1. Introduction
The impact of chemical solvents on the environment is an increasingly debated subject. Indeed, most of these solvents are volatile organic compounds and can, therefore, be easily dispersed in the environment. Risks often accompany this because they are flammable. In addition, they are generally harmful from an ecological and health point of view [
1]. Over the past fifteen years, green chemistry, which has become a priority area for academic and industrial research, has undergone considerable development. In this context, much research is focused on alternative, environmentally friendly solvents that can replace some highly volatile and environmentally harmful solvents [
2].
These include water [
3,
4,
5,
6,
7,
8,
9], supercritical CO
2 [
10,
11,
12,
13,
14], and ionic liquids (ILs) [
15,
16,
17,
18,
19,
20,
21]. Ionic liquids are of particular interest because they are a new class of solvents that offer interesting opportunities as a reaction medium for cleaner chemistry due to their multiple properties.
Ionic liquids are salts consisting solely of ions, whose melting point is below an arbitrary temperature, often 100 °C. They consist of generally bulky organic cations and organic or inorganic anions, which gives them an asymmetry of shape and charge, as well as great flexibility [
22,
23]. In general, cations have a voluminous and asymmetric nature [
15]. The most commonly represented are composed of ammoniums, phosphoniums, or heteroaromatic systems such as imidazolium, pyridinium, pyrrolidinium, oxazolium, thiazolium, pyridazinium, triazolium, and tetraalkylammonium. Different ionic liquids can be formed either by the appropriate combination of cations and anions or by the chemical modification of the cation or anion. This offers a choice of considerable combinations, and the literature now reports the possible synthesis of at least 10
6 different ILs [
19].
Their synthesis is generally carried out in two steps: the formation of the cation followed by an anion exchange step called anionic metathesis [
24].
The properties of the ILs are also very large [
19,
25,
26]: low vapor tension, high decomposition temperature, high solubilizing power, and a wide variety of the structures that allow different polarities [
27,
28].
The ILs are commonly used in the domain of (bio)catalysis [
18,
29], organic synthesis [
30,
31], electrochemistry [
32], or extraction [
33] as recently described in the appropriate reviews [
34,
35,
36]. Catalyses (hydrogenation, oxidation, Pd-catalyzed couplings, Friedel-Crafts, Diels-Alder, etc.) in ILs are also largely developed [
37] and the use of these new solvents generally led to higher kinetics and better selectivity’s.
However, the low biodegradability and the toxicity of the ionic liquids, in general, led the scientific community to reduce their use or to find other greener alternatives by using renewable resources as starting materials such as acids, amino acids, amino alcohols, and sugars which could improve the green character of ILs [
38,
39,
40,
41,
42]. Catalyses (hydrogenation, heck reactions) have also been performed in such ILs [
43,
44,
45].
The Michael reaction is a conjugated nucleophilic addition between a nucleophile and an α,β-unsaturated electrophile. The nucleophiles are thiols, anions, or amines, and the electrophiles are alkenes, alkynes in α and β position of carbonyls, amides, or nitriles. Recently, Yadav et al. reviewed innovative catalysis in Michael addition reactions, including the use of organocatalysts and heterogeneous processes [
46]. Gu et al. in 2019 [
47] also described the role of functionalized quaternary ammonium salt ionic liquids (FQAILs) as an economical and efficient catalyst for the synthesis of glycerol carbonate from glycerol and dimethyl carbonate.
In 2003, Salunkhe and coll. studied the use of hydrophobic or hydrophilic ionic liquids ([bmim]PF
6, [bmim]BF
4, [BuPy]BF
4) as solvents in the conjugated addition reaction of dimethyl malonate to chalcone in the presence of a quaternary ammonium salt derived from quinine (
Scheme 1) [
48]. This last compound used as a chiral phase transfer agent led to moderate ee’s.
The yields were quantitative and the ee up to 56%. Next, in 2005, Ranu and coll. performed the Michael addition of activated methylene compounds on ketones, esters, and conjugated nitriles in the presence of the ionic liquid [Bmim]OH (
Scheme 2). This one was used as a solvent and catalyst for the reaction. The reaction products were obtained with good yields; furthermore, the compound issued from the double addition could also be easily obtained in one step, which was surprising when compared to conventional methods [
49].
In 2005, Rao and Jothilingam described how microwaves could drastically decrease the reaction time for the Michael addition of active methylene compounds to
α,
β-unsaturated carbonyl compounds in the presence of a large excess of K
2CO
3 as a base [
50]. The reaction took place on the surface of potassium carbonate under microwave irradiation (450 W) and led to good to high yields (50% to 90%) in short times (5–10 min).
As previously mentioned, we prepared some biobased ionic liquids with natural carboxylates and used these in hydrogenation and Heck couplings [
38,
39,
40,
41,
42,
43,
44,
45]. Among these ILs, we prepared tetrabutylphosphonium and tetrabutylammonium prolinate or hydroxyprolinate. This paper aims to show how these proline-based ionic liquids could be used as solvents and basic catalysts in a Michael model reaction. Furthermore, this study will be extended to cholinium cation, and we will demonstrate that microwaves could generate greener activation conditions for this reaction in ionic liquids.
2. Results and Discussion
Biosourced ionic liquids were prepared using an acid-base reaction between the tetrabutyl-ammonium or -phosphonium hydroxides (TBAOH and TBPOH, respectively) or cholinium hydroxide ChOH with natural chiral amino acids (
S-proline,
R-proline, and
trans-4-hydroxy-
S-proline) (
Scheme 3). The synthetic methodology was inspired by Ohno’s work [
51] in 2005 and previously reported works for some phosphonium or ammonium derivatives [
38,
39,
40,
41,
42,
43,
44,
45].
Nine ionic liquids were prepared easily with high yields and purity (
Table 1), and the cholinium-based ionic liquids were synthesized following similar procedures previously used with ammonium or phosphonium-based ionic liquids.
The decomposition temperature values obtained by TGA confirmed the good thermal stability of all synthesized ionic liquids (Tdec ≥ 198 °C). We noticed that the thermal stability of ionic liquids was more dependent on the nature of the cation, particularly with phosphonium cations, as described in the literature [
41]. Indeed, the ionic liquid TBP
+ prolinate was the most stable. However, we could remark that the cholinium based ionic liquids were more stable than the ammonium-based ones when prolinate or hydroxyprolinate were present as a counter anion (
Figure 1); this observation is probably due to the formation of hydrogen bonds which could explain this slightly higher thermal stability [
52].
The viscosities also show the importance of the anion (
Table 1). In fact, with (
R)-prolinate or (
S)-prolinate as counter-anions, the viscosities at 60 °C are relatively low between 70 and 100 cp. With the same cation, we observed that the viscosity of choline-based ionic liquids was higher, probably due to the OH group present in this cation involved in hydrogen bonds. The same conclusions could be deduced when
trans-4-hydroxy-
S-prolinate was used as anion following much higher viscosity values due, in this case, to the presence of two OH groups (
Table 1).
These ionic liquids have been used as a catalyst and solvent in Michael reaction models involving dimethyl malonate and chalcone as starting materials. Preliminary results were obtained with a slight excess of dimethyl malonate towards the chalcone (1.2 eq.) at 50 °C for 24 h (
Table 2). DMF was used as a unique solvent or was associated with ionic liquid in a lower amount to reduce the viscosity of the reacting medium.
The results obtained proved the positive impact of the use of ionic liquids for this Michael reaction while higher conversions of chalcone were observed (
Table 2, entries 2–7, 9 and 10 vs. entry 1). Nevertheless, the conversions were not complete, and further experiments were conducted using an excess of dimethylmalonate. Indeed, with 3 or 4 equivalents of dimethylmalonate, the conversions obtained are much higher; furthermore, the reaction time could also be reduced to 4 h (
Table 3, entry 4).
Based on these results, Michael’s reaction was carried out using the various synthesized ILs as solvents (without the addition of DMF) under microwaves activation.
The conditions were adjusted after several experiments; at the beginning, the reactions were carried out with a power of 220 W leading to significant temperature variations depending on the ionic liquid. This aspect was unfavorable to the stability of the reactants or products. In addition, we observed that the presence of water in the ILs could also influence the conversion of the chalcone. Indeed, according to the results obtained in GC, the conversion rate of chalcone was low, and this was probably due to the low solubility of chalcone in water. Therefore, it was important to work with ionic liquids containing as little water as possible. Michael reactions were performed under microwaves during 45 min with a power limited to 100 W, and the ionic liquids were dried under vacuum for 4 h before use.
The reaction conducted under microwaves in DMF with the addition of K
2CO
3 as a base led to a high conversion of chalcone over 45 min (
Table 4, entry 1). Next, the DMF and the base could be substituted by the ionic liquid, which played further roles (
Table 4, entries 2–10). Good to very good conversions of chalcone (60 to 85%) were obtained again in 45 min, proving that the microwaves could decrease the activation energy [
53,
54]. According to the literature concerning the Michael addition [
55], we proposed a mechanism that is described in
Scheme 4.
The anion plays the role of the base towards the dimethyl malonate, the Michael’s donor, which subsequently reacts with the chalcone (the Michael’s acceptor) to produce a stabilized enolate. The latter will recover the proton and regenerate the anion from the ionic liquid to produce the coupling adduct. The pKa of proline (10.64) and the hydroxyproline (9.65) led to suitable basic media when ionic liquids 1–9 were used. The anions of the ionic liquids played the role of the base. The whole ionic liquids played the crucial role of phase transfer agents as explained by Ceccarelli and et al. in 2006 [
56].
Concerning the enantioselectivity of the reaction, all coupling compounds were racemic. The chiral anion of the ionic liquids seemed not to influence the protonation of the enolate species. The presence of residual water in the ILs could also explain the lack of enantioselectivity. The size of our anions was also relatively small contrary to the PCT agents used by Mahajan and al., leading to good enantiomeric excesses in similar Michael’s additions [
57].
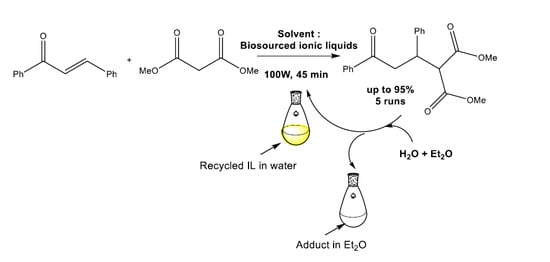
The final work was to prove the possibility of recycling our ILs in the studied Michael’s addition. One of the advantages of ionic liquids is the possibility to recycle them. We studied this possibility with the (
S)-prolinate TBA. We chose this ionic liquid and not the
trans-4-hydroxy-
S-prolinate TBA, which furnished the best conversions because the yields of the first run were high (90%), and secondly, the price of the counter anion (
S-prolinate) was lower than the
trans-4-hydroxy-
S-prolinate one. The procedure is described in
Scheme 5.
The chalcone conversions for the three first cycles were good (81 to 94%). Then, we observed a loss of activity of the reacting medium, probably due to the loss of the anion, which plays a basic role in the Michael’s reaction mechanism and led to the amino acid by protonation. This hypothesis has been confirmed by NMR spectrum of the regenerated IL where a slight modification of its structure has been observed (See
Table 5).
3. Experimental
All reagents were commercially available and used as received. Solvents were dried and distilled under argon before use (CH2Cl2 over CaCl2 and THF over sodium/benzophenone) and stored over molecular sieves. 1H and 13C NMR spectra were recorded on an AC 250 Bruker in CD3OD for 1H and 13C spectra. The infrared spectra were recorded with a Spectrafile IRTM Plus MIDAC. Chromatography was carried out on an SDS Silica 60 (40–63 μm), Art 2050044 (flash-chromatography), or silica 60 F254 (TLC plates).
The GC analyses were recorded on a Hewlett Packard 6890 Series. The conditions used are specified in this
Table 6.
3.1. Synthesis of Phosphonium Based Ionic Liquids-General Procedure [21d]
In a 100 mL two-neck round-bottom flask, one equivalent of a 40% wt aqueous solution of tetrabutylphosphonium hydroxide TBP
+OH
− (30 mmol, 21 mL) and 1.2 equivalents of (
R), (
S)-proline or
trans-4-hydroxy-
S-proline (36 mmol) previously dissolved in 40 mL distilled water were stirred at 100 °C during 24 h. After cooling, the solvent was evaporated, and the resulting mixture was washed with ethyl acetate (4 × 80 mL) to remove the small excess of amine. Finally, the aqueous phase was evaporated.
| tetrabutylphosphonium (S)-prolinate | ![Catalysts 10 00814 i001]() |
3.1.1. General Procedure with 4.14 S-proline (36 mmol). Yield: 78%
δ
H (250.1 MHz; D
2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H
4); 1.31 (m, 16H, H
2 and H
3); 1.6 (m, 3H, H
2′ and H
3′a); 2 (m, 1H, H
3′b); 2.48 (m, 8H, H
1); 2.7 (m, 1H, H
1′a); 2.95 (m, 1H, H
1′b); 3.4 (m, 1H, H
4′). δ
C (62.5 MHz; D
2O) ppm: 13.4 (C4); 18.4 (C1); 23.6 (C2); 23.9 (C1); 25.5 (C2′); 30.9 (C3′); 46.7 (C1′); 62 (C4′); 177.9 (C = O). IR: ν (cm
−1) 1746 (C = O). Analysis: calculated for C
21H
44NO
2P: C 67.52; H 11.87; N 3.75%. Found: C 66.98; H 11.38; N 3.42%.
| tetrabutylphosphonium (R)-prolinate | ![Catalysts 10 00814 i002]() |
3.1.2. General Procedure with 4.14 g (R)-proline (36 mmol). Yield: 89%
δ
H (250.1 MHz; D
2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H
4); 1.32 (m, 16H, H
2 and H
3); 1.6 (m, 3H, H
2′ and H
3′a); 2 (m, 1H, H
3′b); 2.48 (m, 8H, H
1); 2.7 (m, 1H, H
1′a); 2.95 (m, 1H, H
1′b); 3.4 (m, 1H, H
4′). δ
C (62.5 MHz; D
2O) ppm: 13.4 (C4); 18.4 (C1); 23.6 (C2); 23.9 (C1); 25.5 (C2′); 30.9 (C3′); 46.7 (C1′); 62 (C4′); 177.9 (C=O). IR: ν (cm
−1) 1746 (C = O). Analysis: calculated for C
21H
44NO
2P: C 67.52; H 11.87; N 3.75%. Found: C 67.0; H 11.48; N 3.52%.
| tetrabutylphosphonium trans-4-hydroxy-(S)-prolinate | ![Catalysts 10 00814 i003]() |
3.1.3. General Procedure with 4.72 g Trans-4-hydroxy-(S)-prolinate (36 mmol). Yield: 93%
δH (250.1 MHz; D2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H4); 1.31 ppm (m, 16H, H2 and H3); 1.50 (m, 1H, H3′b); 1.71 (m, 1H, H3′a); 2.4 (m, 1H, H1′a); 2.92 (dd, J = 12.3; 5.1 Hz, H1′b); 3.21 (m, 8H, H1); 3.41 (m, 1H, H2′); 4.00 (m, 1H, H4′). δC (62.5 MHz; D2O) ppm: 13.4 (C4); 19.5 (C3); 24.4 (C2); 41.3 (C3′); 55.4 (C1′); 58.7 (C1); 61.9 (C4′); 72.2 (C2′); 177.7 (C=O). IR: ν (cm−1) 1746 (C = O). Analysis: calculated for: C21H44NO3P: C 64.75; H 11.38; N 3.60%. Found: C 64.39; H 10.98; N 3.32%.
3.2. Synthesis of Ammonium Based Ionic Liquids-General Procedure [21c,e]
In a 100 mL two-neck round-bottom flask, one equivalent of a 40% wt aqueous solution of tetrabutylammonium hydroxide TBA
+OH
− (30 mmol, 19.6 mL) and 1.2 equivalents of (
R), (
S)-proline or
trans-4-hydroxy-
S-proline (36 mmol) previously dissolved in 40 mL distilled water were stirred at 100 °C during 24 h. After cooling, the solvent was evaporated, and the resulting mixture was washed with ethyl acetate (4 × 80 mL) to remove the small excess of amine. Finally, the aqueous phase was evaporated.
| Tetrabutylammonium (S)-prolinate | ![Catalysts 10 00814 i004]() |
3.2.1. General Procedure with 4.14 S-proline (36 mmol). Yield: 85%
δ
H (250.1 MHz; D
2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H
4); 1.32 (m, 8H, H
3); 1.55 (m, 8H, H
2); 1.60 (m, 3H, H
2′ and H
3′b); 2.00 (m, 1H, H
3′a); 2.70 (m, 1H, H
1′a); 2.9 (m, 9H, H
1 and H
1′b); 3.40 (m, 1H, H
4′). δ
C (62.5 MHz; D
2O) ppm: 14.1 (C4); 20.6 (C3); 24.7 (C2); 26.7 (C2′); 32.2 (C3′); 47.6 (C1′); 59.4 (C1); 63.04 (C4′); 180.9 (C=O). IR: ν (cm
−1) 1743 (C = O). Analysis: calculated for: C
21H
44N
2O
2: C 70.73; H 12.44; N 7.86%. Found: C 70.29; H 11.98; N 7.37%.
| Tetrabutylammonium (R)-prolinate | ![Catalysts 10 00814 i005]() |
3.2.2. General Procedure with 4.14 R-proline (36 mmol). Yield: 78%
δ
H (250.1 MHz; D
2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H
4); 1.32 (m, 8H, H
3); 1.55 (m, 8H, H
2); 1.60 (m, 3H, H
2′ and H
3′b); 2.00 (m, 1H, H
3′a); 2.70 (m, 1H, H
1′a); 2.9 (m, 9H, H
1 and H
1′b); 3.40 (m, 1H, H
4′).
| tetrabutylammonium trans-4-hydroxy-(S)-prolinate | ![Catalysts 10 00814 i006]() |
3.2.3. General Procedure with 4.72 Trans-4-hydroxy-(S)-prolinate (36 mmol). Yield: 85%
δH (250.1 MHz; D2O) ppm: 0.89 (t, J = 7.3 Hz, 12H, H4); 1.31 ppm (m, 16H, H2 and H3); 1.5 (m, 1H, H3′b); 1.7 (m, 1H, H3′a); 2.4 (m, 1H, H1′a); 2.9 (dd, J = 12.2; 5.1 Hz, H1′b); 3.20 (m, 8H, H1); 3.41 (m, 1H, H2′); 4.00 (m, 1H, H4′). δC (62.5 MHz; D2O) ppm: 13.5 (C4); 19.9 (C3); 24.0 (C2); 41.3 (C3′); 55.4 (C1′); 58.7 (C1); 61.9 (C4′); 72.2 (C2′); 177.7 (C=O). IR: ν (cm−1) 1741 (C = O). Analysis: calculated for: C21H44N2O3: C 67.70; H 11.90; N 7.52%. Found: C 67.31; H 11.48; N 7.23%.
3.3. Synthesis of Cholinium Based Ionic Liquid
In a 100 mL Bicol, a mixture of 46% wt aqueous solution of cholinium hydroxide (1 eq. 30 mmol, 7.37 mL) and proline (1.2 eq., 36 mmol, 14 g) previously dissolved in 40 mL distilled water was stirred under reflux for 24 h at 100 °C. After cooling, the mixture was evaporated, washed (4 × 80 mL) with ethyl acetate, and finally, the aqueous phase was evaporated.
cholinium-(S)-prolinate
Yield: 93%
Decomposition temperature: 230 °C | ![Catalysts 10 00814 i007]() |
δ
H (250.1 MHz; D
2O) ppm: 1.60 (m, 3H, H
2′ and H
3′a); 2.00 (m, 1H, H
3′b); 2.70 (m, 1H, H1′a); 2.80 (m, 1H, H
1′b); 3.11 (s, 9H, H
1); 3.30 (t, 2H, H
2); 3.45 (t, 1H, H
4′); 3.80 (t, 2H, H
3). δ
C (62.5 MHz; D
2O) ppm: (62.5 MHz; D
2O): 22.8 (C2′); 32.3 (C3′); 46.5 (C1′); 55.7 (C1); 64.2 (C3); 67.5 (C4′); 70.2 (C2); 180.4 (C=O). IR: ν (cm
−1) 1741 (C = O). Analysis: calculated for: C
10H
22N
2O
3: C 55.02; H 10.16; N 12.83%. Found: C 54.78; H 9.78; N 12.47%.
cholinium-(R)-prolinate
Yield: 74%
Decomposition temperature: 230 °C | ![Catalysts 10 00814 i008]() |
δ
H (250.1 MHz; D
2O) ppm: 1, 60 (m, 3H, H
2′and H
3′b); 2.00 (m, 1H, H
3′b); 2, 70 ppm (m, 1H, H
1′a); 2.80 (m, 1H, H
1′b); 3.10 (s, 9H, H
1); 3.30 (t, 2H, H
2); 3.45 (s, 1H, H
4′); 3.80 ppm (t, 2H, H
3). δ
C (62.5 MHz; D
2O) ppm: 22.6 (C2′); 32.3 (C3′); 46.7 (C1′); 55.4 (C1); 64.9 (C3); 67.3 (C4′); 70.7 (C2); 180.3 (C=O). Analysis: calculated for: C
10H
22N
2O
3: C 55.02; H 10.16; N 12.83%. Found: C 54.89; H 9.78; N 12.36%.
cholinium-trans-4-hydroxy-(S)-prolinate
Yield: 82%
Decomposition temperature:222 °C | ![Catalysts 10 00814 i009]() |
δH (250.1 MHz; D2O) ppm: 1.50 (m, 1H, H3′b); 1.70 (m, 1H, H3′b); 2,40 ppm (m, 1H, H1′a); 2.90 (dd, J = 12.2 Hz, 5.1 Hz,1H, H1′b); 3.10 (s, 9H, H1); 3.40 (m, 3H, H2′); 3.45 (s, 2H, H3); 3.80 ppm (t, J = 3.43 Hz, 2H, H2), 4 (m, 1H, H4′). δC (62.5 MHz; D2O) ppm: 41.3 (C3′); 54.6(C1′); 55.4 (C1); 61.9 (C4′); 64.9 (C3); 70.2 (C2); 72.2 (C2′); 177.7 (C=O). IR: ν (cm−1) 1738 (C = O). Analysis: calculated for: C10H22N2O4: C 51.26; H 9.46; N 11.96%. Found: C 51.49; H 9.58; N 11.66%.
3.4. Michael Addition Procedures
3.4.1. General Procedure without Ionic Liquids
In a Schlenk tube, the chalcone (1 eq., 2 mmol, 0.42 g), the dimethyl malonate (1.2 eq or 4 eq.) and K2CO3 (1.2 eq.) were mixed in DMF (10 mL). The reaction mixture was stirred for 24 h at 50 °C. The reaction was stopped by adding 15 mL of ice water with the formation of a white precipitate that corresponds to the coupling product. This precipitate was dissolved in diethyl ether. Extractions with diethyl ether were carried out (3 × 60 mL). The organic phases were then dried over sodium sulfate and then evaporated. The resulting compound was purified by chromatography (silica and eluent: Petroleum ether/Ethyl acetate (7/3)).
3.4.2. General Procedure in Ionic Liquids
In a Schlenk tube, 1.2 eq. of ionic liquid (2.4 mmol) was introduced and placed under vacuum for 10 min. Then at atmospheric pressure, 1 eq. of chalcone (2 mmol, 0.42 g) and an excess of dimethyl malonate (4 eq.) were dissolved in DMF (2 mL) in order to fluidify the mixture. The reaction mixture was stirred for 24 h at 50 °C and in the presence of argon. The reaction was stopped by adding 15 mL of ice water. We observed the formation of a white precipitate that corresponded to our product. The latter was dissolved by adding diethyl ether. Extractions with diethyl ether were carried out (3 × 60 mL). The organic phases were then dried over sodium sulfate and then evaporated. The resulting compound was purified by chromatography (silica and eluent: Petroleum ether / Ethyl acetate (7/3)). Finally, the aqueous phase was also evaporated to recover the ionic liquid.
2-(3-oxo-1,3-diphenylpropyl) dimethylmalonate [58]
C20H20O5
White Powder | ![Catalysts 10 00814 i010]() |
δH (500 MHz; CDCl3) ppm: 3.50 (m, 4H, H1b and OMe); 3.55 (dd, 1H, H1a); 3.75 (s, 3H, OMe); 3.85 (d, 1H, H3); 4.25 (m, 1H, H2); 7.15 (m, 1H, Harom); 7.25 (m, 4H, Harom); 7.43 (t, 2H, Harom); 7.50 (m, 1H, Harom); 7.85 (m, 2H, Harom). IR: ν (cm−1) 1735 (C = OEster), 1715 (C = OKetone).
3.4.3. General Procedure in Ionic Liquids under Microwaves
In a balloon, 1.2 eq. of ionic liquid (2.4 mmol) was introduced and placed under vacuum for 10 min. 1 eq. of chalcone (2 mmol, 0.42 g) and an excess of dimethyl malonate (4 eq.) was added, and the mixture was stirred for 45 min under 100 W. The extraction of the Michael’s adduct and the recycling of the ILs was as previously described for the reaction under thermic conditions.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














