Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data

Abstract
:1. Introduction
2. Results and Discussion
2.1. NMR Metabolic Profiles

2.2. Influence of Hypoxia on Metabolic Profiles of MDA-MB-231 Cancer Cells
2.2.1. Temporal Changes in the Metabolic Pattern Induced by Hypoxic Treatment in MDA-MB-231 Breast Cancer Cells


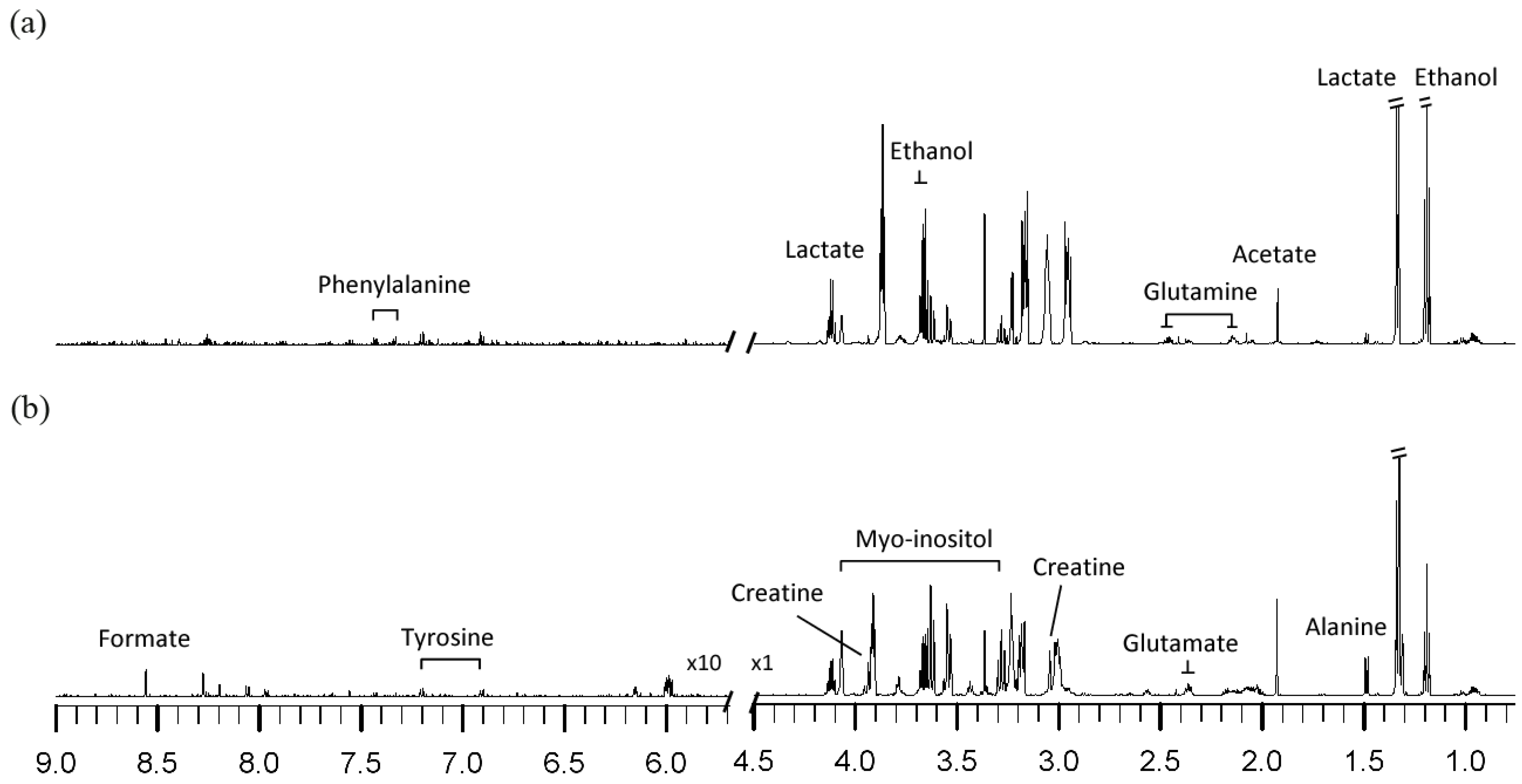{kind=link}
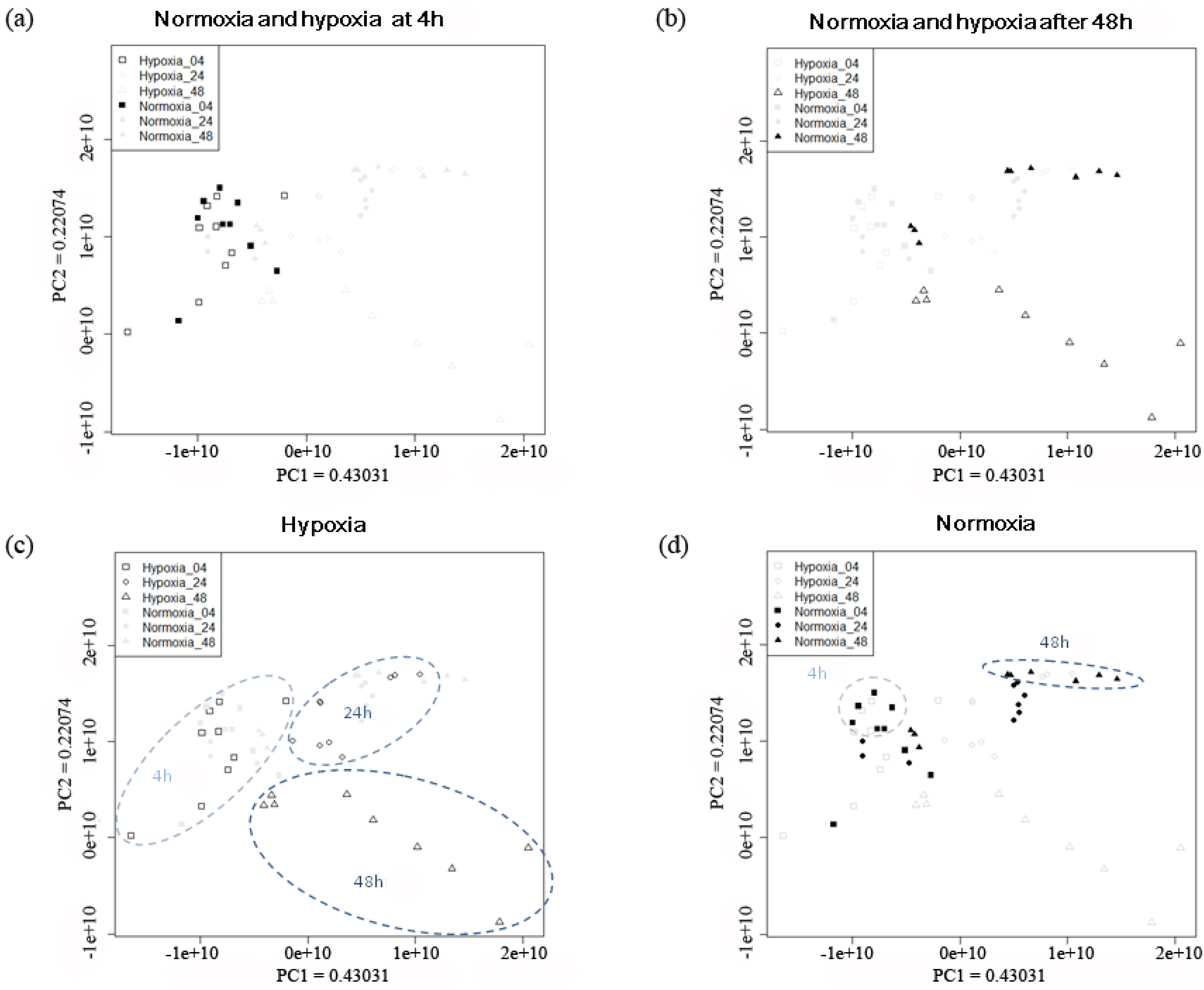{kind=link}
{kind=link}
{kind=link}
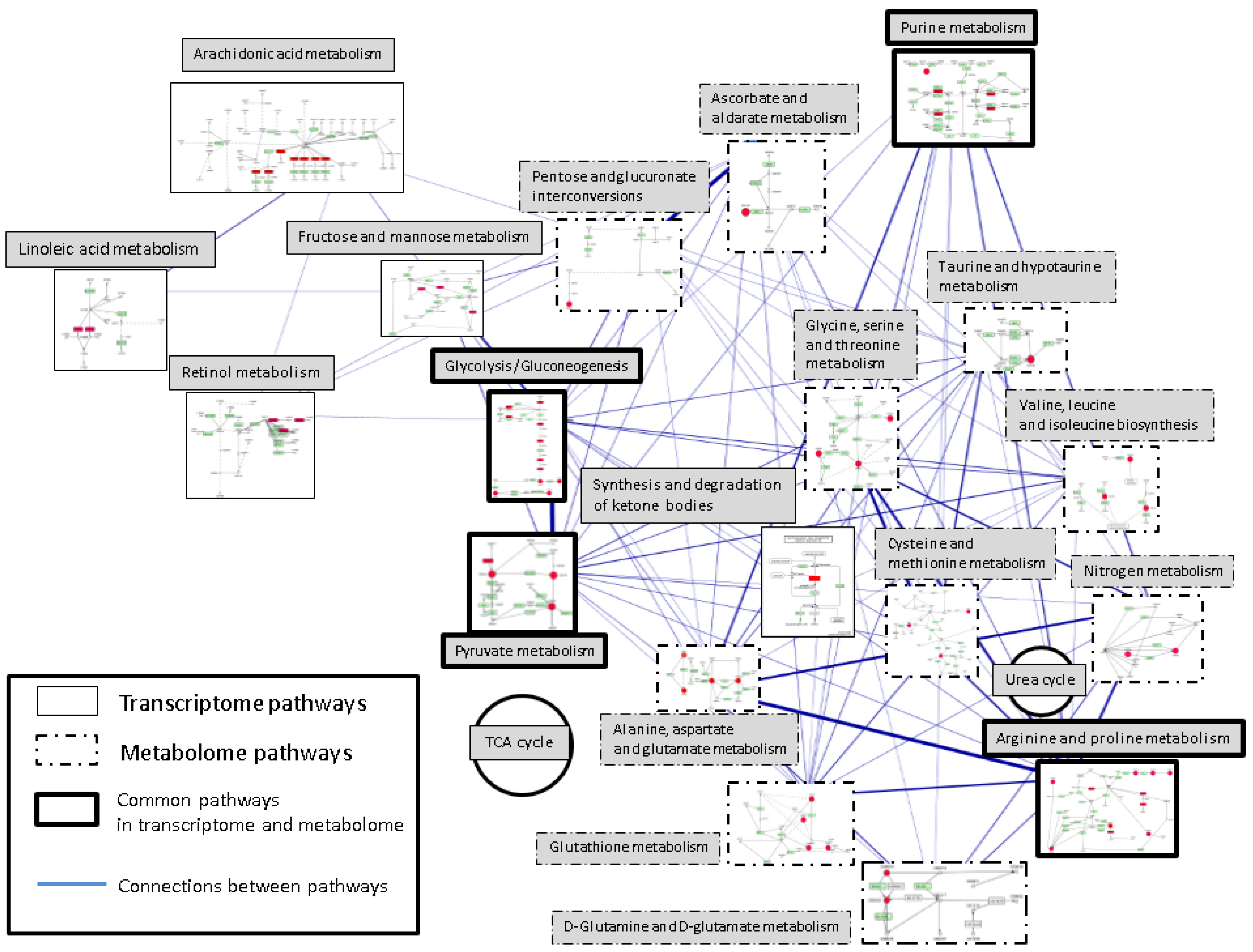{kind=link}
| Training data (LOOCV b) | ||||
|---|---|---|---|---|
| SVM Model a | BAC c | Accuracy d | Specificity e | Sensitivity f |
| 4 h | 50.00% | 33.33% | 33.33% | 33.33% |
| 24 h | 72.86% | 66.67% | 60% | 71.43% |
| 48 h | 83.33% | 83.33% | 83.33% | 83.33% |
| Testing data | ||||
| 4 h | 66.67% | 66.67% | 66.67% | 66.67% |
| 24 h | 66.67% | 66.67% | 33.33% | 100% |
| 48 h | 100% | 100% | 100% | 100% |
2.2.2. Mechanism Discussion of Metabolic Changes

2.3. Inference of Metabolic Pathway Network from Transcriptome and Metabolome
| Pathway | Metabolome | Transcriptome |
|---|---|---|
| Glycolysis/Gluconeogenesis | V | V |
| Purine metabolism | V | V |
| Arginine and proline metabolism | V | V |
| Pyruvate metabolism | V | V |
| Pentose and glucuronate interconversions | V | |
| Ascorbate and aldarate metabolism | V | |
| Alanine, aspartate and glutamate metabolism | V | |
| Glycine, serine and threonine metabolism | V | |
| Cysteine and methionine metabolism | V | |
| Valine, leucine and isoleucine biosynthesis | V | |
| Taurine and hypotaurine metabolism | V | |
| D-Glutamine and D-glutamate metabolism | V | |
| Glutathione metabolism | V | |
| Nitrogen metabolism | V | |
| Fructose and mannose metabolism | V | |
| Synthesis and degradation of ketone bodies | V | |
| Arachidonic acid metabolism | V | |
| Linoleic acid metabolism | V | |
| Retinol metabolism | V | |
| Metabolism of xenobiotics by cytochrome P450 | V | |
| Drug metabolism—cytochrome P450 | V |


2.3.1. Pathways in Common between Metabolome and Transcriptome
2.3.2. Pathways Difference between Metabolic Profiling and Transcriptome
3. Materials and Methods
3.1. Cell Culture and Hypoxia Treatment
3.2. Extraction of Intra-Cellular Metabolite
3.3. Sample Preparation for 1H-NMR Spectroscopy
3.4. Metabolome Analysis
3.4.1. NMR Analysis
3.4.2. Metabolite Identification
3.5. Data Analysis
3.5.1. Statistical Analysis
3.5.2. Classification Evaluation
| 4 h | 24 h | 48 h | |
|---|---|---|---|
| Hypoxia | 9 | 9 | 9 |
| Normoxia | 9 | 9 | 9 |
3.5.3. Construction of Pathway Network from Transcriptome and Metabolome
4. Conclusions
Acknowledgments
References
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2009, 29, 625–634. [Google Scholar] [CrossRef]
- Zhong, H.; de Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1 alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Erb, G.; Elbayed, K.; Piotto, M.; Raya, J.; Neuville, A.; Mohr, M.; Maitrot, D.; Kehrli, P.; Namer, I.J. Toward improved grading of malignancy in oligodendrogliomas using metabolomics. Magn. Reson. Med. 2008, 59, 959–965. [Google Scholar] [CrossRef]
- Chan, E.C.Y.; Koh, P.K.; Mal, M.; Cheah, P.Y.; Eu, K.W.; Backshall, A.; Cavill, R.; Nicholson, J.K.; Keun, H.C. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res. 2009, 8, 352–361. [Google Scholar] [CrossRef]
- Lundgren, K.; Holm, C.; Landberg, G. Hypoxia and breast cancer: Prognostic and therapeutic implications. Cell. Mol. Life Sci. 2007, 64, 3233–3247. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef]
- Brizel, D.M.; Scully, S.P.; Harrelson, J.M.; Layfield, L.J.; Bean, J.M.; Prosnitz, L.R.; Dewhirst, M.W. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996, 56, 941–943. [Google Scholar]
- Raghunand, N.; Gatenby, R.A.; Gillies, R.J. Microenvironmental and cellular consequences of altered blood flow in tumours. Br. J. Radiol. 2003, 76, S11–S22. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism a key to apoptosis resistance? Cell Cycle 2007, 6, 790–792. [Google Scholar] [CrossRef]
- Weidner, N.; Semple, N.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis-correlation in invasive breast-carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef]
- Brahimi-Horn, C.; Berra, E.; Pouyssegur, J. Hypoxia: The tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol. 2001, 11, S32–S36. [Google Scholar]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1 alpha and HIF2 alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Nicholson, J.K. Global systems biology, personalized medicine and molecular epidemiology. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef]
- Lenz, E.M.; Bright, J.; Knight, R.; Westwood, F.R.; Davies, D.; Major, H.; Wilson, I.D. Metabonomics with H-1-NMR spectroscopy and liquid chromatography-mass spectrometry applied to the investigation of metabolic changes caused by gentamicin-induced nephrotoxicity in the rat. Biomarkers 2005, 10, 173–187. [Google Scholar] [CrossRef]
- Hara, T.A.; Bansal, T.R.; de Grado, T.R. Effect of hypoxia on the uptake of [methyl-H-3]choline, [1-C-14] acetate and [F-18]FDG in cultured prostate cancer cells. Nucl. Med. Biol. 2006, 33, 977–984. [Google Scholar] [CrossRef]
- Glunde, K.; Shah, T.; Winnard, P.T.; Raman, V.; Takagi, T.; Vesuna, F.; Artemov, D.; Bhujwalla, Z.M. Hypoxia regulates choline kinase expression through hypoxia-inducible factor-lot signaling in a human prostate cancer model. Cancer Res. 2008, 68, 172–180. [Google Scholar] [CrossRef]
- Yoshii, Y.; Furukawa, T.; Yoshii, H.; Mori, T.; Kiyono, Y.; Waki, A.; Kobayashi, M.; Tsujikawa, T.; Kudo, T.; Okazawa, H.; et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009, 100, 821–827. [Google Scholar] [CrossRef]
- Richardson, A.D.; Yang, C.; Osterman, A.; Smith, J.W. Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res. Treat. 2008, 110, 297–307. [Google Scholar] [CrossRef]
- Morse, D.L.; Carroll, D.; Day, S.; Gray, H.; Sadarangani, P.; Murthi, S.; Job, C.; Baggett, B.; Raghunand, N.; Gillies, R.J. Characterization of breast cancers and therapy response by MRS and quantitative gene expression profiling in the choline pathway. NMR Biomed. 2009, 22, 114–127. [Google Scholar] [CrossRef]
- Weljie, A.M.; Bondareva, A.; Zang, P.; Jirik, F.R. 1H-NMR metabolomics identification of markers of hypoxia-induced metabolic shifts in a breast cancer model system. J. Biomol. NMR 2011, 49, 185–193. [Google Scholar] [CrossRef]
- Troy, H.; Chung, Y.L.; Mayr, M.; Ly, L.; Williams, K.; Stratford, I.; Harris, A.; Griffiths, J.; Stubbs, M. Metabolic profiling of hypoxia-inducible factor-1 beta-deficient and wild type Hepa-1 cells: Effects of hypoxia measured by H-1 magnetic resonance spectroscopy. Metabolomics 2005, 1, 293–303. [Google Scholar]
- Zhu, J.; Sova, P.; Xu, Q.W.; Dombek, K.M.; Xu, E.Y.; Vu, H.; Tu, Z.D.; Brem, R.B.; Bumgarner, R.E.; Schadt, E.E. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012, 10, e1001301. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef]
- Greco, O.; Marples, B.; Joiner, M.C.; Scott, S.D. How to overcome (and exploit) tumor hypoxia for targeted gene therapy. J. Cell. Physiol. 2003, 197, 312–325. [Google Scholar] [CrossRef]
- Graeber, T.G.; Peterson, J.F.; Tsai, M.; Monica, K.; Fornace, A.J.; Giaccia, A.J. Hypoxia induces accumulation of P53 protein, but activation of a G(1)-phase checkpoint by low-oxygen conditions is independent of P53 status. Mol. Cell. Biol. 1994, 14, 6264–6277. [Google Scholar] [CrossRef]
- Koong, A.C.; Chen, E.Y.; Giaccia, A.J. Hypoxia causes the activation of nuclear factor kappa-B through the phosphorylation of I-kappa-B-alpha on tyrosine residues. Cancer Res. 1994, 54, 1425–1430. [Google Scholar]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via denovo protein-synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Freitas, I.; Bertone, V.; Griffini, P.; Accossato, P.; Baronzio, G.F.; Pontiggia, P.; Stoward, P.J. In situ lactate-dehydrogenase patterns as markers of tumor oxygenation. Anticancer Res. 1991, 11, 1293–1299. [Google Scholar]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic-enzymes by hypoxia-inducible factor-1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase-1 and lactate-dehydrogenase-A genes-similarities with the erythropoietin 3' enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Lund, D.D.; Yorek, B.; Grovermckay, M.; Schmid, P.G. Pretranslational regulation of 2 cardiac glucose transporters in rats exposed to hypobaric hypoxia. Am. J. Physiol. 1992, 263, E562–E569. [Google Scholar]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Aberrant glycolytic metabolism of cancer cells: A remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for Type II hexokinase. J. Bioenerg. Biomembr. 1997, 29, 339–343. [Google Scholar] [CrossRef]
- Minchenko, A.; Leshchinsky, I.; Opentanova, I.; Sang, N.L.; Srinivas, V.; Armstead, V.; Caro, J. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene—Its possible role in the Warburg effect. J. Biol. Chem. 2002, 277, 6183–6187. [Google Scholar]
- Airley, R.; Loncaster, J.; Davidson, S.; Bromley, M.; Roberts, S.; Patterson, A.; Hunter, R.; Stratford, I.; West, C. Glucose transporter Glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin. Cancer Res. 2001, 7, 928–934. [Google Scholar]
- Chen, C.H.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1—Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar]
- Kolev, Y.; Uetake, H.; Takagi, Y.; Sugihara, K. Lactate dehydrogenase-5 (LDH-5) expression in human gastric cancer: Association with hypoxia-inducible factor (HIF-1 alpha) pathway, angiogenic factors production and poor prognosis. Ann. Surg. Oncol. 2008, 15, 2336–2344. [Google Scholar] [CrossRef]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1 alpha and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef]
- Shiu, R.P.; Watson, P.H.; Dubik, D. C-myc oncognen expression in estrogen-dependent and estrogen-independent breast-cancer. Clin. Chem. 1993, 39, 353–355. [Google Scholar]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Soh, H.; Wasa, M.; Fukuzawa, M. Hypoxia upregulates amino acid transport in a human neuroblastoma cell line. J. Pediatr. Surg. 2007, 42, 608–612. [Google Scholar] [CrossRef]
- Yoshii, Y.; Waki, A.; Furukawa, T.; Kiyono, Y.; Mori, T.; Yoshii, H.; Kudo, T.; Okazawa, H.; Welch, M.J.; Fujibayashi, Y. Tumor uptake of radiolabeled acetate reflects the expression of cytosolic acetyl-CoA synthetase: Implications for the mechanism of acetate PET. Nucl. Med. Biol. 2009, 36, 771–777. [Google Scholar] [CrossRef]
- Loffler, M.; Schneider, F. Lipogenesis in ehrlich ascites tumor-cells under anaerobic culture conditions. J. Cancer Res. Clin. Oncol. 1979, 95, 115–122. [Google Scholar] [CrossRef]
- Vavere, A.L.; Kridel, S.J.; Wheeler, F.B.; Lewis, J.S. 1-C-11-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J. Nucl. Med. 2008, 49, 327–334. [Google Scholar] [CrossRef]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef]
- Southam, A.D.; Easton, J.M.; Stentiford, G.D.; Ludwig, C.; Arvanitis, T.N.; Viant, M.R. Metabolic changes in flatfish hepatic tumours revealed by NMR-based metabolomics and metabolic correlation networks. J. Proteome Res. 2008, 7, 5277–5285. [Google Scholar]
- Israel, M.; Schwartz, L. The metabolic advantage of tumor cells. Mol. Cancer 2011, 10, 70:1–70:12. [Google Scholar]
- Rofstad, E.K.; Demuth, P.; Fenton, B.M.; Sutherland, R.M. P-31 nuclear magnetic-resonance spectroscopy studies of tumor energy-metabolism and its relationship to intracapillary oxyhemoglobin saturation status and tumor hypoxia. Cancer Res. 1988, 48, 5440–5446. [Google Scholar]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef]
- El-Sayed, S.; Bezabeh, T.; Odlum, O.; Patel, R.; Ahing, S.; MacDonald, K.; Somorjai, R.L.; Smith, I.C.P. An ex vivo study exploring the diagnostic potential of H-1 magnetic resonance spectroscopy in squamous cell carcinoma of the head and neck region. Head Neck 2002, 24, 766–772. [Google Scholar] [CrossRef]
- Moreno, A.; Lopez, L.A.; Fabra, A.; Arus, C. H-1 MRS markers of tumour growth in intrasplenic tumour and liver metastasis induced by injection of HT-29 cells in nude mice spleen. NMR Biomed. 1998, 11, 93–106. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Pastukh, V.; Solodushko, V.; Kramer, J.; Azuma, J. Effect of ischemia, calcium depletion and repletion, acidosis and hypoxia on cellular taurine content. Amino Acids 2002, 23, 395–400. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Beckonert, O.; Monnerjahn, K.; Bonk, U.; Leibfritz, D. Visualizing metabolic changes in breast-cancer tissue using 1H-NMR spectroscopy and self-organizing maps. NMR Biomed. 2003, 16, 1–11. [Google Scholar] [CrossRef]
- Bando, H.; Toi, M.; Kitada, K.; Koike, M. Genes commonly upregulated by hypoxia in human breast cancer cells MCF-7 and MDA-MB-231. Biomed. Pharmacother. 2003, 57, 333–340. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovey. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Wilson, I.D. Understanding “global” systems biology: Metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef]
- R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008.
- Chang, C.; Lin, C. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2001, 2, 27. [Google Scholar]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsai, I.-L.; Kuo, T.-C.; Ho, T.-J.; Harn, Y.-C.; Wang, S.-Y.; Fu, W.-M.; Kuo, C.-H.; Tseng, Y.J. Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data. Cancers 2013, 5, 491-510. https://doi.org/10.3390/cancers5020491
Tsai I-L, Kuo T-C, Ho T-J, Harn Y-C, Wang S-Y, Fu W-M, Kuo C-H, Tseng YJ. Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data. Cancers. 2013; 5(2):491-510. https://doi.org/10.3390/cancers5020491
Chicago/Turabian StyleTsai, I-Lin, Tien-Chueh Kuo, Tsung-Jung Ho, Yeu-Chern Harn, San-Yuan Wang, Wen-Mei Fu, Ching-Hua Kuo, and Yufeng Jane Tseng. 2013. "Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data" Cancers 5, no. 2: 491-510. https://doi.org/10.3390/cancers5020491
APA StyleTsai, I.-L., Kuo, T.-C., Ho, T.-J., Harn, Y.-C., Wang, S.-Y., Fu, W.-M., Kuo, C.-H., & Tseng, Y. J. (2013). Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data. Cancers, 5(2), 491-510. https://doi.org/10.3390/cancers5020491