Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane



Abstract
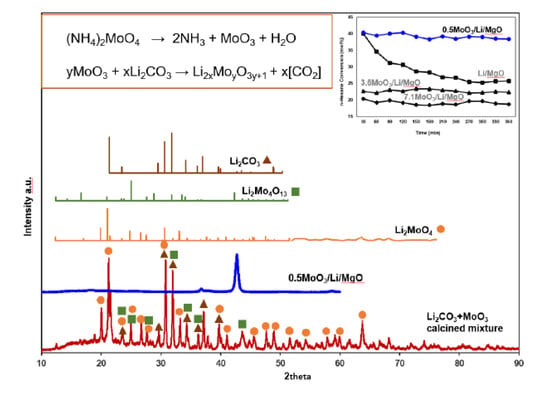
:
1. Introduction
2. Results
2.1. Catalytic Tests
2.2. Surface Area and XRD
2.3. Temperature Programmed Desorption (TPD)
2.4. Raman Spectra
3. Discussion
3.1. Textural Properties and Stability of the Catalyst
3.2. Activity and Selectivity
4. Materials and Methods
4.1. Materials
4.2. Catalyst Preparation
4.3. Catalyst Characterization
4.4. Catalytic Tests
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leveles, L.; Fuchs, S.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of light alkanes to olefins over alkali promoted oxide catalysts. Appl. Catal. A 2002, 227, 287–297. [Google Scholar] [CrossRef]
- Leveles, L.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of propane over lithium-promoted magnesia catalyst II. Active site characterization and hydrocarbon activation. J. Catal. 2003, 218, 307–314. [Google Scholar] [CrossRef]
- Leveles, L.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of propane over lithium-promoted magnesia catalyst I. Kinetics and mechanism. J. Catal. 2003, 218, 296–306. [Google Scholar] [CrossRef]
- Trionfetti, C.; Babich, I.V.; Seshan, K.; Lefferts, L. Formation of high surface area Li/MgO—Efficient catalyst for the oxidative dehydrogenation/cracking of propane. Appl. Catal. A 2006, 310, 105–113. [Google Scholar] [CrossRef]
- Trionfetti, C.; Babich, I.V.; Seshan, K.; Lefferts, L. Presence of Lithium Ions in MgO Lattice: Surface Characterization by Infrared Spectroscopy and Reactivity towards Oxidative Conversion of Propane. Langmuir 2008, 24, 8220–8228. [Google Scholar] [CrossRef]
- Boyadjian, C.; Lefferts, L.; Seshan, K. Catalytic oxidative cracking of hexane as a route to olefins. Appl. Catal. A 2010, 372, 167–174. [Google Scholar] [CrossRef]
- Boyadjian, C.; van der Veer, B.; Babich, I.V.; Lefferts, L.; Seshan, K. Catalytic oxidative cracking as a route to olefins: Oxidative conversion of hexane over MoO3-Li/MgO. Catal. Today 2010, 157, 345–350. [Google Scholar] [CrossRef]
- Gaab, S.; Find, J.; Grasselli, R.K.; Lercher, J.A. Oxidative ethane activation over oxide supported molten alkali metal chloride catalysts. Stud. Surf. Sci. Catal. 2004, 147, 673–678. [Google Scholar]
- Lin, C.-H.; Campbell, K.D.; Wang, J.-X.; Lunsford, J.H. Oxidative dimerizatlon of methane over Lanthanum Oxide. J. Phys. Chem. 1986, 90, 534–537. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H. Oxidative dehydrogenation of propane. React. Kin. Catal. Lett. 1996, 57, 3–11. [Google Scholar] [CrossRef]
- Lunsford, J.H.; Qiu, P.; Rosynek, M.P.; Xu, Z. Catalytic conversion of methane and ethylene to propylene. J. Phys. Chem. 1998, 102, 167–173. [Google Scholar] [CrossRef]
- Morales, E.; Lunsford, J.H. Oxidative dehydrogenation of ethane over a lithium-promoted magnesium oxide catalyst. J. Catal. 1989, 118, 255–265. [Google Scholar] [CrossRef]
- Ito, T.; Wang, J.-X.; Lin, C.-H.; Lunsford, J.H. Oxidative dimerization of methane over a lithium-promoted magnesium oxide catalyst. J. Am. Chem. Soc. 1985, 107, 5062–5068. [Google Scholar] [CrossRef]
- Wang, J.-X.; Lunsford, J.H. Characterization of [Li+O−] centers in lithium-doped MgO catalysts. J. Phys. Chem. 1986, 90, 5883–5887. [Google Scholar] [CrossRef]
- Shi, C.; Hatano, M.; Lunsford, J.H. A kinetic model for the oxidative coupling of methane over Li+/MgO Catalysts. Catal. Today 1992, 13, 191–199. [Google Scholar] [CrossRef]
- Lunsford, J.H. The role of surface-generated gas-phase radicals in catalysis. Langmuir 1989, 5, 12–16. [Google Scholar] [CrossRef]
- Shi, C.; Xu, M.; Rosynek, M.P.; Lunsford, J.H. Origin of kinetic isotope effects during the oxidative coupling of methane over a Li+/MgO catalyst. J. Phys. Chem. 1993, 97, 216–222. [Google Scholar] [CrossRef]
- Xu, M.; Shi, C.; Yang, X.; Rosynek, M.P.; Lunsford, J.H. Effect of carbon dioxide on the activation energy for methyl radical generatlon over Li/MgO catalysts. J. Phys. Chem. 1992, 96, 6395–6398. [Google Scholar] [CrossRef]
- Zavyalova, U.; Geske, M.; Horn, R.; Weinberg, G.; Frandsen, W.; Schuster, M.; Schlögl, R. Morphology and microstructure of Li/MgO catalysts for the oxidative coupling of methane. ChemCatChem 2011, 3, 949–959. [Google Scholar] [CrossRef]
- Kwapien, K.; Paier, J.; Sauer, J.; Geske, M.; Zavyalova, U.; Horn, R.; Schwach, P.; Trunschke, A.; Schlögl, R. Sites for methane activation on lithium-doped magnesium oxide surfaces. Angew. Chem. Int. Ed. 2014, 53, 8774–8778. [Google Scholar] [CrossRef]
- Schwach, P.; Frandsen, W.; Willinger, M.-G.; Schlögl, R.; Trunschke, A. Structure sensitivity of the oxidative activation of methane over MgO model catalysts: I. Kinetic study. J. Catal. 2015, 329, 560–573. [Google Scholar] [CrossRef] [Green Version]
- Schwach, P.; Hamilton, N.; Eichelbaum, M.; Thum, L.; Lunkenbein, T.; Schlögl, R.; Trunschke, A. Structure sensitivity of the oxidative activation of methane over MgO model catalysts: II. Nature of active sites and reaction mechanism. J. Catal. 2015, 329, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Schuh, J.; Sterrer, M.; Diwald, O.; Knözinger, E. Lithium ion induced surface reactivity changes on MgO nanoparticles. J. Catal. 2007, 247, 61–67. [Google Scholar] [CrossRef]
- Sinev, M.Y. Free radicals in catalytic oxidation of light alkanes: Kinetic and thermochemical aspects. J. Catal. 2003, 216, 468–476. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Smyrl, N.R.; Fuller, E.L., Jr.; Powell, G.L. Monitoring the heterogeneous reaction of LiH and LiOH with H2O and CO2 by diffuse reflectance infrared fourier transform spectroscopy. Appl. Spectrosc. 1983, 37, 38–44. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Ferretti, O. Mo/γ-Al2O3 catalysts for the oxidative dehydrogenation of propane: Effect of Mo loading. Appl. Catal. A 2001, 207, 421–431. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Cadus, L.E. Selective oxidation of propane on MgO/γ-Al2O3-supported molybdenum catalyst: Influence of promoters. Catal. Lett. 1998, 53, 185–192. [Google Scholar] [CrossRef]
- Cadus, L.E.; Abello, M.C.; Gomez, M.F.; Rivarola, J.B. Oxidative dehydrogenation of propane over molybdenum supported on MgO−γ-Al2O3. Ind. Eng. Chem. Res. 1996, 35, 14–18. [Google Scholar] [CrossRef]
- Ueda, W.; Lee, K.H.; Yoon, Y.-S.; Moro-oka, Y. Selective oxidative dehydrogenation of propane over surface molybdenum-enriched MgMoO4 catalyst. Catal. Today 1998, 44, 199–203. [Google Scholar] [CrossRef]
- Heracleous, E.; Machli, M.; Lemonidou, A.A.; Vasalos, I.A. Oxidative dehydrogenation of ethane and propane over vanadia and molybdena supported catalysts. J. Mol. Catal A Chem. 2005, 232, 29–39. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Christodoulakis, A.; Boghosian, S. Support effects on structure and activity of molybdenum oxide catalysts for the oxidative dehydrogenation of ethane. Catal. Today 2007, 127, 139–147. [Google Scholar] [CrossRef]
- Dejoz, A.; Lopez Nieto, J.M.; Marquez, F.; Vazquez, M.I. The role of molybdenum in Mo-doped V–Mg–O catalysts during the oxidative dehydrogenation of n-butane. Appl. Catal. A 1999, 180, 83–94. [Google Scholar] [CrossRef]
- Pless, J.D.; Bardin, B.B.; Kim, H.-S.; Ko, D.; Smith, M.T.; Hammond, R.R.; Stair, P.C.; Poeppelmeier, K.R. Catalytic oxidative dehydrogenation of propane over Mg–V/Mo oxides. J. Catal. 2004, 223, 419–431. [Google Scholar] [CrossRef]
- Christodoulakis, A.; Heracleous, E.; Lemonidou, A.A.; Boghosian, S. An operando Raman study of structure and reactivity of alumina-supported molybdenum oxide catalysts for the oxidative dehydrogenation of ethane. J. Catal. 2006, 242, 16–25. [Google Scholar] [CrossRef]
- Christodoulakis, A.; Boghosian, S. Molecular structure and activity of molybdena catalysts supported on zirconia for ethane oxidative dehydrogenation studied by operando Raman spectroscopy. J. Catal. 2008, 260, 178–187. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Ueda, W.; Moro-oka, Y. Oxidative dehydrogenation of propane over magnesium molybdate catalysts. Catal. Lett. 1995, 35, 57–64. [Google Scholar] [CrossRef]
- Vrieland, G.E.; Murchison, C.B. Anaerobic oxidation of butane to butadiene over magnesium molybdate catalysts. I. Magnesia supported catalysts. Appl. Catal. A 1996, 134, 101–121. [Google Scholar] [CrossRef]
- Kim, D.S.; Segawa, K.; Soeya, T.; Wachs, I.E. Surface structures of supported molybdenum oxide catalysts under ambient conditions. J. Catal. 1992, 136, 539–553. [Google Scholar] [CrossRef]
- Vuurman, M.A.; Wachs, I.E. In situ Raman spectroscopy of alumina-supported metal oxide catalysts. J. Phys. Chem. 1992, 96, 5008–5016. [Google Scholar] [CrossRef]
- Chang, S.-C.; Leugers, M.A.; Bare, S. Surface chemlstry of magnesium oxide-supported molybdenum oxide: An in situ Raman spectroscoplc study. J. Phys. Chem. 1992, 96, 10358–10365. [Google Scholar] [CrossRef]
- Bare, S.R.; Mitchell, G.E.; Maj, J.J.; Vrieland, G.E.; Gland, J.L. Local site symmetry of dispersed molybdenum oxide catalysts: XANES at the Mo L2,3-Edges. J. Phys. Chem. 1993, 97, 6048–6053. [Google Scholar] [CrossRef]
- Bare, S.R. Surface structure of highly dispersed MoO3 on MgO using in situ Mo L3-Edge XANES. Langmuir 1998, 14, 1500–1504. [Google Scholar] [CrossRef]
- Tomoyuki, K.; Okazaki, S.; Shishido, T.; Teramura, K.; Tanaka, T. Brønsted acid generation of alumina-supported molybdenum oxide calcined at high temperatures: Characterization by acid-catalyzed reactions and spectroscopic methods. J. Mol. Catal. A Chem. 2013, 371, 21–28. [Google Scholar]
- El-Sharkawy, E.A.; Khder, A.S.; Ahmed, A.I. Structural characterization and catalytic activity of molybdenum oxide supported zirconia catalysts. Microporous Mesoporous Mater. 2007, 102, 128–137. [Google Scholar] [CrossRef]
- Valigi, M.; Cimino, A.; Cordischi, D.; De Rossi, C.; Ferraris, G.; Gazzoli, D.; Indovina, V.; Occhiuzzi, M. Molybdenum (VI) interaction with zirconia surface and its influence on the crystallization and sintering. Solid State Ion. 1993, 63–65, 136–142. [Google Scholar] [CrossRef]
- Afanasiev, P.; Geantet, C.; Breysse, M. Preparation of high surface area Mo/ZrO2 catalysts by a molten salt method: Aplication to Hydrosulfurization. J. Catal. 1995, 153, 17–24. [Google Scholar] [CrossRef]
- Ulla, A.A.; Spretz, R.; Lombardo, E.; Daniell, W.; Knözinger, H. Catalytic combustion of methane on Co/MgO: Characterisation of active cobalt sites. Appl. Catal. B 2001, 29, 217–229. [Google Scholar] [CrossRef]
- Payen, E.; Grimblot, J.; Kasztelan, S. Study of oxidic and reduced alumina-supported molybdate and heptamolybdate species by in situ laser Raman spectroscopy. J. Phys. Chem. 1987, 91, 6642–6648. [Google Scholar] [CrossRef]
- Brooker, M.H.; Wang, J. Raman and infrared studies of lithium and cesium carbonates. Spectrochim. Acta A 1992, 48, 999–1008. [Google Scholar] [CrossRef]
- Li, G.; Li, H.; Mo, Y.; Chen, L.; Huang, X. Further identification to the SEI film on Ag electrode in lithium batteries by surface enhanced Raman scattering (SERS). J. Power Sources 2002, 104, 190–194. [Google Scholar] [CrossRef]
- Erdoheyli, A.; Fodor, K.; Nemeth, R.; Hancz, A.; Oszko, A. Partial oxidation of methane on silica-supported different alkali metal molybdates. J. Catal. 2001, 199, 328–337. [Google Scholar] [CrossRef]
- Moser, M.; Klimm, D.; Ganschow, S.; Kwasniewski, A.; Jacobs, K. Re-determination of the pseudobinary system Li2O—MoO3. Cryst. Res. Technol. 2008, 43, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhang, B.; Yao, Y.; Zheng, G.; Zhang, S.; You, J. Raman and density functional theory studies of Li2Mo4O13 structures in crystalline and molten states. Inorg. Chem. 2017, 56, 14129–14134. [Google Scholar] [CrossRef] [PubMed]
- Gatehouse, B.M.; Miskin, B.K. Structural studies in the Li2MoO4−MoO3 system: Part 2. The high-temperature form of lithium tetramolybdate, H-Li2Mo4O13. J. Solid State Chem. 1975, 15, 274–282. [Google Scholar] [CrossRef]
- Perrichon, V.; Durupty, M.C. Thermal stability of alkali metals deposited on oxide supports and their influence on the surface area of the support. Appl. Catal. 1988, 42, 217–227. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LiMgO | 0.5MoO3/Li/MgO | 3.6MoO3/Li/MgO | 7.1MoO3/Li/MgO | |
|---|---|---|---|---|
| Conversion (mol %) | ||||
| n-Hexane | 40.0 | 40.4 | 22.7 | 20.38 |
| Oxygen | 65.2 | 94.7 | 99.6 | 99.6 |
| Selectivity based on C (mol %) | ||||
| CO | 9.6 | 11.4 | 18.7 | 13.4 |
| CO2 | 15.0 | 13.5 | 28.4 | 35.0 |
| CH4 | 1.9 | 1.9 | 1.3 | 1.0 |
| C2–C5 alkanes | 5.2 | 8.2 | 2.1 | 3.2 |
| C2H4 | 23.2 | 22.6 | 11.4 | 7.2 |
| C3H6 | 25.9 | 24.2 | 12.4 | 11.3 |
| C4 = (butenes) | 11.5 | 11.5 | 12.1 | 6.1 |
| C5 = (pentenes) | 7.3 | 6.7 | 3.6 | 2.6 |
| C6 = (hexenes) | 0.4 | 0.0 | 10.0 | 20.2 |
| Catalyst | BET Surface Area (m2/g) | MoO3 Loading (wt %) | θ c (%) | Mo/Li d |
|---|---|---|---|---|
| Li/MgO a | 106 | - | - | - |
| Li/MgO b | 15 | - | - | - |
| 0.5MoO3/Li/MgO b | 70 | 0.51 | 7 | 0.03 |
| 3.6MoO3/Li/MgO b | 76 | 3.60 | 45 | 0.20 |
| 7.1MoO3/Li/MgO b | 82 | 7.11 | 86 | 0.40 |
| MgO a | 195 | - | - | - |
| MgO b | 148 | - | - | - |
| 0.5MoO3/MgO b | 144 | 0.53 | 3 | - |
| 3.3MoO3/MgO b | 178 | 3.26 | 17 | - |
| 7.9MoO3/MgO b | 189 | 7.94 | 42 | - |
| Mode Assignments | Compound | Reference | Raman Band Number | ||
|---|---|---|---|---|---|
| 0.5MoO3/MgO | 3.3MoO3/MgO | 7.9MoO3/MgO | |||
| lattice vibration | Mg(OH)2 | Bare et al. [41] | 275 | 275 | |
| Mo–Ot bending | [MoO4]2− | Bare et al. [41] | 320 | 325 | |
| Mg–O stretching | Mg(OH)2 | Bare et al. [41] | 445 | 445 | 445 |
| Mo–O-Mg vibrations | Mo–O–Mg | Bare et al. [41] | 813 | 813 | |
| Mo–O-Mg vibrations | Mo–O–Mg | Bare et al. [41] | 860 | 860 | |
| Mo–Ot asym stretching | [MoO4]2− | Bare et al. [41] | 874 | 874 | |
| Mo–Ot sym stretching | [MoO4]2− | Bare et al. [41] | 909 | 912 | 917 |
| Mode Assignments | Compound | Reference | Raman Band Number | ||
|---|---|---|---|---|---|
| 0.5MoO3/Li/MgO | 3.6MoO3/Li/MgO | 7.1MoO3/Li/MgO | |||
| lattice vibration | Li2CO3 | G. Li et al. [51] | 122 | 122 | 122 |
| lattice vibration | Li2CO3 | G. Li et al. [51] | 155 | 155 | 155 |
| lattice vibration | Li2CO4 | G. Li et al. [51] | 193 | 193 | 193 |
| lattice vibration | Mg(OH)2 | Bare et al. [41] | 275 | 275 | - |
| Mo–Ot bending | Li2Mo4O13 | Wan et al. [54] | - | - | 289 |
| Mo–Ot bending | Li2MoO4 | Erdöhelyi et al. [52] | - | - | 309 |
| Mo–Ot bending | [MoO4]2- | Bare et al. [41] | - | 320 | 321 |
| Mo–Ot bending | Li2MoO4 | Erdöhelyi et al. [52] | - | 321 | |
| Mg–O stretching | Mg(OH)2 | Bare et al. [41] | 445 | 445 | 445 |
| Mo–Ot asym stretching | Li2MoO4 | Erdöhelyi et al. [52] | - | - | 820 |
| Mo–Ot asym stretching | Li2MoO4 | Erdöhelyi et al. [52] | - | - | 846 |
| Mo–Ot asym stretching | Li2MoO4 | Erdöhelyi et al. [52] | - | - | 878 |
| Mo–Ot sym stretching | Li2Mo4O13 | Wan et al. [54] | - | - | 891 |
| Mo–Ot sym stretching | Li2MoO4 | Erdöhelyi et al. [52] | - | - | 904 |
| Mo–Ot sym stretching | [MoO4]2− | Bare et al. [41] | 909 | 912 | 917 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyadjian, C.; Lefferts, L. Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane. Catalysts 2020, 10, 354. https://doi.org/10.3390/catal10030354
Boyadjian C, Lefferts L. Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane. Catalysts. 2020; 10(3):354. https://doi.org/10.3390/catal10030354
Chicago/Turabian StyleBoyadjian, Cassia, and Leon Lefferts. 2020. "Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane" Catalysts 10, no. 3: 354. https://doi.org/10.3390/catal10030354
APA StyleBoyadjian, C., & Lefferts, L. (2020). Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane. Catalysts, 10(3), 354. https://doi.org/10.3390/catal10030354