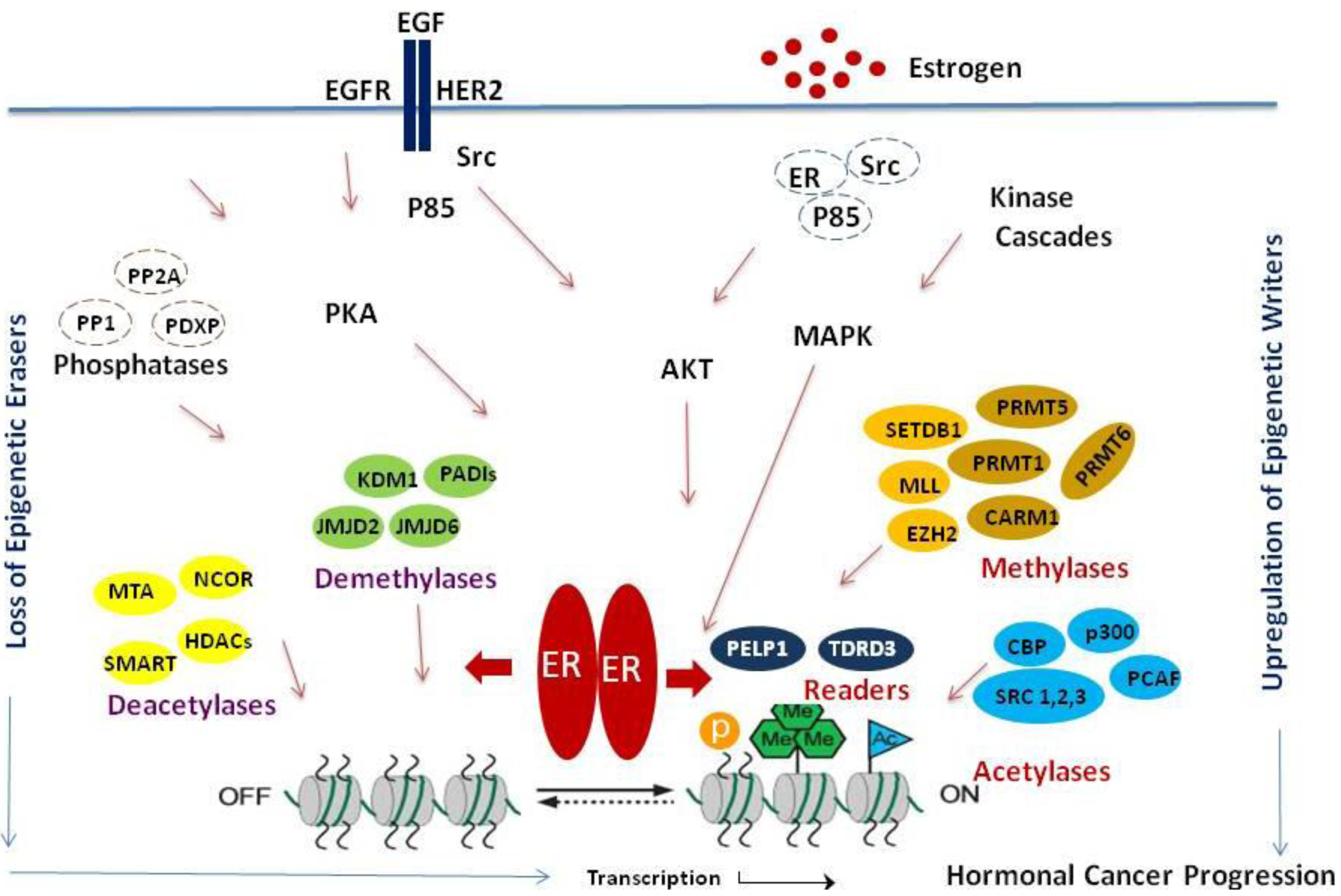
The steroid hormone estradiol plays an important role in the initiation and progression of breast cancer. The biological effects of estrogen are mediated by its binding to the structurally and functionally distinct estrogen receptors (ERα and ERβ) [1]. ERα is implicated as a key transcriptional regulator in breast cancer biology [2,3]. ERα functions as a ligand-dependent transcription factor that modulates gene transcription via direct recruitment to target gene chromatin. The transcription functions of ERα are shown to be influenced by several coactivators, including SRC1, SRC2, AIB1, PELP1, CBP, p300, PCAF, CARM1, PRMT1 and corepressors such as NCoR, SMRT and MTA1 [4-6]. ERα transcriptional outcome is regulated by a dynamic interaction of histone modifying enzymes, which are frequently associated with coregulators [7,8]. Evidence suggests that multiprotein complexes containing ERα, coactivators and histone modifying enzymes assemble in response to hormone binding leading to activation of transcription [9]. Estrogen stimulation induces several histone modifications at the ERα target gene promoters including acetylation, phosphorylation and methylation. The ‘histone-code hypothesis’ proposes combinatorial and/or sequential post-translational modification of histones, that is written by specific enzymes (‘writers’) and removed by others (‘erasers’) can be read by nuclear factors (‘readers’) to promote a variety of cellular processes by regulating gene expression [10]. The histone code hypothesis suggests that post-translational modifications confer specific functions and the modified histones recruit specialized proteins that facilitate defined functions [11]. The mechanism by which ERα targets and coordinates activities of kinases/phosphatases and histone modifying enzymes is poorly understood. Estrogen-ERα signaling has been traditionally implicated in the stimulation of transcription. Several acetylases/deacetylases and methylases/demethylases interact with ERα directly or indirectly and facilitate the necessary histone modifications. ERα's ability to modulate epigenetic changes by regulating writers, erasers and readers of epigenetic modifications and the reversible nature of these modifications provides a unique therapeutic opportunity to design novel drugs/small molecular inhibitors for treating hormonal cancers. In this review, we summarized the key evidence that links ERα to the regulation of histone modifying enzymes and readers of histone modifications and discuss the possibility of targeting these pathways for therapeutics.
2. Mechanisms of ERα-mediated Histone Modifications
2.1. Acetylation/deacetylation
Acetylation and deacetylation of conserved lysine residues present in histone tails have been suggested as a mechanism by which ERα modifies chromatin structure [12]. Acetylated histones are usually associated with transcriptionally active chromatin, while deacetylated histones are associated with inactive chromatin [13]. ERα coactivators like SRC1, and AIB1 have been shown to possess histone acetyltransferase activity [14]. In addition, ERα associates with and modulates functions of general acetyltransferases including p300/CBP and p300/CBP-associated factor (PCAF) [2]. ERα -mediated deacetylation is accomplished by recruitment of histone deacetylases (HDACs), which are indirectly recruited to ERα target genes through multi-subunit corepressor complexes. The HDAC1/2-containing corepressor complex is the main route by which deacetylation of chromatin-associated histones takes place; the key adaptor protein in this complex is Sin3 [15]. ERα also utilizes corepressor complexes such as nuclear receptor corepressor (NCOR), silencing mediator of retinoid and thyroid hormone receptors (SMART) and the metastasis-associated 1 (MTA1) protein that associate with histone deacetylases [16]. Ligand-dependent corepressor (LCOR) is recruited to ERα target genes and interacts with ERα and HDAC6. Studies employing siRNA targeting HDAC6 and LCOR indicated that HDAC6 may function with LCoR on some ERα target genes (such as IGFBP4, ADORA1 and CYP26B1) as part of a feedback loop to regulate estrogen-dependent gene regulation in breast cancer cells [17]. A recent report showed that HDAC7 and FoxA1 interactions play a role in estrogen mediated repression of a subset of ERα target genes [18]. Collectively, these results suggest that ERα achieves histone acetylation modifications at target gene promoters using several coregulators.
2.2. Phosphorylation/dephosphorylation
In addition to its well established role in nuclear actions, ERα signaling also activates a number of kinases in the extranuclear compartment including protein kinase B (AKT) and extracellular signal-regulated protein kinase (ERK) [19]. Hormonal stimulation promotes alterations in the phosphorylation of specific residues in histone tails via modulation of these extranuclear kinases. Estrogen-ERα signaling activates mitogen-activated protein kinase (MAPK) cascades that transmit and amplify signals involved in cellular proliferation and apoptosis. Of the three major MAPK pathways in human tissues, the one involving ERK-1 and -2 is most relevant to breast cancer [20]. Estrogen-ERα signaling also activates Src-MAPK and Src-AKT pathways [21,22]. Since kinases that phosphorylate core histones (Msk1 and Msk2) and histone H1 (Cdk2) are downstream substrates of MAPK and AKT, ERα-mediated extranuclear signals can influence their downstream chromatin targets [23,24]. Estrogen-ERα signaling also regulates the expression and function of several phosphatases including PP1, PP2A and PDXP [25,26]. These results suggest that ERα-extranuclear signaling has the potential to modulate epigenetic modifications.
2.3. Methylation/demethylation
Histone methylation is crucial for regulating chromatin structure, gene transcription and the overall epigenetic state of the cell. The methylation of histones is a key regulatory signal in ERα-mediated gene expression and histone methylation-dependent mechanisms impose ligand dependency for gene activation [27]. Unlike acetylation, which is associated with gene activation, the consequence of histone methylation appears to be site dependent. For example, H3K4 methylation is linked with activation, while H3K9 methylation correlates with repression [28,29]. The recent discovery of lysine-specific demethylase 1 (KDM1, also designated as LSD1) suggests that histone methylation is reversible [30]. Recent studies showed that KDM1, which demethylates H3K4 and H3K9, is recruited to a significant fraction of ERα target genes and is required to demethylate proximal histones to enable ERα-mediated transcription [27]. Enhancer of Zeste homolog 2 (EZH2) is a histone methyltransferase and a polycomb group protein that catalyzes the histone modification of H3K27me3. Arginine methylation of histone tails occurs by the transfer of a methyl group to guanidine nitrogen of arginine catalyzed by the PRMT family of arginine methyltransferases. Type I PRMTs catalyze the formation of asymmetric dimethylarginine (me2a) while Type II PRMTs form symmetric dimethylarginine (me2s). Some of the modifications include the activation mark H3R2me2a by PRMT1, H3R17me2a and H3R26me2a by CARM1/PRMT4, as well as repressive marks H3R2me2a by PRMT6, H4R3me2s and H3R8me2s by PRMT5 [31,32]. H3R3me2a is antagonistic to the activation mark H3K4me3 which is catalyzed by the MLL1 complex. This is a very stable mark which at this point is not clear whether it can be completely enzymatically reversed [32]. Jumonji domain-containing protein (JMJD6) was believed to be a demethylase of asymmetric and symmetric H3R2me2 and H4R3me2 [33]. However, recently it was identified as a lysine hydroxylase without any detectable demethylase activity [34]. The methylarginine residue can also undergo a nonreversible conversion to citrulline through deamination by the peptidylarginine deiminases (PADIs) [35]. PADI4 is a novel transcriptional repressor that targets multiple sites including H3R17 and H4R3 catalyzed by CARM1 and PRMT1 respectively [36]. The activity of PADI4 is associated with the transcriptional regulation of estrogen responsive genes in MCF7 cells by recruitment to ERα promoters resulting in a decrease of ERα mediated gene induction [37]. PADI4 and HDAC work together to create a repressive chromatin state at the TFF1 promoter [35]. Collectively, these studies suggest that ERα interacts with a variety of methylases and demethylases and deregulation of these enzymes may have implications on ERα target gene activation.
3. ERα Regulation of Histone Modifiers and Readers
3.1. ERα regulation of acetylases and deacetylases
ERα transcriptional outcome is shown to be regulated by a dynamic interaction of histone acetyltransferases and histone deacetylases, which are generally associated with coactivators and corepressors, respectively [7]. ERα signaling induces dramatic hyperacetylation at endogenous target genes through HAT activity utilizing p300/CBP to acetylate ERα coregulators [38]. ERα exerts a positive feedback role promoting induction of hCYP19 gene transcription contributing to local estrogen synthesis by promoting increased acetylation in the hCYP promoter [39]. ERα also enhances the recruitment of MTA-1, a component of the histone deacetylase and nucleosome remodeling complex (NuRD), in a ligand and growth factor signaling dependent manner and such recruitment may involve an attenuation of ERα signaling [40]. Although estrogen-ERα signaling has been traditionally implicated in the stimulation of transcription, several recent studies using microarray and ChIP methodology indicated that more than half of the ERα transcriptome is repressed [41-43]. The mechanism by which ERα achieves differential regulation is elusive; however, it is suspected that differential cofactor recruitment and local chromatin modifications including deacetylation may play a role. EZH2 is a binding partner of REA (repressor of estrogen receptor activity) and this interaction is needed for its recruitment to specific target genes and repression of estrogen-dependent transcription. The inhibition of EZH2 by siRNA results in an increase of estrogen-dependent transcription [44]. The activity of PADI4 is associated with the transcriptional regulation of ERα responsive genes by being recruited to ERα promoters and modifying arginine residues on histones causing repression of ERα-mediated gene induction [37]. PADI4 and HDAC work together to create a repressive chromatin state at the TFF1 promoter [35]. HDAC6, an estrogen target gene, expression levels correlate with better prognosis and response to endocrine therapy in breast cancer patients [45]. Class I and II HDACs can reverse p300-mediated acetylation in ERα, thereby inhibiting ERα-dependent gene transcription [46]. SIRT1, a class III HDAC, regulates ERα repression as well as ERα target gene expression [47].
3.2. ERα regulation of kinases and phosphatases
Using estrogen dendrimers and microarray analysis, it was demonstrated that around 25% of estrogen-regulated genes could be activated by ERα-extranuclear signaling pathways emphasizing the importance of these pathways in the activation of ERα target genes [48]. Cell cycle-dependent phosphorylation of histone H3 in both ovarian granulosa and breast cancer cells is driven by estrogen, acting through the oncogenic kinase, Aurora B [49]. Membrane-associated ERα signaling regulates EZH2 via phosphorylation at S21 by constitutively activated AKT resulting in a decrease of H3K27me3 in hormone-responsive cells [50]. Estrogen is implicated in the overexpression of Aurora A/B and the deregulation of Aurora kinase protein substrates is involved in eliciting the alterations observed during oncogenesis [51]. ERα activates ERK2, resulting in colocalization at chromatin binding sites across the genome of breast cancer cells and enables ERK2 modulation of estrogen-dependent gene expression and proliferation programs. This study revealed a novel paradigm with convergence of ERK2 and ERα at the chromatin level that positions this kinase to support nuclear receptor activities in a direct manner [52].
3.3. ERα regulation of methylases and demethylases
Mixed lineage leukemia histone methylases (MLL1-4) are H3K4 methyltransferases [53] and H3K4 trimethylation correlates with ERα transcriptional activation [54]. ERα also recruits MLLs to the HOXC13 promoter and the knockdown of MLLs suppresses the estrogen-induced activation of HOXC13 [55]. Another study showed direct interaction of ERα with MLL2 plays a central role in the growth of ERα positive cells [56]. SMYD3 directly interacts with the ligand binding domain of ERα and is recruited to its target gene promoters TFF1 and GREB1C upon gene induction, catalyzing H3K4me3 [57]. Optimal ERα transcription requires removal of methyl marks such as H3K9 facilitated by demethylase KDM1 and the addition of methyl marks such as H3K4me2 [58]. KDM1 is widely recruited to active promoters in estrogen-stimulated cells and opposes the silencing function of H3K9 methyltransferase SETDB1 [59]. Hormonal stimulation also induce the cyclic recruitment of CARM1 and PRMT1 to the ERα target genes and both are coactivators of ERα [60]. PADI4 is recruited to the TFF1 promoter before the loss of H3R17me2a and deamination of methylarginines at R2, R8, R17 and R26 [35,37]. CARM1 is essential for estrogen-induced cell cycle progression in MCF7 cells and the cell cycle transcriptional regulator E2F1. However, H3R17me2 of E2F1 by CARM1 is dependent on the oncogenic coactivator AIB1 [61]. CARM1 is recruited to the TFF1 promoter during transcriptional activation and deamination by PADI4 inhibits methylation by CARM1.
3.4. ERα regulation of readers of histone modifications
A recent study using a protein domain microarray approach identified the Tudor domain-containing protein (TDRD3) as a reader of histone arginine methyl marks. Importantly, hormonal treatment induces TDRD3 recruitment to ERα target genes, and enhances ERα transcriptional activation. These results suggest that TDRD3 serves as an effector molecule that promotes ERα transcription by binding methylarginine marks on histone tails [62]. Menin, a component of the MLL complex, is a transcriptional coactivator of ERα serving as a link between MLL recruitment and ERα-mediated transcription. Menin is recruited by the activation of TFF1 resulting in increased H3K4 methylation [63]. Recent study results indicate that ERα-coregulator proline glutamic acid leucine rich protein-1 (PELP1) is a novel KDM1-interacting protein. PELP1 functions as a reader of dimethyl histone modifications. Hormonal stimulation enhances PELP1 interactions with KDM1 and PELP1-KDM1 interactions play an essential role in histone methyl modifications at ERα target genes [64]. Additionally, a recent study identified PELP1 as a component of the MLL1 methyltransferase complex [65].
4. Deregulation of Histone Modifying Enzymes in Hormonal Cancers
The human ERα is implicated in hormonal cancer initiation and progression [2,8,66]. Current endocrine therapy for ERα-positive hormonal cancer involves modulating the ERα-pathway using either antiestrogens (AEs) or aromatase inhibitors (AIs). Despite the positive effects, de novo and/or acquired resistance to endocrine therapies frequently occurs. While mechanisms for hormonal therapy resistance remain elusive, emerging data suggest that resistance can be caused by acquisition of epigenetic modifications on ERα and its target gene promoters [67-69]. Some evidence also implicates activation of MAPK- and AKT-signaling pathways in activating ERα and its downstream pathways in the absence of estrogen and thus these pathways represent new targets for drug therapy [70,71]. Estrogen stimulation promotes histone modifications at ERα target gene promoters [72]. SMYD3, the H3K4me methyltransferase, is overexpressed in breast cancer causing dysregulation of the WNT signaling pathway [73]. The MLL complex also plays a role in breast cancer. The oncogenic transformation caused by this complex can be blocked by knockdown of the MLL component PRMT1 [74].
EZH2 overexpression in breast cancer is associated with larger tumors, higher histological grade and reduced overall patient survival [50,75]. High expression of EZH2 is sufficient to induce oncogenic potential in a xenograft model [76]. There is a strong correlation between EZH2 expression levels and increased tumor cell proliferation [77]. EZH2 is essential for promoting chemotherapy resistance in cancer cells in vitro and in vivo, and EZH2 could be a potential novel epigenetic target to overcome drug resistance [78]. Also, the frequent overexpression of EZH2 in human epithelial ovarian cancer cells promotes cellular proliferation and invasive ability, further supporting its possibility as a novel therapeutic target [79]. CARM1 is an essential part of the estrogen-stimulated breast cancer growth downstream of ERα and its aberrant expression as well as PRMT1 overexpression is linked to breast cancer [61,80]. PADI4 is overexpressed in malignant tumors but not in benign tumors [37]. ERα is critical for demethylase JMJD2B induction in hypoxia which is critical for breast cancer cell survival, is highly expressed in ERα-positive primary breast cancers and is an adverse prognostic factor in hypoxic breast cancers [81].
Deregulation of AIB1, an ERα coregulator with intrinsic acetylase activity, was reported in breast tumors [82,83]. Elevated amounts of coregulators SRC-2 and CBP have been reported in intraductal carcinomas compared to normal mammary tissue [84]. ERα coregulatory proteins such as SRC1 with histone modifying activity have also been suggested to play a role in the generally observed tissue-specific effects of tamoxifen [85]. AIB1 knockout mice studies demonstrated that normal expression of coactivator AIB1 is required for the initiation of tumorigenesis by carcinogens and oncogenes [86,87]. Overexpression of AIB1 in the mouse mammary gland promotes tumorigenesis suggesting that ERα coregulators have oncogenic potential [88]. Similarly, in mammary epithelium dysregulation of another ERα coregulator MTA1 that associates with deacetylase complexes caused increased cell proliferation, hyper-branched ductal structure formation and precocious development. It also resulted in the development of hyperplastic nodules and mammary gland tumors in virgin mice [89]. PELP1, an ERα coregulator and reader of epigenetic modifications plays an important role in ERα signaling [90]. PELP1 is a recently discovered proto-oncogene [91] that exhibits aberrant expression in many hormonal cancers [90] and is a prognostic indicator of decreased survival in breast cancer and disease-free intervals when over-expressed [92].
5. Therapeutic Potential of Targeting Histone Modifying Enzymes for Hormonal Driven Cancers
Epigenetic modifications are reversible, and drugs targeting epigenetic modifiers which are often overexpressed in hormonal cancers, can be potentially used for therapeutic targeting. Inhibition of SIRT1 deacetylase activity by either pharmacological inhibitors or genetic depletion impairs ERα-mediated signaling pathways [47]. Research performed over the last decade has highlighted the role of HDAC inhibitors as modulators of transcriptional activity. A new class of therapeutic agents in ERα-driven as well as ERα-therapy resistant cells [93] has led to the initiation of several clinical trials combining HDAC inhibitors with hormonal therapy [94,95]. Clinical trials show that HDAC inhibitors have varying antitumor activity, the FDA-approved HDAC inhibitors (SAHA/vorinostat and depsipeptide) had significant clinical benefits [96]. SAHA promotes ERα degradation by a proteasome-mediated mechanism implicating SAHA as a suitable pharmacological agent for the depletion of ERα in breast tumors [97]. Currently SAHA is given in combination with tamoxifen for patients with advanced breast cancer for whom anti-hormonal therapy has been ineffective. Recent studies also showed that HDAC inhibitors (LBH589/panobinostat) function as potent inhibitors of aromatase expression. Results from this study demonstrated synergistic interaction between LBH589 and letrozole to suppress proliferation of hormone-responsive breast cancer cells [98]. Entinostat (ENT), another HDAC inhibitor is shown to trigger re-expression of ERα and aromatase in breast cancer cells. Preclinical studies showed that ENT treatment restores letrozole responsiveness of ERα-negative tumors providing a strong rationale for clinical evaluation of combinatorial therapy of ENT and letrozole to treat ERα-negative and endocrine-resistant breast cancers [99].
Recent studies also showed combination therapy involving HDAC inhibitors with DNA methyltransferase-1 (DNMT1) inhibition is synergistically effective in inducing apoptosis, differentiation and/or cell growth arrest in many cancers including breast cancer [100]. DNMT1 inhibitor 5-aza-2′-deoxycytidine (AZA) and HDACi trichostatin A (TSA) induces ERα expression in ERα-negative breast cancer cells. Preclinical studies indicate AZA and TSA could restore sensitivity of the ERα-negative breast cancer cells to endocrine therapy in vitro and in vivo [101]. Zebularine, another DNMT inhibitor, inhibits the growth of breast cancer cells. Combination therapy with HDAC inhibitors (decitabine or vorinostat) significantly inhibited cellular proliferation and colony formation in breast cancer cells compared to either drug alone [102].
Preclinical studies also suggest that drugs targeting histone methylation would have therapeutic benefits in treating hormonal cancers. Dysregulation of arginine methylation or enzymes responsible for these modifications could be key events in estrogen-dependent cancers and thus these enzymes represent novel therapeutic targets [4]. Small molecule regulator of protein arginine methyltransferases (AMI-1) is shown to modulate nuclear receptor-regulated transcription from estrogen and androgen response elements, thus operating as a brake on certain hormone actions [103]. Recent studies demonstrated that F- and Cl-amidine, two potent PADI4 inhibitors, display micromolar cytotoxic effects towards several cancerous cell lines (HL-60, MCF7 and HT-29) with no effect on noncancerous lines implicating PADI4 inhibition as a novel epigenetic approach for the treatment of hormonal cancer [37]. A recent study showed that targeting EZH2 via siRNA could be used to reduce angiogenesis and ovarian cancer growth proving the feasibility for using EZH2 as an important therapeutic target [104]. Histone methyltransferases such as G9a are required to perpetuate the malignant phenotype [105] and G9a inhibitor BIX-01294 (diazepin-quinazolin-amine derivative) may have therapeutic utility in breast cancer cells overexpressing the methyltransferases [106,107].
Histone methylation is shown to play a key role in ERα-mediated transactivation of target genes. Recent studies showed the histone demethylase KDM1 and ERα coregulator PELP1 play a role in regulating histone methyl marks at ERα target genes [64]. PELP1 deregulation alters histone methylation at ERα target genes, contributing to hormone-driven tumor progression [64] and therapy resistance [108]. In a recent study, we examined the therapeutic efficacy of treating breast tumor cells with Pargyline, an FDA approved drug to block KDM1 functions. The results from this study suggested that histone methyl modifications play a role in therapy resistance and targeting the KDM1 axis with Pargyline in combination with current endocrine therapies will improve therapeutic efficacy [109]. Polycomb-repressive complex 2 (PRC2)-mediated histone methylation plays an important role in aberrant cancer gene silencing. Preclinical studies using S-adenosyl homocysteine hydrolase inhibitor 3-Deazaneplanocin A (DZNep) induces efficient apoptotic cell death in cancer cells but not in normal cells. Mechanistic studies showed that DZNep effectively depletes cellular levels of PRC2 components EZH2, SUZ12 and EED and inhibits histone H3K27 methylation suggesting a unique feature of DZNep as a novel chromatin remodeling compound that could be used as a novel cancer therapeutic [110].
6. Conclusions and Future Directions
The emerging evidence strongly implicates the importance of ERα-mediated epigenetic modifications to ERα. Histone tail modifications play a significant role in ERα–mediated physiological functions as well as in cancer progression (Figure 1). In this review, we focused on summarizing recent findings that relate to ERα-mediated histone tail modifications, enzymes that mediate ERα-mediated epigenetic modifications on histone tails, deregulation of these enzymes in hormonal cancers and its implications in ERα signaling. Because of the evolving evidence connecting histone modifications and hormonal cancer progression/therapy resistance, the epigenetic enzymes that play a role in ERα signaling represent promising targets for hormonal cancer treatment. Further, combinatorial treatment of HDACi and/or histone methylase inhibitors along with currently used hormonal therapy regimen(s) may be a useful tool in treating therapy resistant cancers. However, future improvements in therapeutic targeting of ERα epigenetic modifications will depend upon a better understanding of: (1) molecular basis by which epigenetic modifications play a role in cancer progression; (2) epigenetic code during progression and therapy resistance using the emerging Chip-Seq methodologies; (3) prognostic significance of the epigenetic marks in hormonal cancer progression and (4) development of specific molecular inhibitors targeting epigenetic modifiers.
Acknowledgments
This work was supported by NIH-CA095681 and Komen-KG090447 grants.
References
- Hall, J.M.; McDonnell, D.P. Coregulators in nuclear estrogen receptor action: From concept to therapeutic targeting. Mol. Interv. 2005, 5, 343–357. [Google Scholar]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; Gustafsson, J.A. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar]
- Teyssier, C.; Le, R.M.; Sentis, S.; Jalaguier, S.; Corbo, L.; Cavailles, V. Protein arginine methylation in estrogen signaling and estrogen-related cancers. Trends Endocrinol. MeTable 2010, 21, 181–189. [Google Scholar]
- Tsai, M.J.; O'Malley, B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar]
- Barnes, C.J.; Vadlamudi, R.K.; Kumar, R. Novel estrogen receptor coregulators and signaling molecules in human diseases. Cell Mol. Life Sci. 2004, 61, 281–291. [Google Scholar]
- Collingwood, T.N.; Urnov, F.D.; Wolffe, A.P. Nuclear receptors: Coactivators, corepressors and chromatin remodeling in the control of transcription. J. Mol. Endocrinol. 1999, 23, 255–275. [Google Scholar]
- McDonnell, D.P.; Norris, J.D. Connections and regulation of the human estrogen receptor. Science 2002, 296, 1642–1644. [Google Scholar]
- McKenna, N.J.; Lanz, R.B.; O'Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, part I: Covalent histone modifications. Trends Mol. Med. 2007, 13, 363–372. [Google Scholar]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar]
- Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 1997, 9, 222–232. [Google Scholar]
- Grant, P.A. A tale of histone modifications. Genome Biol. 2001, 2. REVIEWS0003. [Google Scholar]
- Tsai, M.J.; O'Malley, B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar]
- Silverstein, R.A.; Ekwall, K. Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 2005, 47, 1–17. [Google Scholar]
- Kumar, R.; Gururaj, A.E.; Vadlamudi, R.K.; Rayala, S.K. The clinical relevance of steroid hormone receptor corepressors. Clin. Cancer Res. 2005, 11, 2822–2831. [Google Scholar]
- Palijan, A.; Fernandes, I.; Bastien, Y.; Tang, L.; Verway, M.; Kourelis, M.; Tavera-Mendoza, L.E.; Li, Z.; Bourdeau, V.; Mader, S.; Yang, X.J.; White, J.H. Function of histone deacetylase 6 as a cofactor of nuclear receptor coregulator LCoR. J. Biol. Chem. 2009, 284, 30264–30274. [Google Scholar]
- Malik, S.; Jiang, S.; Garee, J.P.; Verdin, E.; Lee, A.V.; O'Malley, B.W.; Zhang, M.; Belaguli, N.S.; Oesterreich, S. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol. Cell Biol. 2010, 30, 399–412. [Google Scholar]
- Losel, R.; Wehling, M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003, 4, 46–56. [Google Scholar]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar]
- Chakravarty, D.; Nair, S.S.; Santhamma, B.; Nair, B.; Wang, L.; Bandyopadhyay, A.; Agyin, J.; Brann, D.; Sun, L.; Yeh, I.; Lee, F.; Tekmal, R.; Kumar, R.; Vadlamudi, R.K. Extranuclear functions of ER impact invasive migration and metastases of breast cancer cells. Cancer Res. 2010, 70, 4092. [Google Scholar]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar]
- Espino, P.S.; Li, L.; He, S.; Yu, J.; Davie, J.R. Chromatin modification of the trefoil factor 1 gene in human breast cancer cells by the Ras/mitogen-activated protein kinase pathway. Cancer Res. 2006, 66, 4610–4616. [Google Scholar]
- Vicent, G.P.; Nacht, A.S.; Zaurin, R.; Ballare, C.; Clausell, J.; Beato, M. Minireview: Role of kinases and chromatin remodeling in progesterone signaling to chromatin. Mol. Endocrinol. 2010, 24, 2088–2098. [Google Scholar]
- Li, C.; Liang, Y.Y.; Feng, X.H.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol. Cell 2008, 31, 835–849. [Google Scholar]
- Yi, K.D.; Simpkins, J.W. Protein phosphatase 1, protein phosphatase 2A, and calcineurin play a role in estrogen-mediated neuroprotection. Endocrinology. 2008, 149, 5235–5243. [Google Scholar]
- Garcia-Bassets, I.; Kwon, Y.S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; Rose, D.W.; Glass, C.K.; Fu, X.D.; Rosenfeld, M.G. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007, 128, 505–518. [Google Scholar]
- Litt, M.D.; Simpson, M.; Gaszner, M.; Allis, C.D.; Felsenfeld, G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 2001, 293, 2453–2455. [Google Scholar]
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155. [Google Scholar]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar]
- Tang, J.; Gary, J.D.; Clarke, S.; Herschman, H.R. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 1998, 273, 16935–16945. [Google Scholar]
- Di, L.A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2010. [Google Scholar] [CrossRef]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R., III; Delahunty, C.M.; Hahn, P.; Lengeling, A.; Mann, M.; Proudfoot, N.J.; Schofield, C.J.; Bottger, A. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93. [Google Scholar]
- Denis, H.; Deplus, R.; Putmans, P.; Yamada, M.; Metivier, R.; Fuks, F. Functional connection between deimination and deacetylation of histones. Mol. Cell Biol. 2009, 29, 4982–4993. [Google Scholar]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; Roeder, R.G.; Clarke, S.; Stallcup, M.R.; Allis, C.D.; Coonrod, S.A. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar]
- Slack, J.L.; Causey, C.P.; Thompson, P.R. Protein arginine deiminase 4: A target for an epigenetic cancer therapy. Cell Mol. Life Sci. 2010, 68, 709–720. [Google Scholar]
- Chen, H.; Lin, R.J.; Xie, W.; Wilpitz, D.; Evans, R.M. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999, 98, 675–686. [Google Scholar]
- Kumar, P.; Kamat, A.; Mendelson, C.R. Estrogen Receptor α(ERα) Mediates Stimulatory Effects of Estrogen on Aromatase (CYP19) Gene Expression in Human Placenta. Mol. Endocrinol. 2009, 23, 784–793. [Google Scholar]
- Mazumdar, A.; Wang, R.A.; Mishra, S.K.; Adam, L.; Bagheri-Yarmand, R.; Mandal, M.; Vadlamudi, R.K.; Kumar, R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat. Cell Biol. 2001, 3, 30–37. [Google Scholar]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar]
- Levenson, A.S.; Svoboda, K.M.; Pease, K.M.; Kaiser, S.A.; Chen, B.; Simons, L.A.; Jovanovic, B.D.; Dyck, P.A.; Jordan, V.C. Gene expression profiles with activation of the estrogen receptor alpha-selective estrogen receptor modulator complex in breast cancer cells expressing wild-type estrogen receptor. Cancer Res. 2002, 62, 4419–4426. [Google Scholar]
- Monroe, D.G.; Getz, B.J.; Johnsen, S.A.; Riggs, B.L.; Khosla, S.; Spelsberg, T.C. Estrogen receptor isoform-specific regulation of endogenous gene expression in human osteoblastic cell lines expressing either ERalpha or ERbeta. J. Cell Biochem. 2003, 90, 315–326. [Google Scholar]
- Hwang, C.; Giri, V.N.; Wilkinson, J.C.; Wright, C.W.; Wilkinson, A.S.; Cooney, K.A.; Duckett, C.S. EZH2 regulates the transcription of estrogen-responsive genes through association with REA, an estrogen receptor corepressor. Breast Cancer Res. Treat. 2008, 107, 235–242. [Google Scholar]
- Saji, S.; Kawakami, M.; Hayashi, S.; Yoshida, N.; Hirose, M.; Horiguchi, S.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; Toi, M. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.; Homenko, D.R.; Kraus, W.L. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 2006, 20, 1479–1493. [Google Scholar]
- Yao, Y.; Li, H.; Gu, Y.; Davidson, N.E.; Zhou, Q. Inhibition of SIRT1 deacetylase suppresses estrogen receptor signaling. Carcinogenesis 2010, 31, 382–387. [Google Scholar]
- Madak-Erdogan, Z.; Kieser, K.J.; Kim, S.H.; Komm, B.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol. Endocrinol. 2008, 22, 2116–2127. [Google Scholar]
- Ruiz-Cortes, Z.T.; Kimmins, S.; Monaco, L.; Burns, K.H.; Sassone-Corsi, P.; Murphy, B.D. Estrogen mediates phosphorylation of histone H3 in ovarian follicle and mammary epithelial tumor cells via the mitotic kinase, Aurora B. Mol. Endocrinol. 2005, 19, 2991–3000. [Google Scholar]
- Bredfeldt, T.G.; Greathouse, K.L.; Safe, S.H.; Hung, M.C.; Bedford, M.T.; Walker, C.L. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol. Endocrinol. 2010, 24, 993–1006. [Google Scholar]
- Hontz, A.E.; Li, S.A.; Salisbury, J.L.; Lingle, W.L.; Li, J.J. Expression of selected Aurora A kinase substrates in solely estrogen-induced ectopic uterine stem cell tumors in the Syrian hamster kidney. Adv. Exp. Med. Biol. 2008, 617, 411–418. [Google Scholar]
- Madak-Erdogan, Z.; Lupien, M.; Stossi, F.; Brown, M.; Katzenellenbogen, B.S. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell Biol. 2011, 31, 226–236. [Google Scholar]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar]
- Dreijerink, K.M.; Mulder, K.W.; Winkler, G.S.; Hoppener, J.W.; Lips, C.J.; Timmers, H.T. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar]
- Ansari, K.I.; Kasiri, S.; Hussain, I.; Mandal, S.S. Mixed lineage leukemia histone methylases play critical roles in estrogen-mediated regulation of HOXC13. FEBS J. 2009, 276, 7400–7411. [Google Scholar]
- Mo, R.; Rao, S.M.; Zhu, Y.J. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J. Biol. Chem. 2006, 281, 15714–15720. [Google Scholar]
- Kim, H.; Heo, K.; Kim, J.H.; Kim, K.; Choi, J.; An, W. Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J. Biol. Chem. 2009, 284, 19867–19877. [Google Scholar]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar]
- Garcia-Bassets, I.; Kwon, Y.S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; Rose, D.W.; Glass, C.K.; Fu, X.D.; Rosenfeld, M.G. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007, 128, 505–518. [Google Scholar]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Brand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003, 115, 751–763. [Google Scholar]
- Frietze, S.; Lupien, M.; Silver, P.A.; Brown, M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res. 2008, 68, 301–306. [Google Scholar]
- Yang, Y.; Lu, Y.; Espejo, A.; Wu, J.; Xu, W.; Liang, S.; Bedford, M.T. TDRD3 Is an Effector Molecule for Arginine-Methylated Histone Marks. Mol. Cell 2010, 40, 1016–1023. [Google Scholar]
- Dreijerink, K.M.; Mulder, K.W.; Winkler, G.S.; Hoppener, J.W.; Lips, C.J.; Timmers, H.T. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar]
- Nair, S.S.; Nair, B.C.; Cortez, V.; Chakravarty, D.; Metzger, E.; Schule, R.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor-alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep. 2010, 11, 438–444. [Google Scholar]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; Gustafsson, J.A. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar]
- Kurebayashi, J. Endocrine-resistant breast cancer: Underlying mechanisms and strategies for overcoming resistance. Breast Cancer 2003, 10, 112–119. [Google Scholar]
- Pathiraja, T.N.; Stearns, V.; Oesterreich, S. Epigenetic regulation in estrogen receptor positive breast cancer--role in treatment response. J. Mammary. Gland. Biol. Neoplasia. 2010, 15, 35–47. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Gururaj, A.E.; Rayala, S.K.; Vadlamudi, R.K.; Kumar, R. Novel mechanisms of resistance to endocrine therapy: Genomic and nongenomic considerations. Clin. Cancer Res. 2006, 12, 1001s–1007s. [Google Scholar]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Arpino, G.; Rimawi, M.; Osborne, C.K. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: Implicated role of growth factor signaling and estrogen receptor coregulators. Cancer Chemother. Pharmacol. 2005, 56 Suppl. 1, 10–20. [Google Scholar]
- Kovalchuk, O.; Tryndyak, V.P.; Montgomery, B.; Boyko, A.; Kutanzi, K.; Zemp, F.; Warbritton, A.R.; Latendresse, J.R.; Kovalchuk, I.; Beland, F.A.; Pogribny, I.P. Estrogen-induced rat breast carcinogenesis is characterized by alterations in DNA methylation, histone modifications and aberrant microRNA expression. Cell Cycle 2007, 6, 2010–2018. [Google Scholar]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar]
- Cheung, N.; Chan, L.C.; Thompson, A.; Cleary, M.L.; So, C.W. Protein arginine-methyltransferase-dependent oncogenesis. Nat. Cell Biol. 2007, 9, 1208–1215. [Google Scholar]
- Reijm, E.A.; Jansen, M.P.; Ruigrok-Ritstier, K.; van Staveren, I.L.; Look, M.P.; van Gelder, M.E.; Sieuwerts, A.M.; Sleijfer, S.; Foekens, J.A.; Berns, E.M. Decreased expression of EZH2 is associated with upregulation of ER and favorable outcome to tamoxifen in advanced breast cancer. Breast Cancer Res. Treat. 2011, 125, 387–394. [Google Scholar]
- Croonquist, P.A.; Van, N.B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar]
- Collett, K.; Eide, G.E.; Arnes, J.; Stefansson, I.M.; Eide, J.; Braaten, A.; Aas, T.; Otte, A.P.; Akslen, L.A. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin. Cancer Res. 2006, 12, 1168–1174. [Google Scholar]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 2010, 8, 1610–1618. [Google Scholar]
- El, M.S.; Fabbrizio, E.; Rodriguez, C.; Chuchana, P.; Fauquier, L.; Cheng, D.; Theillet, C.; Vandel, L.; Bedford, M.T.; Sardet, C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc. Natl. Acad. Sci. USA 2006, 103, 13351–13356. [Google Scholar]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar]
- List, H.J.; Reiter, R.; Singh, B.; Wellstein, A.; Riegel, A.T. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res. Treat. 2001, 68, 21–28. [Google Scholar]
- Azorsa, D.O.; Cunliffe, H.E.; Meltzer, P.S. Association of steroid receptor coactivator AIB1 with estrogen receptor-alpha in breast cancer cells. Breast Cancer Res. Treat. 2001, 70, 89–101. [Google Scholar]
- Kurebayashi, J.; Otsuki, T.; Kunisue, H.; Tanaka, K.; Yamamoto, S.; Sonoo, H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin. Cancer Res. 2000, 6, 512–518. [Google Scholar]
- Smith, C.L.; O'Malley, B.W. Coregulator function: A key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004, 25, 45–71. [Google Scholar]
- Kuang, S.Q.; Liao, L.; Wang, S.; Medina, D.; O'Malley, B.W.; Xu, J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005, 65, 7993–8002. [Google Scholar]
- Kuang, S.Q.; Liao, L.; Zhang, H.; Lee, A.V.; O'Malley, B.W.; Xu, J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004, 64, 1875–1885. [Google Scholar]
- Torres-Arzayus, M.I.; Font de, M.J.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004, 6, 263–274. [Google Scholar]
- Bagheri-Yarmand, R.; Talukder, A.H.; Wang, R.A.; Vadlamudi, R.K.; Kumar, R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development 2004, 131, 3469–3479. [Google Scholar]
- Vadlamudi, R.K.; Kumar, R. Functional and biological properties of the nuclear receptor coregulator PELP1/MNAR. Nucl. Recept. Signal 2007, 5, e004. [Google Scholar]
- Rajhans, R.; Nair, S.; Holden, A.H.; Kumar, R.; Tekmal, R.R.; Vadlamudi, R.K. Oncogenic Potential of the Nuclear Receptor Coregulator Proline-, Glutamic Acid-, Leucine-Rich Protein 1/Modulator of the Nongenomic Actions of the Estrogen Receptor. Cancer Res. 2007, 67, 5505–5512. [Google Scholar]
- Habashy, H.O.; Powe, D.G.; Rakha, E.A.; Ball, G.; Macmillan, R.D.; Green, A.R.; Ellis, I.O. The prognostic significance of PELP1 expression in invasive breast cancer with emphasis on the ER-positive luminal-like subtype. Breast Cancer Res. Treat. 2009, 120, 603–12. [Google Scholar]
- Margueron, R.; Duong, V.; Castet, A.; Cavailles, V. Histone deacetylase inhibition and estrogen signalling in human breast cancer cells. Biochem. Pharmacol. 2004, 68, 1239–1246. [Google Scholar]
- Sigalotti, L.; Fratta, E.; Coral, S.; Cortini, E.; Covre, A.; Nicolay, H.J.; Anzalone, L.; Pezzani, L.; Di Giacomo, A.M.; Fonsatti, E.; Colizzi, F.; Altomonte, M.; Calabro, L.; Maio, M. Epigenetic drugs as pleiotropic agents in cancer treatment: Biomolecular aspects and clinical applications. J. Cell Physiol. 2007, 212, 330–344. [Google Scholar]
- Thomas, S.; Munster, P.N. Histone deacetylase inhibitor induced modulation of anti-estrogen therapy. Cancer Lett. 2009, 280, 184–191. [Google Scholar]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar]
- Yi, X.; Wei, W.; Wang, S.Y.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Histone deacetylase inhibitor SAHA induces ERalpha degradation in breast cancer MCF-7 cells by CHIP-mediated ubiquitin pathway and inhibits survival signaling. Biochem. Pharmacol. 2008, 75, 1697–1705. [Google Scholar]
- Chen, S.; Ye, J.; Kijima, I.; Evans, D. The HDAC inhibitor LBH589 (panobinostat) is an inhibitory modulator of aromatase gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 11032–11037. [Google Scholar]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar]
- Zhu, W.G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer Agents 2003, 3, 187–199. [Google Scholar]
- Fan, J.; Yin, W.J.; Lu, J.S.; Wang, L.; Wu, J.; Wu, F.Y.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol. 2008, 134, 883–890. [Google Scholar]
- Billam, M.; Sobolewski, M.D.; Davidson, N.E. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res. Treat. 2010, 120, 581–592. [Google Scholar]
- Cheng, D.; Yadav, N.; King, R.W.; Swanson, M.S.; Weinstein, E.J.; Bedford, M.T. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. [Google Scholar]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; Ravoori, M.K.; Shahzad, M.M.; Lee, J.W.; Mora, E.; Langley, R.R.; Carroll, A.R.; Matsuo, K.; Spannuth, W.A.; Schmandt, R.; Jennings, N.B.; Goodman, B.W.; Jaffe, R.B.; Nick, A.M.; Kim, H.S.; Guven, E.O.; Chen, Y.H.; Li, L.Y.; Hsu, M.C.; Coleman, R.L.; Calin, G.A.; Denkbas, E.B.; Lim, J.Y.; Lee, J.S.; Kundra, V.; Birrer, M.J.; Hung, M.C.; Lopez-Berestein, G.; Sood, A.K. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar]
- Kondo, Y.; Shen, L.; Ahmed, S.; Boumber, Y.; Sekido, Y.; Haddad, B.R.; Issa, J.P. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One 2008, 3, e2037. [Google Scholar]
- Kubicek, S.; O'Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; Kelly, T.A.; Jenuwein, T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. [Google Scholar]
- Vallabhaneni, S.; Nair, B.C.; Cortez, V.; Challa, R.; Chakravarty, D.; Tekmal, R.R.; Vadlamudi, R.K. Significance of ER-Src axis in hormonal therapy resistance. Breast Cancer Res. Treat. 2010. [Google Scholar] [PubMed]
- Cortez, V.; Nair, S.S.; Nair, B.; Tekmal, R.; Vadlamudi, R.K. Potential Role of Pargyline in Overcoming Adaptive Resistance in Breast Cancer Cells. Cancer Res. 2011, 69, 409. [Google Scholar]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy. Cancers 2011, 3, 1691-1707. https://doi.org/10.3390/cancers3021691
Mann M, Cortez V, Vadlamudi RK. Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy. Cancers. 2011; 3(2):1691-1707. https://doi.org/10.3390/cancers3021691
Chicago/Turabian StyleMann, Monica, Valerie Cortez, and Ratna K. Vadlamudi. 2011. "Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy" Cancers 3, no. 2: 1691-1707. https://doi.org/10.3390/cancers3021691
APA StyleMann, M., Cortez, V., & Vadlamudi, R. K. (2011). Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy. Cancers, 3(2), 1691-1707. https://doi.org/10.3390/cancers3021691