Simple Environmentally-Friendly Reduction of 4-Nitrophenol




Abstract
:1. Introduction
2. Results and Discussion
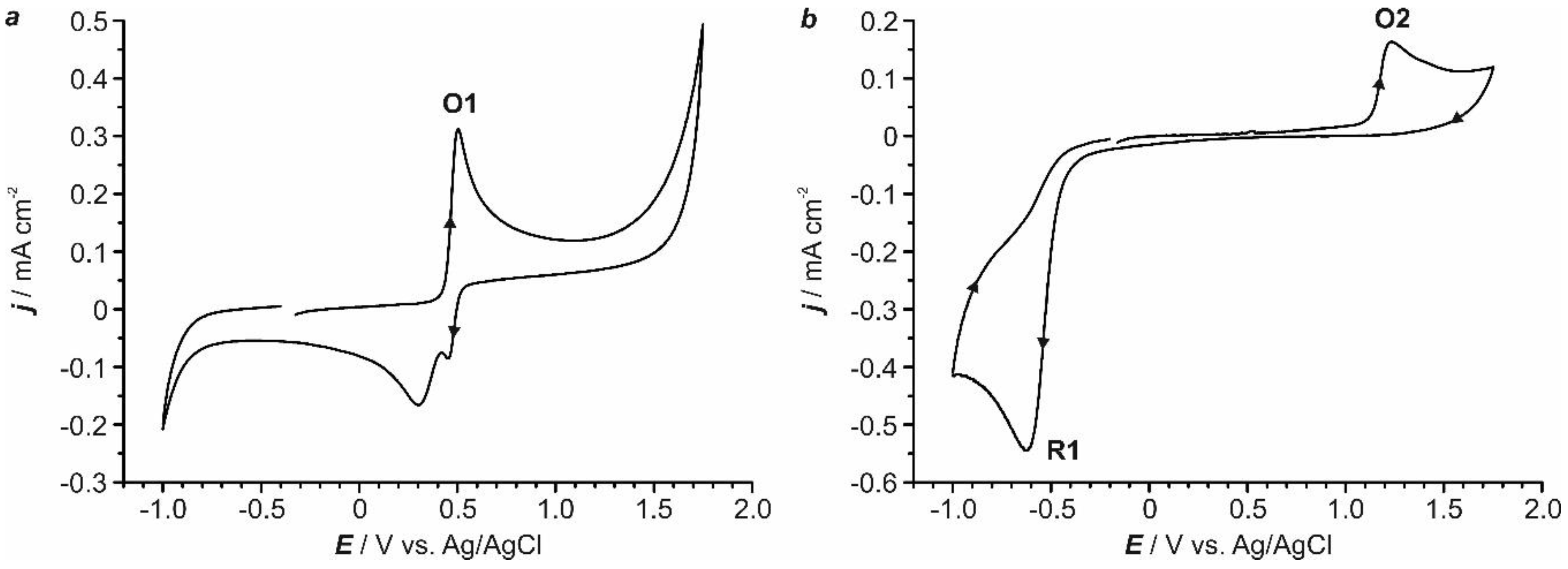
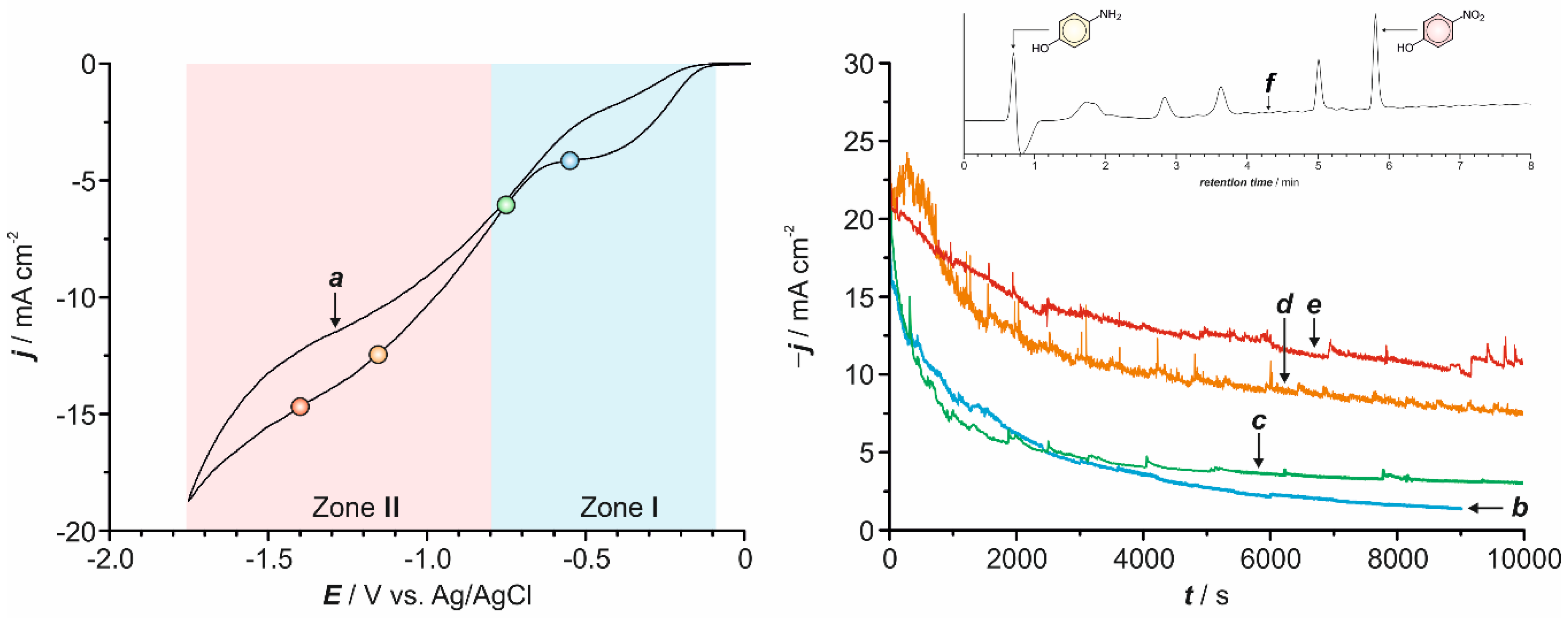
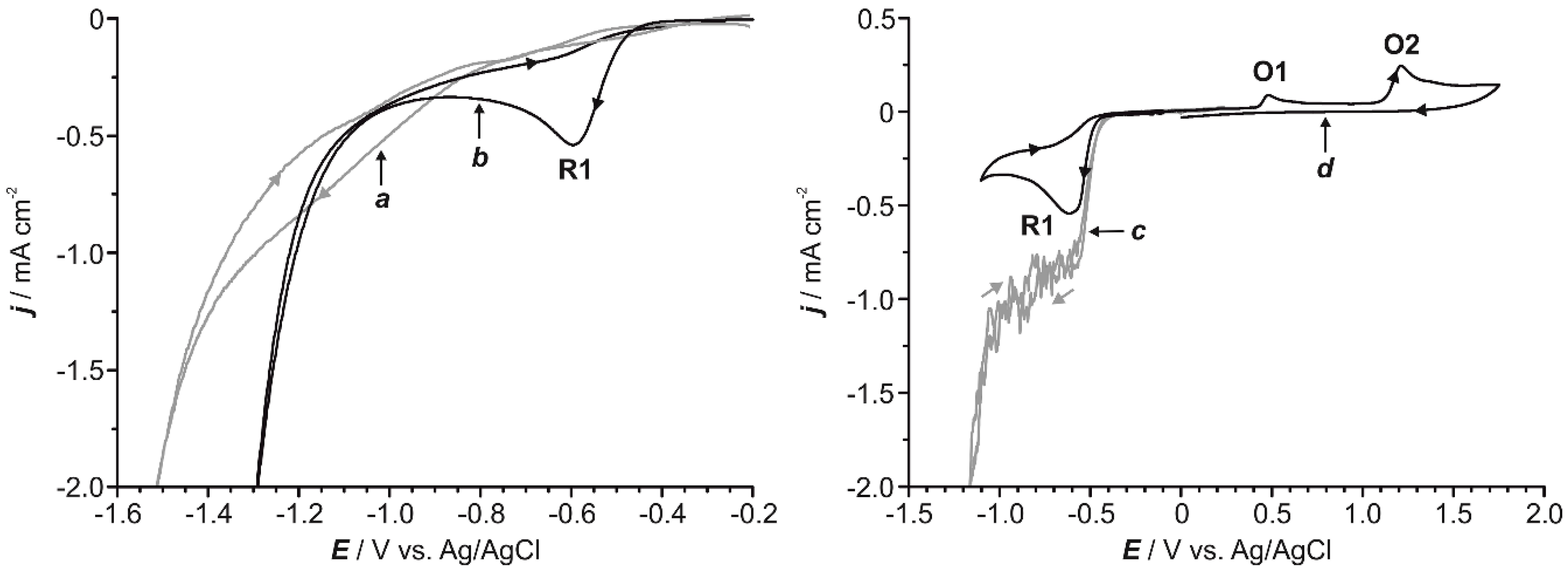
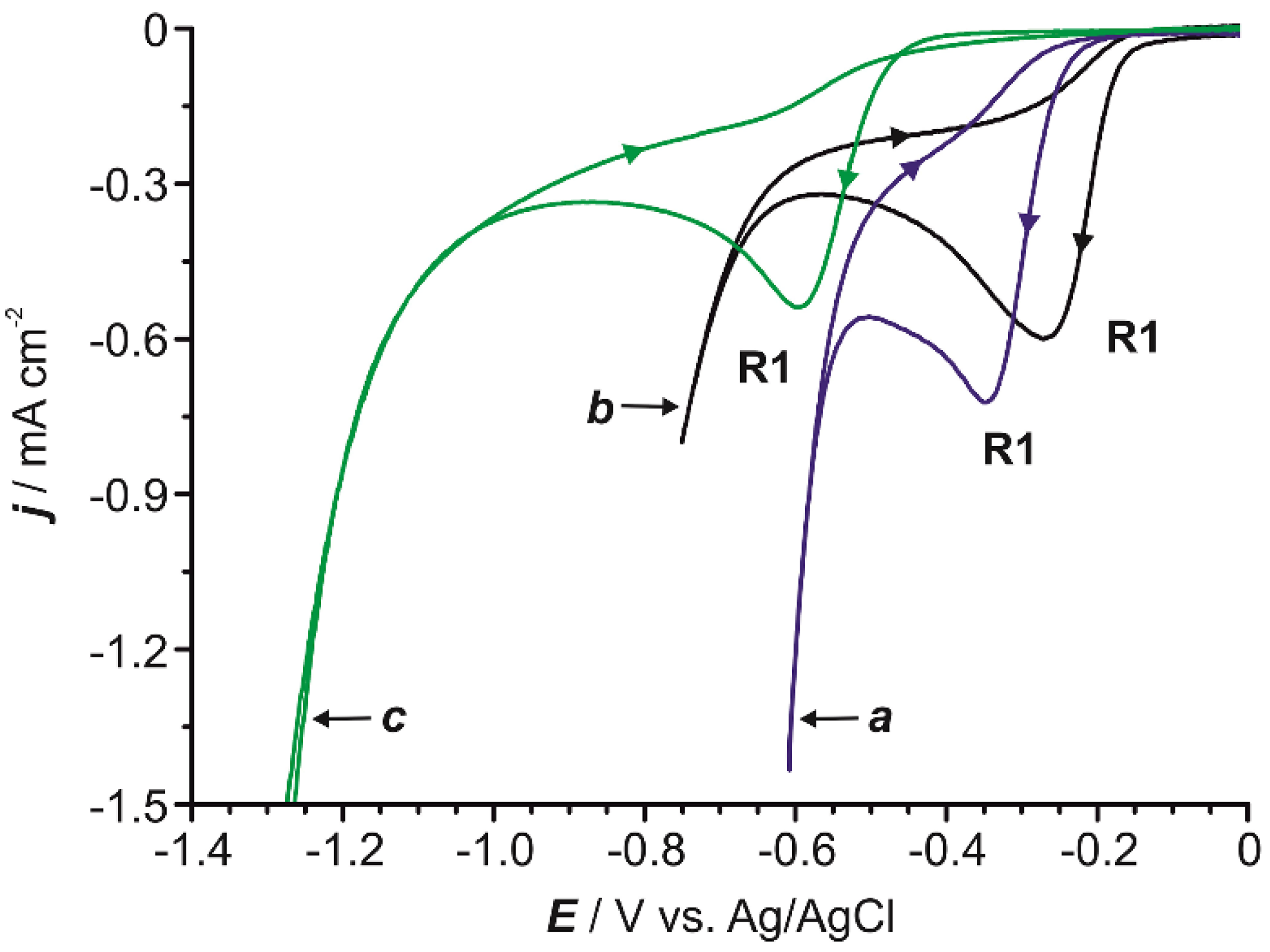
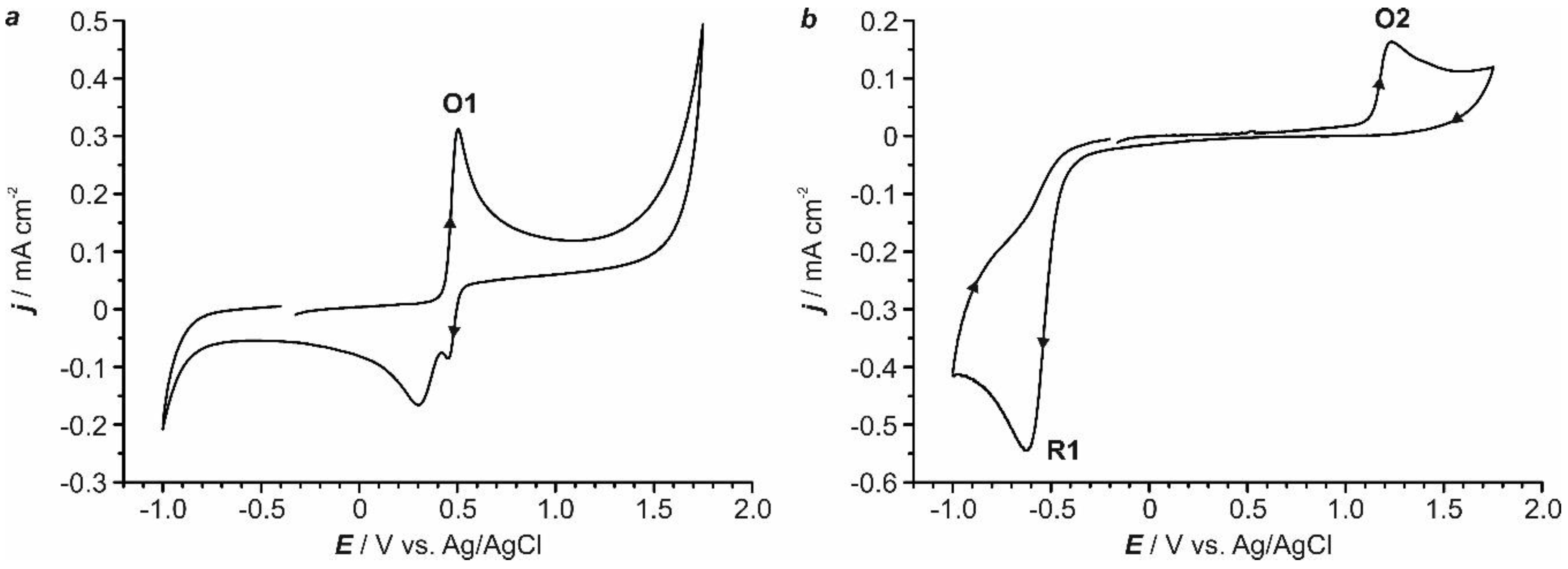
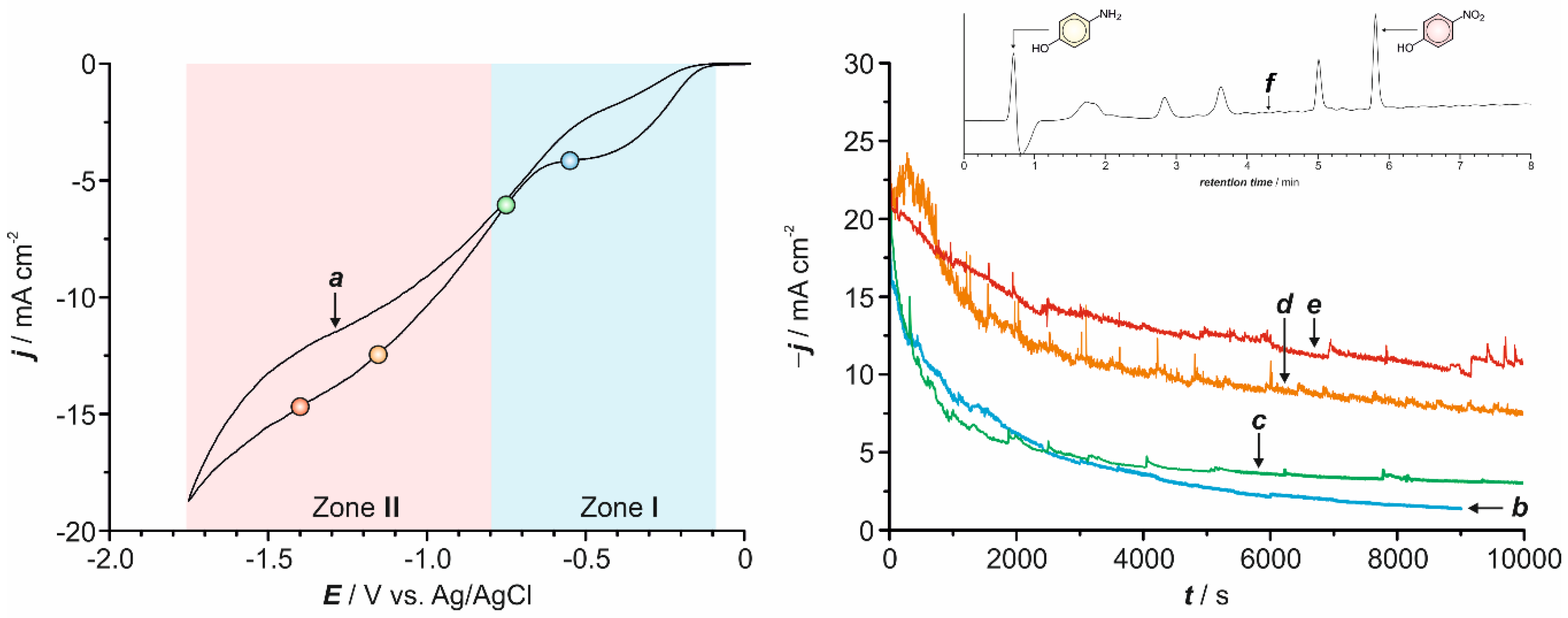
2.1. Preliminary Voltammetric Study
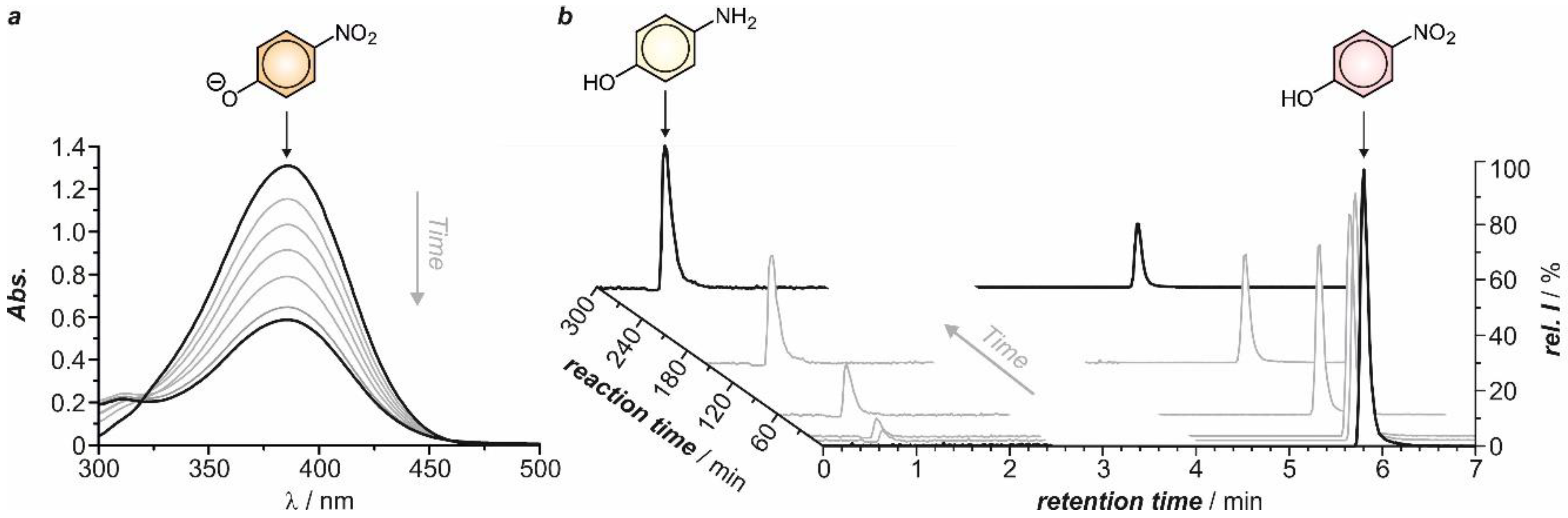
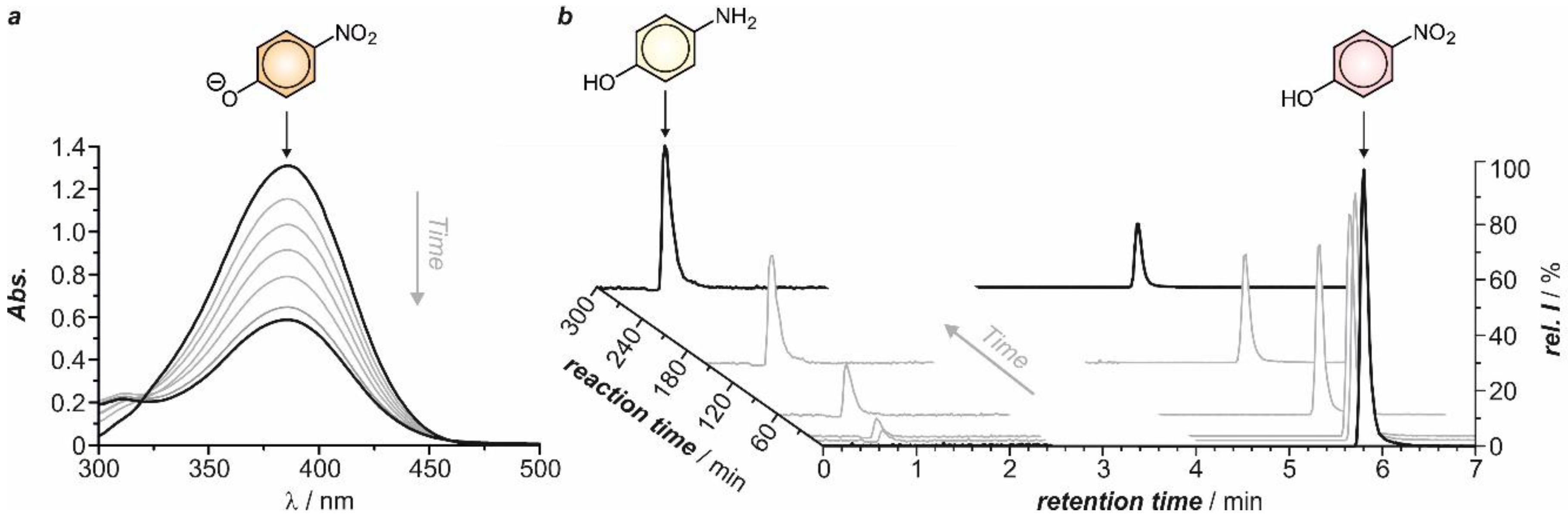
2.2. Electrochemical Synthesis
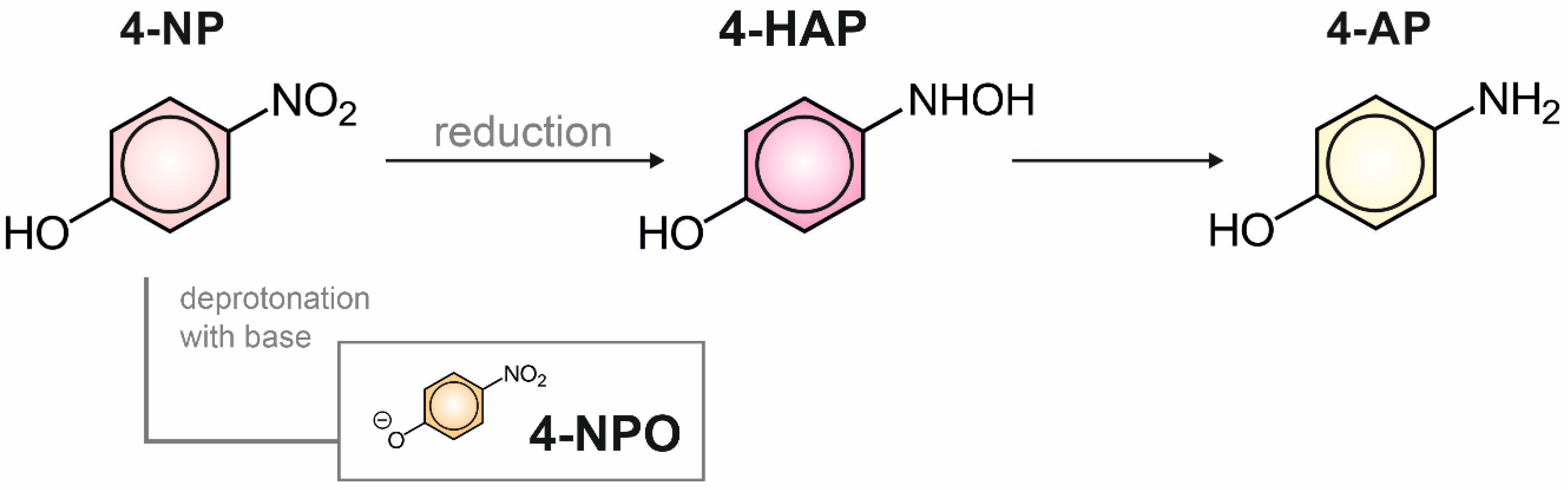
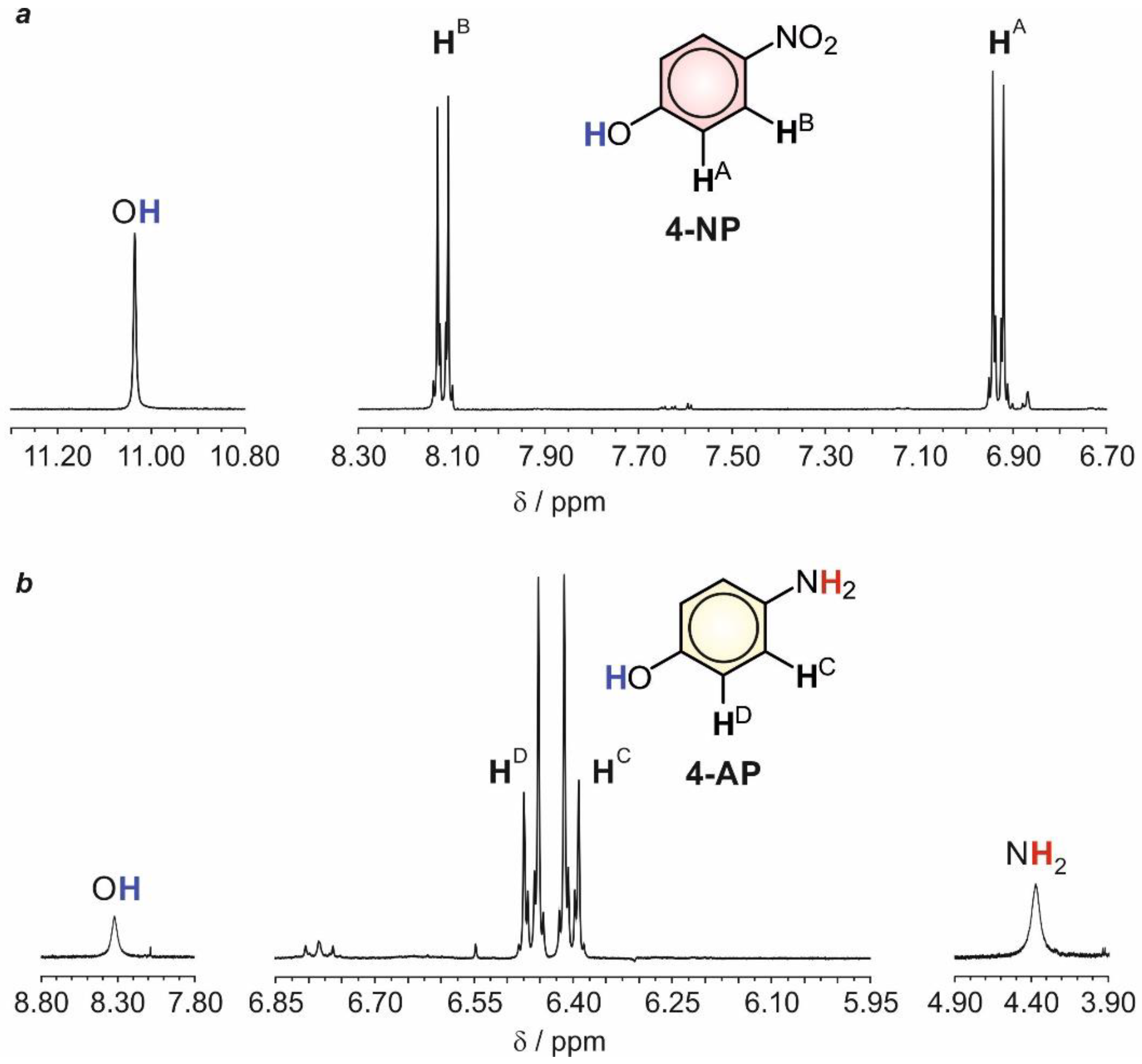
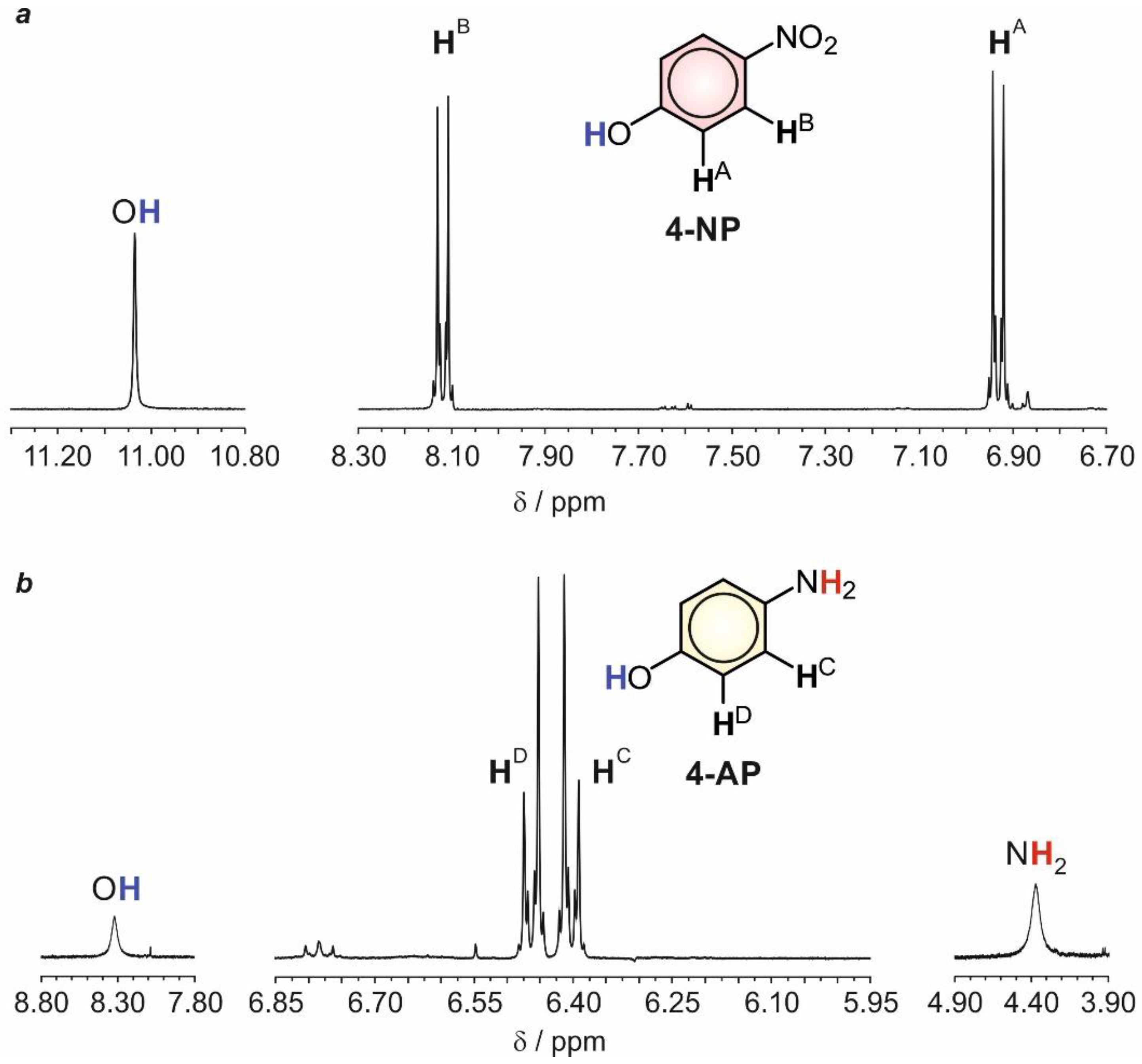
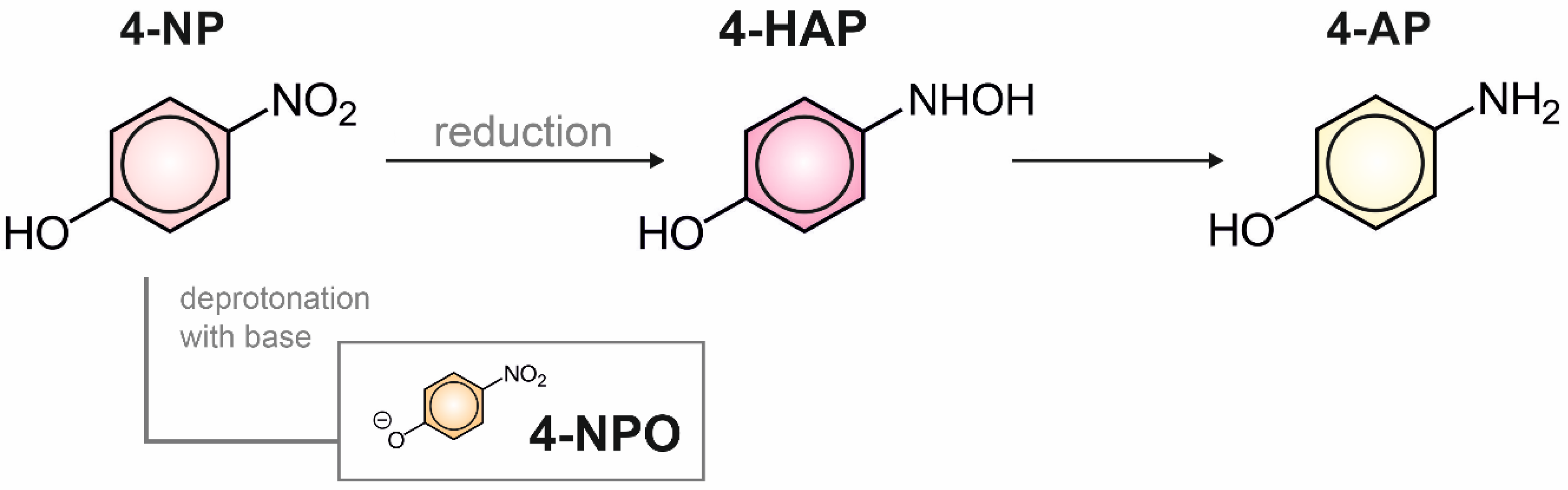
2.3. Structural and Electrochemical Characterization of the Formed Products
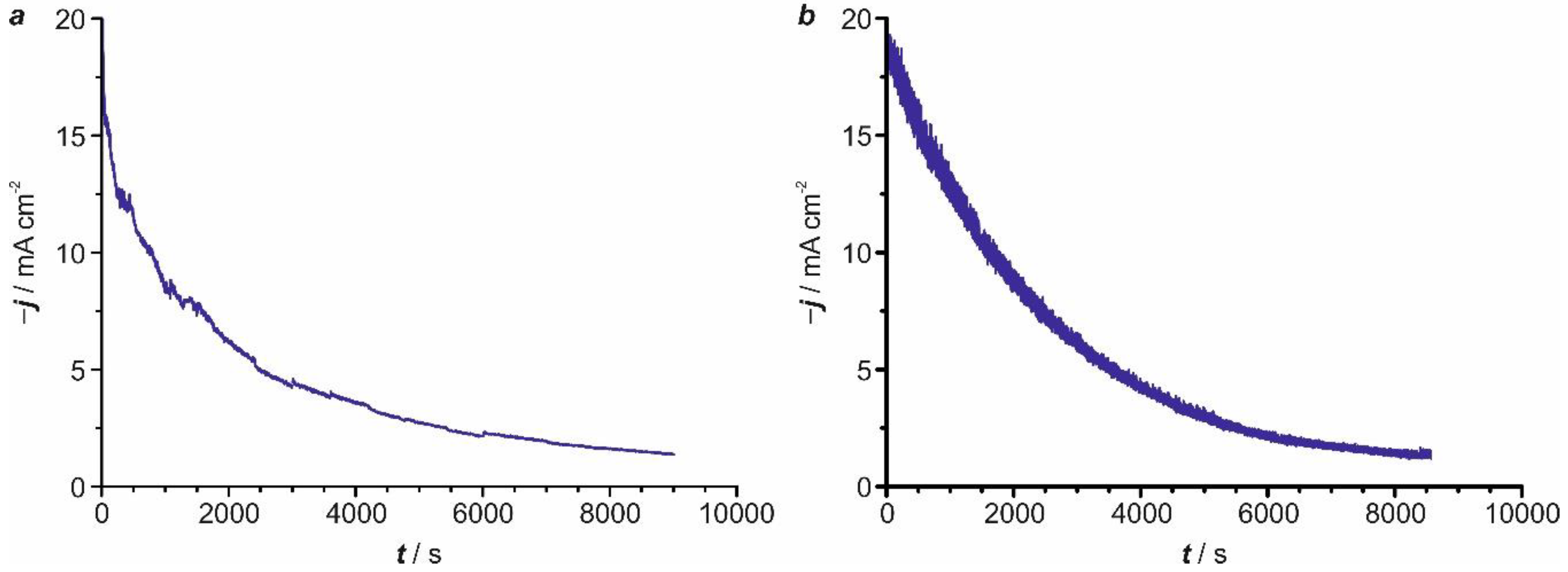
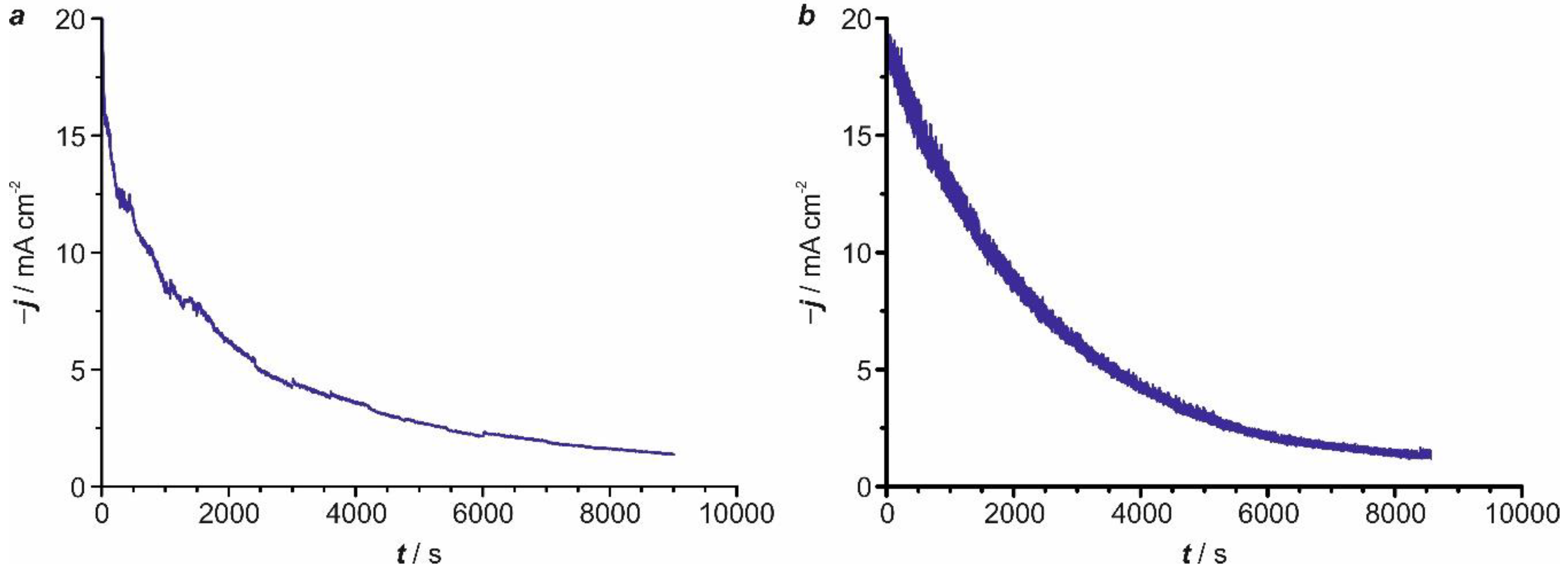
2.4. Kinetic Analysis
2.5. Influence of the Electrode Nature
2.6. Influence of the Temperature
2.7. Influence of the Applied Potential
3. Materials and Methods
3.1. Materials
3.2. General Methods and Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Payra, S.; Challagulla, S.; Chakraborty, C.; Roy, S. A hydrogen evolution reaction induced unprecedentedly rapid electrocatalytic reduction of 4-nitrophenol over ZIF-67 compare to ZIF-8. J. Electroanal. Chem. 2019, 853, 113545. [Google Scholar] [CrossRef]
- Li, J.; Kuang, D.; Feng, Y.; Zhang, F.; Xu, Z.; Liu, M. A graphene oxide-based electrochemical sensor for sensitive determination of 4-nitrophenol. J. Hazard. Mater. 2012, 201, 250–259. [Google Scholar] [CrossRef]
- Serrà, A.; Gómez, E.; Philippe, L. Bioinspired ZnO-based solar photocatalysts for the efficient decontamination of persistent organic pollutants and hexavalent chromium in wastewater. Catalysts 2019, 9, 974. [Google Scholar] [CrossRef] [Green Version]
- WWAP (UNESCO World Water Assessment Programme). The United Nations World Water Development Report 2019: Leaving No One Behind; UNESCO: Paris, France, 2019. [Google Scholar]
- Zheng, Y.; Liu, D.; Liu, S.; Xu, S.; Yuan, Y.; Xiong, L. Kinetics and mechanisms of p-nitrophenol biodegradation by Pseudomonas aeruginosa HS-D38. J. Environ. Sci. 2009, 21, 1194–1199. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Health and Environmental Effect Profiles; United States Environmental Protection Agency: Washington, DC, USA, 1980.
- United States Environmental Protection Agency. Health Effects Assessment for Nitrophenols; United States Environmental Protection Agency: Washington, DC, USA, 1987.
- Keith, L.H.; Telliard, W.A. Priority pollutants. I. A perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar] [CrossRef]
- Min, J.; Xu, L.; Fang, S.; Chen, W.; Hu, X. Molecular and biochemical characterization of 2-chloro-4-nitrophenol degradation via the 1,2,4-benzenetriol pathway in a Gram-negative bacterium. Appl. Microbiol. Biotechnol. 2019, 103, 7741–7750. [Google Scholar] [CrossRef]
- Sj, S. The gene cluster for para-nitrophenol catabolism is responsible for 2-chloro-4-nitrophenol degradation in Burkholderia sp. Strain SJ98. Appl. Environ. Microbiol. 2014, 80, 6212–6222. [Google Scholar]
- Wang, J.C.; Li, Y.; Li, H.; Cui, Z.H.; Hou, Y.; Shi, W.; Jiang, K.; Qu, L.; Zhang, Y.P. A novel synthesis of oleophylic Fe2O3/polystyrene fibers by Γ-Ray irradiation for the enhanced photocatalysis of 4-chlorophenol and 4-nitrophenol degradation. J. Hazard. Mater. 2019, 379, 120806. [Google Scholar] [CrossRef]
- Serrà, A.; Artal, R.; García-Amorós, J.; Sepúlveda, B.; Gómez, E.; Nogués, J.; Philippe, L. Hybrid Ni@ZnO@ZnS-Microalgae for Circular Economy: A Smart Route to the Efficient Integration of Solar Photocatalytic Water Decontamination and Bioethanol Production. Adv. Sci. 2020, 7, 1–9. [Google Scholar] [CrossRef]
- Osin, O.A.; Yu, T.; Cai, X.; Jiang, Y.; Peng, G.; Cheng, X.; Li, R.; Qin, Y.; Lin, S. Photocatalytic degradation of 4-nitrophenol by C, N-TiO2: Degradation efficiency vs. embryonic toxicity of the resulting compounds. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Salehi, Z.; Rasouli, A.; Doosthosseini, H. p-nitrophenol Degradation Kinetics and Mass Transfer Study by Ralstonia eutropha as a Whole Cell Biocatalyst. Polycycl. Aromat. Compd. 2019, 5, 1–14. [Google Scholar] [CrossRef]
- Kulkarni, M.; Chaudhari, A. Biodegradation of p-nitrophenol by P. putida. Bioresour. Technol. 2006, 97, 982–988. [Google Scholar] [CrossRef]
- Tomei, M.C.; Annesini, M.C. 4-Nitrophenol biodegradation in a sequencing batch reactor operating with aerobic-anoxic cycles. Environ. Sci. Technol. 2005, 39, 5059–5065. [Google Scholar] [CrossRef]
- Oturan, M.A.; Peiroten, J.; Chartrin, P.; Acher, A.J. Complete destruction of p-Nitrophenol in aqueous medium by electro-fenton method. Environ. Sci. Technol. 2000, 34, 3474–3479. [Google Scholar] [CrossRef]
- Kiwi, J.; Pulgarin, C.; Peringer, P. Effect of Fenton and photo-Fenton reactions on the degradation and biodegradability of 2 and 4-nitrophenols in water treatment. Appl. Catal. B Environ. 1994, 3, 335–350. [Google Scholar] [CrossRef]
- Xie, F.; Xu, Y.; Xia, K.; Jia, C.; Zhang, P. Alternate pulses of ultrasound and electricity enhanced electrochemical process for p-nitrophenol degradation. Ultrason. Sonochem. 2016, 28, 199–206. [Google Scholar] [CrossRef]
- Serrà, A.; Alcobé, X.; Sort, J.; Nogués, J.; Vallés, E. Highly efficient electrochemical and chemical hydrogenation of 4-nitrophenol using recyclable narrow mesoporous magnetic CoPt nanowires. J. Mater. Chem. A 2016, 4, 15676–15687. [Google Scholar] [CrossRef] [Green Version]
- Chen, G. Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 2004, 38, 11–41. [Google Scholar] [CrossRef]
- Arora, P.K.; Srivastava, A.; Singh, V.P. Bacterial degradation of nitrophenols and their derivatives. J. Hazard. Mater. 2014, 266, 42–59. [Google Scholar] [CrossRef]
- Yuan, S.; Tian, M.; Cui, Y.; Lin, L.; Lu, X. Treatment of nitrophenols by cathode reduction and electro-Fenton methods. J. Hazard. Mater. 2006, 137, 573–580. [Google Scholar] [CrossRef]
- Wang, T.C.; Lu, N.; Li, J.; Wu, Y. Plasma-TiO2 catalytic method for high-efficiency remediation of p-nitrophenol contaminated soil in pulsed discharge. Environ. Sci. Technol. 2011, 45, 9301–9307. [Google Scholar] [CrossRef]
- Ruan, M.; Song, P.; Liu, J.; Li, E.; Xu, W. Highly Efficient Regeneration of Deactivated Au/C Catalyst for 4-Nitrophenol Reduction. J. Phys. Chem. C 2017, 121, 25882–25887. [Google Scholar] [CrossRef]
- Wang, C.; Salmon, L.; Li, Q.; Igartua, M.E.; Moya, S.; Ciganda, R.; Ruiz, J.; Astruc, D. From Mono to Tris-1,2,3-triazole-Stabilized Gold Nanoparticles and Their Compared Catalytic Efficiency in 4-Nitrophenol Reduction. Inorg. Chem. 2016, 55, 6776–6780. [Google Scholar] [CrossRef]
- Dutta, S.; Sarkar, S.; Ray, C.; Roy, A.; Sahoo, R.; Pal, T. Mesoporous gold and palladium nanoleaves from liquid-liquid interface: Enhanced catalytic activity of the palladium analogue toward hydrazine-assisted room-temperature 4-nitrophenol reduction. ACS Appl. Mater. Interfaces 2014, 6, 9134–9143. [Google Scholar] [CrossRef]
- Song, H.; Chen, T.S. P-Aminophenol-induced liver toxicity: Tentative evidence of a role for acetaminophen. J. Biochem. Mol. Toxicol. 2001, 15, 34–40. [Google Scholar] [CrossRef]
- SCCS (Scientific Committee on Consumer Safety). Opinion on P-Aminophenol; European Commission: Brussels, Belgium, 2011. [Google Scholar]
- United States Environmental Protection Agency. Provisional Peer Reviewed Toxicity Values for p-Aminophenol (CASRN 123-30-8); United States Environmental Protection Agency: Washington, DC, USA, 2005.
- Megharaj, M.; Pearson, H.W.; Venkateswarlu, K. Toxicity of p-aminophenol and p-nitrophenol to Chlorella vulgaris and two species of Nostoc isolated from soil. Pestic. Biochem. Physiol. 1991, 40, 266–273. [Google Scholar] [CrossRef]
- Saran, S.; Manjari, G.; Devipriya, S.P. Synergistic eminently active catalytic and recyclable Ag, Cu and Ag-Cu alloy nanoparticles supported on TiO2 for sustainable and cleaner environmental applications: A phytogenic mediated synthesis. J. Clean. Prod. 2018, 177, 134–143. [Google Scholar] [CrossRef]
- Corbett, J.F. An historical review of the use of dye precursors in the formulation of commercial oxidation hair dyes. Dye. Pigment. 1999, 41, 127–136. [Google Scholar] [CrossRef]
- He, Q.; Tian, Y.; Wu, Y.; Liu, J.; Li, G.; Deng, P.; Chen, D. Facile and ultrasensitive determination of 4-nitrophenol based on acetylene black paste and graphene hybrid electrode. Nanomaterials 2019, 9, 429. [Google Scholar] [CrossRef] [Green Version]
- Marquez, J.; Pletcher, D. A study of the electrochemical reduction of nitrobenzene to p-aminophenol. J. Appl. Electrochem. 1980, 10, 567–573. [Google Scholar] [CrossRef]
- Cyr, A.; Huot, P.; Marcoux, J.F.; Belot, G.; Laviron, E.; Lessard, J. The electrochemical reduction of nitrobenzene and azoxybenzene in neutral and basic aqueous methanolic solutions at polycrystalline copper and nickel electrodes. Electrochim. Acta 1989, 34, 439–445. [Google Scholar] [CrossRef]
- Wu, T.; Wang, G.; Zhang, Y.; Kang, S.; Zhang, H. Electrochemical deposition of Pt on carbon fiber cloth utilizing Pt mesh counter electrode during hydrogen evolution reaction for electrocatalytic hydrogenation reduction of p-nitrophenol. New J. Chem. 2017, 41, 7012–7019. [Google Scholar] [CrossRef]
- Silvester, D.S.; Wain, A.J.; Aldous, L.; Hardacre, C.; Compton, R.G. Electrochemical reduction of nitrobenzene and 4-nitrophenol in the room temperature ionic liquid [C4dmim][N(Tf)2]. J. Electroanal. Chem. 2006, 596, 131–140. [Google Scholar] [CrossRef]
- Karthik, R.; Hou, Y.S.; Chen, S.M.; Elangovan, A.; Ganesan, M.; Muthukrishnan, P. Eco-friendly synthesis of Ag-NP s using Cerasus serrulata plant extract—Its catalytic, electrochemical reduction of 4-NPh and antibacterial activity. J. Ind. Eng. Chem. 2016, 37, 330–339. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k/min−1 cm−2 | ||||
|---|---|---|---|---|
| Electrode | T/°C | E/V | UV-vis Spectroscopy | HPLC |
| Ag | 25 | −0.550 | 1.2 × 10−3 | 1.2 × 10−3 |
| 25 | −0.750 | 1.9 × 10−3 | 1.8 × 10−3 | |
| 25 | −1.150 | 3.2 × 10−3 * | 4.0 × 10−3 * | |
| 25 | −1.400 | 3.9 × 10−3 * | 2.9 × 10−3 * | |
| Au | 25 | −0.465 | 1.2 × 10−3 | 1.4 × 10−3 |
| 40 | −0.465 | 2.5 × 10−3 | 2.5 × 10−3 | |
| 50 | −0.465 | 4.2 × 10−3 | 4.3 × 10−3 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrà, A.; Artal, R.; Pozo, M.; Garcia-Amorós, J.; Gómez, E. Simple Environmentally-Friendly Reduction of 4-Nitrophenol. Catalysts 2020, 10, 458. https://doi.org/10.3390/catal10040458
Serrà A, Artal R, Pozo M, Garcia-Amorós J, Gómez E. Simple Environmentally-Friendly Reduction of 4-Nitrophenol. Catalysts. 2020; 10(4):458. https://doi.org/10.3390/catal10040458
Chicago/Turabian StyleSerrà, Albert, Raül Artal, Maria Pozo, Jaume Garcia-Amorós, and Elvira Gómez. 2020. "Simple Environmentally-Friendly Reduction of 4-Nitrophenol" Catalysts 10, no. 4: 458. https://doi.org/10.3390/catal10040458
APA StyleSerrà, A., Artal, R., Pozo, M., Garcia-Amorós, J., & Gómez, E. (2020). Simple Environmentally-Friendly Reduction of 4-Nitrophenol. Catalysts, 10(4), 458. https://doi.org/10.3390/catal10040458