Abstract
The subject of the research was to determine the ability of the filamentous fungi to biotransform bicyclic halolactones containing two methyl groups in their structure. By chemical synthesis three bicyclic halolactones with two methyl groups, one in the cyclohexane ring and one in the lactone ring, were obtained: 2-chloro-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one, 2-bromo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one, and 2-iodo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one. These compounds were formed as mixtures of two diastereoisomers. The obtained halolactones (as mixture of two diastereoisomers) were subjected to screening biotransformation with the use of eight strains of filamentous fungi: Fusarium culmorum AM10, F. avenaceum AM12, F. semitectum AM20, F. solani AM203, Absidia coerulea AM93, A. cylindrospora AM336, Penicillium chermesinum AM113, P. frequentans AM351. Two of the substrates, 2-bromo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one and 2-iodo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one, were hydroxylated without removing the halogen atom from the molecule, giving 2-bromo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one, 2-bromo-5-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one, and 2-iodo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one as products. The hydroxylation capacity was demonstrated by strains of Absidia cylindrospora AM336, Fusarium avenaceum AM12, and F. solani AM203. The structures of all lactones were determined on the basis spectroscopic data.
1. Introduction
Biotransformations are a useful tool for obtaining interesting new derivatives of organic compounds. They can be used as an alternative to classical chemical synthesis [1,2,3,4,5]. The undoubted advantage of this method is the relatively low cost and the possibility of conducting transformations of compounds in mild conditions. An equally important aspect is the possibility of obtaining new compounds, which are not available through classical synthesis or require the use of specific catalysts. Such catalysts can be biocatalysts, which include whole cells of filamentous fungi or yeast or enzymes isolated from them. Biotransformations using whole fungal cells are often used to transform bicyclic lactones. These are usually lactones containing in their structure, apart from methyl groups, halogen atoms, or double bond. The most common products of such reactions are hydroxyl derivatives of lactones [6,7,8,9,10,11,12].
Hydroxylation reactions are mainly catalyzed by monooxygenases of cytochrome P450 (CYP). These enzymes are present in all kingdoms of life, with fungi being particularly diverse in terms of CYPs. Using data from genome sequencing projects, more than 300,000 CYPs have been identified to date [13]. Diversification of their forms results from the key function of these enzymes in the biosynthesis of primary and secondary metabolites, but primarily in the detoxification and biodegradation of xenobiotics [14,15].
However, hydroxylation reactions can also be catalyzed by unspecific peroxygenases (UPO), which are also widespread in the fungal kingdom [16]. The analysis of the genomes of basidiomycetes and saccharides revealed over a thousand putative peroxygenase genes [17].
In our research, we used lactones containing halogen atoms in the molecule of substrates. Previous studies carried out so far showed [18,19,20,21,22,23,24] that filamentous fungi, especially those belonging to the genera Fusarium and Absidia, can carry out the hydrolytic dehalogenation of halolactones. As a result, hydroxylactones with a different conformation than halolactones obtained by chemical synthesis were obtained. It is also possible to obtain halo-hydroxylactones, compounds in which the halogen atom remains intact, and additionally a hydroxyl group is introduced [20,25,26]. It is worth emphasizing that, due to the variety of enzymes capable of catalyzing the hydroxylation of xenobiotics, it is not possible to predict the position of hydroxylation without experimental basic research. Additionally, the disadvantage of isolated hydroxylases are their instability and the requirement to use expensive cofactors or auxiliary enzymes [16].
2. Results and Discussion
2.1. Obtaining of Substrates for Biotransformation and Analysis of Their Structures
The known 5-methylcyclohex-2-en-1-ol 1 [27] was used as a substrate to obtain of three halolactones. Alcohol 1 was subjected in the first stage the reaction of Claisen’s rearrangement with orthopropionate modification [28]. Ethyl ester of (4,7-dimethylcyclohex-2-en-1-yl)acetic acid 2 was obtained in the form of a pair of diastereoisomers, designated A and B, in a ratio of 41%:59%, respectively. The formation of ester 2 in the form of two diastereoisomers results from presence of additional methyl group in the side chain. Ethyl ester of (4,7-dimethylcyclohex-2-en-1-yl)acetic acid 2 was then subjected to alkaline hydrolysis to obtain (4,7-dimethylcyclohex-2-en-1-yl)acetic acid 3, also occurring as a pair of diastereoisomers A and B, in a ratio of 41%:59%. In the next step, 2-chloro-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one (chlorolactone 4) 2-bromo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one (bromolactone 5) and (2-iodo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one (iodolactone 6), were obtained from acid 3. These lactones were also mixtures of diastereoisomers A and B in the proportion of 54%:46% (chlorolactone 4), 48%:52% (bromolactone 5), and 47%:53% (iodolactone 6), respectively. The scheme of halolactone synthesis is presented in Figure 1.
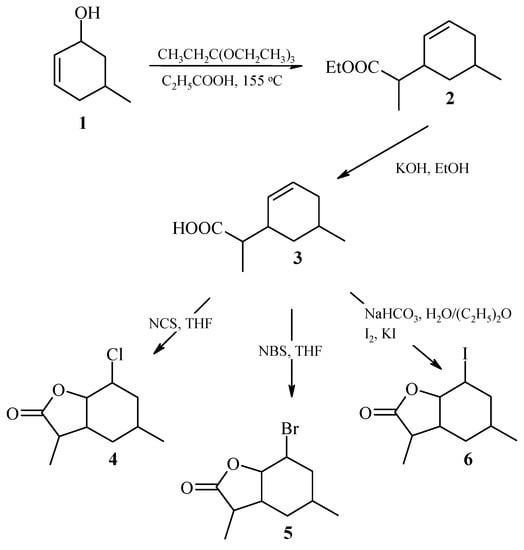
Figure 1.
Synthesis of (±)-halolactones 4–6.
The analysis of 1H NMR spectra of both isomers A and B of ester 2 and acid 3 showed that in both cases, the methyl group located at C-5 carbon took an equatorial position. This was indicated by the signals from H-5 protons of both A and B isomers, being wide multiplets in the range of 2.45–2.58 ppm (ester 2) and 2.53–2.63 ppm (acid 3). The side chain also occupied an equatorial position, as evidenced by wide multiplets in the range 1.97–2.06 and 1.99–2.09 ppm derived from H-1 protons of both isomers A and B ester 2 and acid 3, respectively (Figure 2).
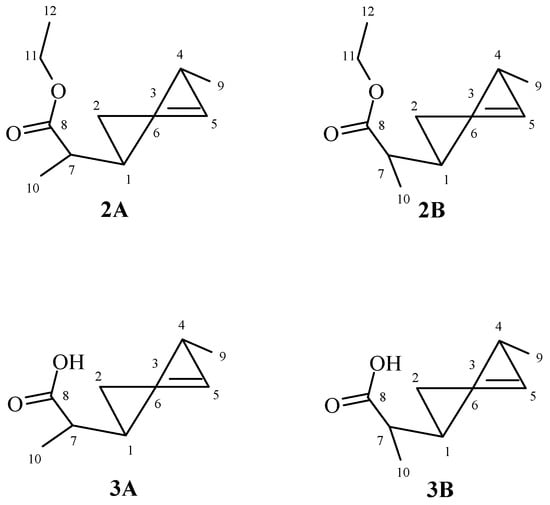
Figure 2.
The structures of ester 2 and acid 3.
Analysis of 1H NMR spectra of both diastereoisomers A and B chloro-4, bromo-5, and iodolactone 6 allowed to conclude that their spatial structures are similar. The cyclohexane ring was in the chair conformation. The methyl group lying on C-4 carbon took the equatorial position. This was evidenced by signals from H-4 protons belonging to both isomers, being broad multiplets located in the range of 1.92–1.99 ppm (chlorolactone 4), 1.95–2.04 ppm (bromolactone 5), and 1.97–2.02 ppm (iodolactone 6). Signals coming from the H-1B protons formed narrow multiplets in the range of 4.35–4.39 ppm (chlorolactone 4), 4.48–4.51 ppm (bromolactone 5), and 4.58–4.60 ppm (iodolactone 6), which indicated their equatorial orientation. In turn, in the range of 4.49–4.54, 4.60–4.65, and 4.78–4.81 ppm, there were narrow multiplets originating from H-2A and H-2B protons, indicating their equatorial orientation. The H-1A protons from all three lactones 4–6 also occupied an equatorial position, as indicated by the narrow multiplets lying at 4.57–4.59, 4.69–4.72, and 4.78–4.81 ppm, respectively. The equatorial position of the H-1 and H-2 protons causes the axial position to be taken by both the C-O bond of the lactone ring and the halogen atom in both A and B isomers of lactones 4–6. The signals derived from the protons H-6A and H-6B were broad multiplets. They were in the ranges of 2.32–2.37, 2.39–2.44, and 2.46–2.53 ppm for the A isomer (compounds 4, 5, and 6, respectively). For isomer B, these signals were at 2.67 (dddd, J = 12.4, 10.0, 6.0 and 4.0 Hz), 2.70–2.76, and 2.81–2.89 ppm (compounds 4, 5, and 6, respectively). This means that in both isomers A and B, the C-C bond of the lactone ring assumed the equatorial orientation.
The comparison of the chemical shifts of the signals coming from the H-7A and H-7B protons allowed to determine the arrangement of the CH3-10 methyl group in the lactone ring. In the case of the A isomer of lactones 4, 5, and 6, the signal coming from the H-7 proton was at 2.34, 2.37, and 2.33 ppm, respectively. For the B isomer of lactones 4, 5, and 6, the signals from the H-7 proton were at 2.77, 2.77, and 2.78 ppm, respectively. This means that the H-7 protons of the A isomers was lying across the plane of the lactone ring, and the H-7 protons of the B isomers—in the plane of the lactone ring. This implied the appropriate position of the CH3-10 methyl group in the plane of the lactone ring (isomer A) and across the plane of the lactone ring (isomer B) (Figure 3).
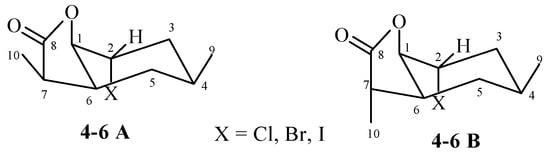
Figure 3.
The structures of halolactones 4–6.
2.2. Screening Biotransformations of Halolactones 4–6
Filamentous fungi strains belonging to the collections of the Department of Chemistry: Fusarium culmorum AM10, F. avenaceum AM12, F. semitectum AM20, F. solani AM203, Absidia coerulea AM93, A. cylindrospora AM336, Penicillium chermesinum AM113, and P. frequentans AM351 were used for screening biotransformations. The substrates were bicyclic halolactones 4–6 obtained by chemical synthesis. The duration of biotransformation was 7 days. The results are presented in Table 1.

Table 1.
Results obtained from screening biotransformation of lactones 4–6 after 7 days of incubation (in percentage according to GC).
Analyzing the results presented in Table 1, it can be concluded that the type of halogen was especially important for the course of transformation. Chlorolactone 4 was not transformed by any of the eight strains. Iodolactone 6 was converted to one derivative—2-jodo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 9, and bromolactone 5 to two derivatives—2-bromo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 7 and 2-bromo-5-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 8. These products were obtained as a result of transformation carried out with A. cylindrospora AM336 strain. The formation of lactone 8 from lactone 5 was also observed in the cultures of F. avenaceum AM12 and F. solani AM203 strains.
2.3. Preparative Biotransformation of Halolactones 5 and 6. Analysis of the Structures of Obtained Derivatives
After analyzing the results of the screening tests, it was found that that the microorganism transforming substrates with the highest efficiency was strain A. cylindrospora AM336. In order to determine the structure of the resulting products, a preparative biotransformation was carried out using strain A. cylindrospora AM336 as a biocatalyst. Bromolactone 5 and iodolactone 6 were used as substrates in this stage. The results of the preparative biotransformation are presented in Table 2.
Table 2.
Results obtained from preparative scale biotransformation of lactones 5 and 6 by A. cylindrospora AM336 after 7 days of incubation (in percentage according to GC).
The biotransformation resulted in three new lactones. Two of them, compounds 7 and 8, were the products of hydroxylation of bromolactone 5, while compound 9 was formed by hydroxylation of iodolactone 6. For substrates (lactones 4–6), two peaks were observed on GC chromatograms for each compound, corresponding to two diastereoisomers A and B. In the case of biotransformation products (lactones 7–9), only single peaks were observed. Analysis of the NMR spectra confirmed that each of the biotransformation products was one compound. Therefore, optical rotation of lactones 7–9 was measured. In all three cases, the values obtained were different from zero. In the next step, the enantiomeric excesses of these compounds were determined using a column with chiral filling (Supplementary Materials, Figures S41–S43). The retention times of the analyzed compounds were consistent with our previous experience with structurally similar compounds [20,26]. The results are presented in Table 3.
Table 3.
The values of enantiomeric excesses and optical rotation of halo-hydroxylactones 7–9.
Analyzing the data presented in Table 3, the enantioselectivity of the hydroxylation process is dependent on the place where the hydroxyl group was introduced. In case of hydroxylation of secondary C-5 carbon being a fragment of cyclohexane ring, the process was highly enantioselective. On the other hand, when the hydroxyl group was introduced to the tertiary C-7 carbon fragment, a part of the lactone ring, the products were characterized by a slight enantiomeric excess.
Analysis of the 1H NMR spectra of the obtained biotransformation products showed that strain A. cylindrospora AM336 preferred hydroxylation, while the hydrolytic dehalogenation characteristic for filamentous fungi was not observed. In the case of bromolactone 5, hydroxylation of carbon C-7 in the lactone ring (compound 7) or C-5 carbon in the cyclohexane ring (compound 8) took place. In the case of iodolactone 6, the hydroxyl group was introduced into the lactone ring only (compound 9). The structures of the obtained compounds are presented in Figure 4.
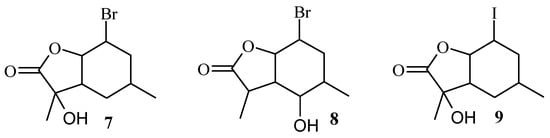
Figure 4.
The structures of halo-hydroxylactones 7–9.
Analysis of the 1H NMR spectra of the biotransformation products showed that these compounds were formed from the B isomer of bromolactone 5 or iodolactone 6. In the case of bromolactone, it was observed that tertiary carbon C-7 (lactone 7) or secondary carbon C-5 (lactone 8) were hydroxylated, while in the case of iodolactone, only the carbon C-7 (lactone 9) was hydroxylated. In all three lactones, the chair conformation of the cyclohexane ring was preserved. The H-6 proton retained its axial orientation, while H-1 and H-2 protons retained their equatorial orientation. The disappearance of the signal from the H-7 proton observed in the NMR spectra of compounds 7 and 9 indicates hydroxylation of C-7 carbon. It is known from previous considerations that in the case of the B isomer, the CH3-10 methyl group lied across the plane of the lactone ring. This means that the hydroxyl group in products 7 and 9 was introduced into an equatorial position. In the case of lactone 8, the hydroxyl group was also introduced into an equatorial position, but this time on the C-5 carbon. This was evidenced by the position of the signal coming from the H-5 proton lying at 2.97 ppm and its shape (a triplet with a coupling constant of 10.0 Hz) and the differences in the position of the H-1 proton and the CH3-10 methyl group, which shifted towards the lower field as well as H-6 proton that shifted towards the higher field.
The formation of halo-hydroxylactones during biotransformation is not common. During our previous studies, the formation of halo-hydroxylactones was observed in two cases only. This was the case when halolactones with one methyl group located at C-6 carbon were used as substrates and the microorganism was the filamentous fungus Penicillium vermiculatum AM30 [20]. The second time, the microorganisms inducing the formation of such compounds were edible fungi of the genus Pleurotus ostreatus. At that time, the only effectively transformed substrate was chlorolactone with four methyl groups, two at C-4 carbon and one each at C-6 and C-7 carbon [26].
Based on our experience with the biotransformation of halolactones with whole cells of filamentous fungi, the formation of products such as hydroxylactone depends on the type of halogen. Typically, filamentous fungi slightest transforms lactones with chlorine atom in the molecule. The lack of products derived from chlorolactone 4 was, therefore, not surprising. Bromo- and iodolactones are much more frequently transformed by filamentous fungi [19,20,21,22]. Such a dependence can be seen here as well. Three strains were able to hydroxylate bromolactone 5: A. cylindrospora AM336, F. avenaceum AM12, and F. solani AM203, and for hydroxylation of iodolactone 6—only strain A. cylindrospora AM336. It should be emphasized that out of two diastereoisomers of halolactones 4–6, only one from them was hydroxylated, the B isomer. This means that the presence of a methyl group in the lactone ring has a great influence on the course of biotransformation. The presence of this methyl group prevents the process of hydrolytic dehalogenation, which was observed many times during the biotransformation of halolactones with Fusarium fungi. In the situation discussed here, the A isomer remained unreacted, only the B isomer, in which the methyl group lies across the plane of the lactone ring, was hydroxylated. This orientation of the methyl group allows the hydroxyl group to approach in the plane of the lactone ring. According to our experience, the hydroxyl group introduced into the cyclohexane ring of the lactone molecule usually took the equatorial position. This trend has been maintained here as well.
3. Materials and Methods
3.1. General Methods
The progress of subsequent steps of synthesis and the course of biotransformation was analyzed on GC on CP-3380 instrument (Varian, Agilent Technologies, Santa Clara, CA, USA) using DB-1 column (cross-linked methyl silicone gum 30 m × 0.25 mm × 0.25 µm). The following temperature program was used: injector 250 °C and detector (FID) 300 °C, initial column temperature 140 °C (hold for 1 min), ramp 140–180 °C at 15 °C/min, ramp 180–300 °C at 40 °C/min and hold 1 min at 300 °C. For purification of products obtained from the synthesis and biotransformation reactions, column chromatography was used, using as a carrier silica gel (Kieselgel 60, grid 230–400) and as an eluent a mixture of hexane and acetone in a ratio of 3:1. The enantiomeric excess was analyzed on GC on 6890N instrument (Agilent Technologies, Santa Clara, CA, USA) using a chiral column CP-Chirasil-B-325 (25 m × 0.25 mm × 0.25 μm, Supelco, Bellefonte, PA, USA). The temperature program for lactones 7 and 9 was as follows: injector 200 °C, detector (FID) 200 °C, initial column temperature 140 °C, ramp 140–200 °C at the rate of 1.0 °C/min, and hold at 200 °C for 1 min. For lactone 8, a slightly different temperature program was used: 200 °C injector, 200 °C detector (FID), 140 °C initial column temperature, ramp 140–160 °C at the rate of 0.5 °C/min, ramp 160–200 °C at the rate of 20 °C/min, and hold at 200 °C for 1 min. To confirm the molar masses of the compounds obtained, a high-resolution mass spectrometry analysis was performed using the Waters LCT Premier XE instrument (ESI ionization, Waters, Milford, MA, USA). The NMR spectra were recorded on the JEOL DeltaTM 400 MHz spectrometer (JEOL USA, Inc., Peabody, MA, USA) or on the Bruker AvanceTM 600 MHz spectrometer, using CDCl3 as a solvent. To determine the optical rotation of biotransformation products, the Jasco P-2000 polarimeter (Jasco, Easton, PA, USA) was used. Measurements were performed for chloroform solutions, in concentrations expressed in g/100 mL.
3.2. Organic Synthesis
Ethyl ester of (4,7-dimethylcyclohex-2-en-1-yl)acetic acid 2A + 2B
The known allylic alcohol 1 [27] was used as a starting substrate. This compound was subjected to Claisen rearrangement with orthopropionate modification [28]. Briefly, 3.0 g, 0.027 mol of alcohol 1 was heated with 18 mL, 0.1 mol of triethyl orthopropionate and catalytic amount of propionic acid. After completion of the reaction (30 h), the product was purified by column chromatography giving 3.1 g (59%) of ester 2 as a pair of diastereoisomers A and B with the following spectral properties: 1H NMR (400 MHz, CDCl3): 0.93 (d, J = 3.2 Hz, 3H, CH3-9A), 0.94 (d, J = 3.2 Hz, 3H, CH3-9B), 1.08 (d, J = 7.2 Hz, 3H, CH3-10A), 1.11 (d, J = 7.2 Hz, 3H, CH3-10B), 1.24 (t, J = 7.2 Hz, 3H, CH3-12B), 1.25 (t, J = 6.8 Hz, 3H, CH3-12A), 1.51–1.58 (m, 2H, H-4A, H-4B), 1.59–1.72 (m, 4H, CH2-6A, CH2-6B), 1.97–2.06 (m, 2H, H-1A, H-1B), 2.32 (quintet, J = 7.2 Hz, 2H, CH2-7B), 2.36 (quintet, J = 7.2 Hz, 2H, CH2-7A), 2.45–2.58 (m, 2H, H-5A, H-5B), 4.08–4.16 (m, 4H, CH2-11A and CH2-11B), 5.40–5.43 (dm, J = 10.0 Hz, 1H, H-2A), 5.53–5.57 (dm, J = 10.0 z, 1H, H-2B), 5.67–5.73 (m, 2H, H-3A, H-3B),13C NMR (100 MHz, CDCl3): 13.19 (C-10B), 13.46 (C-10A), 13.38 (C-12A and C-12B), 22.44 (C-9A), 22.51 (C-9B), 28.99 (C-6B), 29.12 (C-6A), 34.02 (C-1B), 34.09 (C-1A), 34.32 (C-4B), 36.04 (C-4A), 39.47 (C-5B), 39.70 (C-5A), 44.05 (C-7A), 44.36 (C-7B), 60.20 (C-11B), 60.24 (C-11A), 127.98 (C-3B), 128.22 (C-3A), 128.48 (C-2A), 129.34 (C-2B), 176.05 (C-8A), 176.11 (C-8B), ESIHRMS: calcd for C12H20O2, m/z 197.1542 (M + H)+, found 197.1534. (Supplementary Materials, Figures S1–S5).
(4,7-dimethylcyclohex-2-en-1-yl)acetic acid 3A + 3B
Basic hydrolysis of the obtained mixture of two diastereoisomers A and B of ester 2 was carried out according to the procedure described previously [27]. Briefly, 3.1 g of ester 2 was heated with 5% potassium hydroxide solution in methanol for 4 h. After evaporation of methanol, the residue was dissolved in water and acidified with 1 molar hydrochloric acid, and the product was extracted with diethyl ether. Then, 1.9 g (yield 72%) of acid 3 also as two diastereoisomers A and B was obtained. The spectral data of acid are as follows: 1H NMR (400 MHz, CDCl3): 0.94 (d, J=10.2 Hz, 3H, CH3-9B), 0.96 (d, J = 9.6 Hz, 3H, CH3-9A), 1.11 (d, J = 10.2 Hz, 3H, CH3-10A), 1.15 (d, J = 10.2 Hz, 3H, CH3-10B), 1.54–1.61 (m, 2H, H-4A, H-4B), 1.64–1.71 (m, 4H, CH2-6A, CH2-6B), 1.99–2.09 (m, 2H, H-1A, H-1B), 2.35–2.48 (m, 2H, H-7A, H-7B), 2.53–2.63 (m, 2H, H-5A, H-5B), 5.44–5.47 (dm, J = 0.0 Hz, 1H, H-2A), 5.55–5.58 (dm, J = 10.4 z, 1H, H-2B), 5.70–5.77 (m, 2H, H-3A, H-3B),13C NMR (100 MHz, CDCl3): 12.74 (C-10A), 13.18 (C-10B), 22.43 (C-9), 22.45 (C-9A), 28.95 (C-6A), 29.11 (C-6B), 34.00 (C-1B), 34.04 (C-1A), 36.05 (C-4A), 34.06 (C-4B), 39.22 (C-5A), 39.43 (C-5B), 43.77 (C-7A), 43.95 (C-7B), 127.56 (C-3B), 128.58 (C-3A), 128.90 (C-2B), 129.11 (C-2A), 181.31 (C-8B), 181.31 (C-8A), ESIHRMS: calcd for C10H16O2, m/z 169.1228 (M + H)+, found 169.1235. (Supplementary Materials, Figures S6–S10).
2-Chloro-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 4A +4B
Chlorolactonization of acid 3 was carried out according to the procedure of Grabarczyk and Białońska [29]. Briefly, 0.6 g, 0.0036 mol of THF solution of acid 3 was stirred with 1.1 g of N-chlorosuccinimide for 24 h. After completion of the reaction, product was extracted with diethyl ether giving 0.31 g (43%) of chlorolactone 2 as a pair of diastereoisomers A and B with the following physical and spectral properties: 1H NMR (400 MHz, CDCl3): 0.63 and 0.70 (two d, J = 12.4 Hz, 1H, one of CH2-5B), 0.75 and 0.81 (two d, J = 12.0 Hz, 1H, one of CH2-5A), 0.89 (d, J = 6.4 Hz, 3H, CH3-9A), 0.90 (d, J = 6.8 Hz, 3H, CH3-9B), 1.14 (d, J = 7.2 Hz, 3H, CH3-10B), 1.28 (d, J = 7.6 Hz, 3H, CH3-10A), 1.55–1.64 (m, 3H, one of CH2-3A, one of CH2-3B, one of CH2-5A), 1.72–1.76 (m, 1H, one of CH2-5B), 1.84–1.88 (m, 1H, one of CH2-3A), 1.88–1.91 (m, 1H, one of CH2-3B), 1.92–1.99 (m, 2H, H-4A, H-4B), 2.32–2.37 (m, 1H, H-6B), 2.34 (q, J = 7.2 Hz, 1H, H-7A), 2.67 (dddd, J = 12.4, 10.0, 6.0 and 4.0 Hz, 1H, H-6A), 2.77 (m, 1H, H-7B), 4.35–4.39 (m, 1H, H-1B), 4.49–4.54 (m, 2H, H-2B, H-2A), 4.57–4.59 (m, 1H, 1A), 13C NMR (100 MHz, CDCl3): 8.95 (C-10B), 14.05 (C-10A), 21.53 (C-9A), 21.70 (C-9B), 23.11 (C-4B), 23.40 (C-4A), 31.16 (C-5B), 36.00 (C-3A), 36.00 (C-3B), 36.18 (C-5A), 36.43 (C-6A), 38.72 (C-6B), 41.81 (C-7B), 44.66 (C-7A), 55.31 (C-2B), 55.34 (C-2A), 78.09 (C-1A), 78.29 (C-1B), 178.38 (C-8B), 179.16 (C-8A), ESIHRMS: calcd for C10H15ClO2, m/z 203.0839 (M + H)+, found 203.0832. (Supplementary Materials, Figures S11–S15).
2-Bromo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 5A + 5B
Bromolactonization of acid 1 was carried out according to the known method Grabarczyk and Białońska [29]. Briefly, 0.6 g (0.0036 mol) of tetrahydrofuran solution of acid 1 was stirred with 0.8 g N-bromosuccinimide for 24 h. After completion of the reaction product was extracted with diethyl ether giving 0.49 g (56%) of bromolactone 5 also as a pair of diastereoisomers A and B was obtained. The physical and spectral data of this products are as follows: 1H NMR (400 MHz, CDCl3): 0.65 and 0.72 (two d, J = 12.4 Hz, 1H, one of CH2-5B), 0.77 and 0.83 (two d, J = 12.4 Hz, 1H, one of CH2-5A), 0.91 (d, J = 6.4 Hz, 3H, CH3-9A), 0.94 (d, J = 6.4 Hz, 3H, CH3-9B), 1.15 (d, J = 7.2 Hz, 3H, CH3-10B), 1.28 (d, J = 7.6 Hz, 3H, CH3-10A), 1.59–1.68 (m, 3H, one of CH2-3A, one of CH2-3B, one of CH2-5A), 1.72–1.79 (m, 1H, one of CH2-5B), 1.92–1.94 (m, 2H, one of CH2-3A, one of CH2-3B), 1.95–2.04 (m, 2H, H-4A, H-4B), 2.37 (q, J = 7.6 Hz, 1H, H-7A), 2.39–2.44 (m, 1H, H-6B), 2.70–2.76 (m, 1H, H-6A), 2.77 (q, J = 7.2 Hz, 1H, H-7B), 4.48–4.51 (m, 1H, H-1B), 4.60–4.65 (m, 2H, H-2A, H-2B), 4.69–4.72 (m, 1H, 1A), 13C NMR (100 MHz, CDCl3): 8.97 (C-10B), 14.05 (C-10A), 21.48 (C-9A), 21.66 (C-9B), 24.14 (C-4B), 24.43 (C-4A), 31.25 (C-5B), 36.06 (C-5A), 36.27 (C-6A), 36.47 (C-3A), 36.47 (C-3B), 38.72 (C-6B), 42.11 (C-7B), 44.93 (C-7A), 47.81 (C-2A), 47.87 (C-2B), 78.43 (C-1A), 78.64 (C-1B), 178.56 (C-8B), 179.31 (C-8A), ESIHRMS: calcd for C10H15BrO2, m/z 247.0334 and 247.0331 (M + H)+, found 247.0331 and 247.0344, calcd for C10H15BrO2Na, m/z 269.0153 and 271.0133 (M + H)+, found 269.0144 and 271.0069. (Supplementary Materials, Figures S16–S20).
2-Iodo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 6A + 6B
Iodolactonization of acid 3 was carried out according to the known procedure [27]. Briefly, 0.6 g (0.0036 mol) of acid 3 dissolved in diethyl ether was stirred with 1 g of sodium bicarbonate for an hour, and then 1 g of iodine in 2 g of potassium iodide was added. After 24 h, product was extracted with diethyl ether giving 0.39 g (37%) of iodolactone 6 as a pair of diastereoisomers A and B with the spectral properties: 1H NMR (400 MHz, CDCl3): 0.67 and 0.74 (two d, J = 12.0 Hz, 1H, one of CH2-5B), 0.77 and 0.83 (two d, J = 12.0 Hz, 1H, one of CH2-5A), 0.93 (d, J = 6.8 Hz, 3H, CH3-9A), 0.96 (d, J = 6.8 Hz, 3H, CH3-9B), 1.14 (d, J = 6.8 Hz, 3H, CH3-10B), 1.27 (d, J = 7.6 Hz, 3H, CH3-10A), 1.42–1.48 (m, 2H, one of CH2-3A, one of CH2-3B), 1.62–1.64 (m, 1H, one of CH2-5B), 1.72–1.77 (m, 1H, one of CH2-5A), 1.88–1.94 (m, 2H, one of CH2-3A, one of CH2-3B), 1.97–2.02 (m, 2H, H-4A, H-4B), 2.33 (q, J = 7.6 Hz, 1H, H-7A), 2.46–2.53 (m, 1H, H-6B), 2.78 (q, J = 6.8 Hz, 1H, H-7B), 2.81–2.89 (m, 1H, H-6A), 4.58–4.60 (m, 1H, H-1B), 4.78–4.81 (m, 3H, H-1A, H-2A, H-2B), 13C NMR (100 MHz, CDCl3): 8.95 (C-10B), 14.00 (C-10A), 21.36 (C-9A), 21.54 (C-9B), 26.20 (C-4B), 26.52 (C-4A), 27.43 (C-2A), 27.56 (C-2B), 31.47 (C-5B), 36.27 (C-5A), 36.59 (C-6A), 37.24 (C-3B), 37.42 (C-3A), 38.82 (C-6B), 42.61 (C-7B), 45.39 (C-7A), 80.22 (C-1A), 80.45 (C-1B), 178.87 (C-8B), 179.55 (C-8A), ESIHRMS: calcd for C10H15IO2, m/z 295.0195 (M + H)+, found 295.0202. (Supplementary Materials, Figures S21–S25).
3.3. Biotransformation
3.3.1. Microorganisms
The biotransformations were carried out with the use of eight strains of filamentous fungi belonging to the own collection of the Department of Chemistry of Wrocław University of Life Sciences. These were Fusarium culmorum AM10, F. avenaceum AM12, F. semitectum AM20, F. solani AM203, Absidia coerulea AM93, A. cylindrospora AM336, Penicillium chermesinum AM113, P. frequentans AM351. These strains were cultured on Sabouraud’s agar (0.5 g aminobac, 0.5 g peptone, 4 g glucose, and 1.5 g agar dissolved in 100 mL of water) at 28 °C, and after growing, stored at 4 °C.
3.3.2. Screening Biotransformation
Erlenmeyer flasks with a capacity of 300 mL containing 100 mL of Sabouraud medium, consisting of 3 g of glucose and 1 g of peptone dissolved in 100 mL of water, were used for biotransformation. Three days after inoculation of the medium with the given microorganism, 10 mg of substrate (halolactone 4–6) dissolved in 1 mL of acetone was added to each of the flasks with the mycelium growth. Shaking cultures were incubated with the substrate for the next 7 days. After 3, 5, and 7 days, samples were taken (about 30 mL each). The medium and mycelium were extracted with dichloromethane (15 mL), dried with magnesium sulfate, and analyzed on GC (DB-1 column). Each biotransformation was performed in two repetitions.
3.3.3. Preparative Biotransformation
For this, 100 mg of bromo-5 or iodolactone 6 dissolved in 10 mL of acetone was added to 10 Erlenmeyer flasks (300 mL) containing 3-day cultures of A. cylindrospora AM336 strain, prepared as described above. After 7 days of incubation of the shaking culture, the entire contents of 10 flasks were extracted with dichloromethane (3 × 40 mL). The combined organic fractions were dried with anhydrous magnesium sulphate. After evaporation of the solvent in vacuo, the products were purified on the chromatographic column. Spectral data of the obtained compounds are presented below.
2-bromo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 7
1H NMR (400 MHz, CDCl3): 0.57 and 0.63 (two d, J = 12.8 Hz, 1H, one of CH2-5), 0.94 (d, J = 6.8 Hz, 3H, CH3-9), 1.39 (s, 3H, CH3-10), 1.55–1.64 (m, 2H, one of CH2-3, one of CH2-5), 1.92–1.97 (m, 1H, one of CH2-3), 1.98–2.09 (m, 1H, H-4), 2.61 (ddd, J = 12.8, 6.4 and 3.6 Hz, 1H, H-6), 4.63–4.65 (m, 1H, H-2), 5.02–5.04 (m, 1H, H-1), 13C NMR (100 MHz, CDCl3): 18.85 (C-10), 21.67 (C-9), 24.29 (C-4), 31.42 (C-5), 36.16 (C-3), 42.68 (C-6), 47.30 (C-2), 78.15 (C-7), 77.90 (C-1), 176.55 (C-8), ESIHRMS: calcd for C10H15BrO3Na, m/z 285.0102 and 287.0083 (M + H)+, found 285.0106 and 287.0120. (Supplementary Materials, Figures S26–S30).
2-bromo-5-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 8
1H NMR (400 MHz, CDCl3): 1.04 (d, J = 6.4 Hz, 3H, CH3-9), 1.34 (d, J = 7.6 Hz, 3H, CH3-10), 1.80 (ddd, J = 15.2, 11.6 and 3.2 Hz, 1H, one of CH2-3), 1.98–2.00 (m, 1H, one of CH2-3), 2.01–2.10 (m, 1H, H-4), 2.22–2.25 (m, 1H, H-6), 2.87 (q, J = 7.6 Hz, 1H, H-7), 2.97 (t, J = 10.0 Hz, 1H, H-5), 4.55–4.57 (m, 1H, H-2), 4.83–4.85 (m, 1H, H-1), 13C NMR (100 MHz, CDCl3): 14.32 (C-10), 17.33 (C-9), 32.16 (C-4), 35.51 (C-3), 42.47 (C-7), 46.17 (C-2), 47.05 (C-6), 76.35 (C-5), 80.95 (C-1), 178.72 (C-8), ESIHRMS: calcd for C10H15BrO3Na, m/z 285.0102 and 287.0083 (M + H)+, found 285.0104 and 287.0118. (Supplementary Materials, Figures S31–S35).
2-iodo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 9
1H NMR (600 MHz, CDCl3): 0.64 and 0.68 (two d, J = 12.6 Hz, one of CH2-5), 1.01 (d, J = 6.6 Hz, 3H, CH3-9), 1.43 (s, 3H, CH3-10), 1.45–1.47 (m, 1H, one of CH2-3), 1.63–1.67 (m, 1H, one of CH2-5), 1.94–1.96 (m, 1H, one of CH2-3), 2.03–2.10 (m, 1H, H-4), 2.72–2.76 (ddd, J = 12.6, 4.0 and 2.4 Hz, 1H, H-6), 4.85–4.87 (m, 1H, H-2), 5.16–5.17 (m, 1H, H-1), 13C NMR (151 MHz, CDCl3): 18.75 (C-10), 21.50 (C-9), 26.28 (C-4), 26.65 (C-2), 31.54 (C-5), 37.05 (C-3), 42.51 (C-6), 78.46 (C-7), 79.66 (C-1), 176.92 (C-8), ESIHRMS: calcd for C10H15IO3Na, m/z 332.9964 (M + H)+, found 332.9960. (Supplementary Materials, Figures S36–S40).
4. Conclusions
Chemical synthesis made it possible to obtain halolactones containing two methyl groups in the molecule, one in the cyclohexane ring and the other in the lactone ring. These compounds were formed as a mixture of two diastereoisomers, named “A and B,” differing in the spatial orientation of the methyl group located in the lactone ring. This difference significantly influenced the course of halolactone transformation when filamentous fungi were used as biocatalysts. The only reactions observed were` the hydroxylation of the tertiary (C-7) or secondary (C-5) carbon in the bromolactone molecule and the C-7 carbon in the iodolactone molecule. Moreover, only the one isomer in which the methyl group was positioned across the plane of the lactone ring was hydroxylated.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/11/1/73/s1, Figure S1: 1H NMR (400 MHz, CDCl3) spectrum of ester 2, Figure S2. COSY (100 MHz, CDCl3) spectrum of ester 2, Figure S3. HMQC (100 MHz, CDCl3) spectrum of ester 2, Figure S4. 13C NMR (100 MHz, CDCl3) spectrum of ester 2, Figure S5. HRMS spectrum of ester 2, Figure S6. 1H NMR (400 MHz, CDCl3) spectrum of acid 3, Figure S7. COSY (100 MHz, CDCl3) spectrum of acid 3, Figure S8. HMQC (100 MHz, CDCl3) spectrum of acid 3, Figure S9. 13C NMR (100 MHz, CDCl3) spectrum of acid 3, Figure S10. HRMS spectrum of acid 3, Figure S11. 1H NMR (400 MHz, CDCl3) spectrum of chlorolactone 4, Figure S12. COSY (100 MHz, CDCl3) spectrum of chlorolactone 4, Figure S13. HMQC (100 MHz, CDCl3) spectrum of chlorolactone 4, Figure S14. 13C NMR (100 MHz, CDCl3) spectrum of chlorolactone 4, Figure S15. HRMS spectrum chlorolactone 4, Figure S16. 1H NMR (400 MHz, CDCl3) spectrum of bromolactone 5, Figure S17. COSY (100 MHz, CDCl3) spectrum of bromolactone 5, Figure S18. HMQC (100 MHz, CDCl3) spectrum of bromolactone 5, Figure S19. 13C NMR (100 MHz, CDCl3) spectrum bromolactone 5, Figure S20. HRMS spectrum bromolactone 5, Figure S21. 1H NMR (400 MHz, CDCl3) spectrum of iodolactone 6, Figure S22. COSY (100 MHz, CDCl3) spectrum of iodolactone 6, Figure S23. HMQC (100 MHz, CDCl3) spectrum of iodolactone 6, Figure S24. 13C NMR (100 MHz, CDCl3) spectrum of iodolactone 6, Figure S25. HRMS spectrum of iodolactone 6, Figure S26. 1H NMR (400 MHz, CDCl3) spectrum of bromo-hydroxylactone 7, Figure S27. COSY (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 7, Figure S28. HMQC (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 7, Figure S29. 13C NMR (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 7, Figure S30. HRMS spectrum of bromo-hydroxylactone 7, Figure S31. 1H NMR (400 MHz, CDCl3) spectrum of bromo-hydroxylactone 8, Figure S32. COSY (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 8, Figure S33. HMQC (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 8, Figure S34. 13C NMR (100 MHz, CDCl3) spectrum of bromo-hydroxylactone 8, Figure S35. HRMS spectrum of bromo-hydroxylactone 8, Figure S36. 1H NMR (600 MHz, CDCl3) spectrum of iodo-hydroxylactone 9, Figure S37. COSY (151 MHz, CDCl3) spectrum of iodo-hydroxylactone 9, Figure S38. HMQC (151 MHz, CDCl3) spectrum of iodo-hydroxylactone 9, Figure S39. 13C NMR (151 MHz, CDCl3) spectrum of iodo-hydroxylactone 9, Figure S40. HRMS spectrum of iodo-hydroxylactone 9, Figure S41. Chiral chromatogram of bromo-hydroxylactone 7, Figure S42. Chiral chromatogram of bromo-hydroxylactone 8, Figure S43. Chiral chromatogram of iodo-hydroxylactone 9.
Author Contributions
Conceptualization, M.G.; formal analysis, M.G. and G.M.; investigation, M.G., W.M., and K.W.; methodology, M.G.; supervision, K.W.; writing—original draft, M.G.; Writing—review and editing, W.M. and K.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Faber, K. Biotransformations in Organic Chemistry, 7th ed.; Springer International Publishing: Graz, Austria, 2018. [Google Scholar]
- Braga, A.; Guerreiro, C.; Belo, I. Generation of Flavors and Fragrances through Biotransformation and De Novo Synthesis. Food. Bioprocess Technol. 2018, 11, 2217–2228. [Google Scholar] [CrossRef]
- Mangas-Sánchez, J.; Busto, E.; Gotor-Fernández, V.; Gotor, V. Straightforward Synthesis of Enantiopure 2,3-Dihydrobenzofurans by a Sequential Stereoselective Biotransformation and Chemical Intramolecular Cyclization. Org. Lett. 2010, 12, 3498–3501. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Ahmed, R.; Khan, R. Biotransformation: A green and efficient way of antioxidant synthesis. Free Radic. Res. 2016, 50, 939–948. [Google Scholar] [CrossRef] [PubMed]
- de Souza, G.G.; Oliveira, T.S.; Takahashi, J.A.; Collado, I.G.; Macías-Sánchez, A.J.; Hernández-Galán, R. Biotransformation of clovane derivatives. Whole cell fungi mediated domino synthesis of rumphellclovane A. Org. Biomol. Chem. 2012, 10, 3315–3320. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z. Cascade Biotransformations via Enantioselective Reduction, Oxidation, and Hydrolysis: Preparation of (R)-δ-Lactones from 2-Alkylidenecyclopentanones. ACS Catal. 2013, 3, 908–911. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Sutherland, J.B. The use of Aspergillus niger cultures for biotransformation of terpenoids. Process Biochem. 2014, 49, 2086–2100. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Yang, X.W. Four new eudesmane-type sesquiterpenoid lactones from atractylenolide II by biotransformation of rat hepatic microsomes. J. Asian Nat. Prod. Res. 2013, 15, 344–356. [Google Scholar] [CrossRef]
- Cano-Flores, A.; Delgado, G. Transformations of Some Sesquiterpene Lactones by Filamentous Fungi and Cytotoxic Evaluations. Chem. Biodivers. 2017, 14, e1700211. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Wawrzeńczyk, C. Microbial Asymmetric Functionalization of β-Cyclocitral-Derived Tetramethyl-Substituted γ-Lactone. Molecules 2019, 24, 666. [Google Scholar] [CrossRef]
- Ghasemi, S.; Habibi, Z.; Mohajeri, M.; Yousefi, M. Biotransformation of two furanocoumarins by the fungi species Aspergillus sp. PTCC 5266 and Aspergillus niger PTCC 5010. Nat. Prod. Res. 2019, 3, 835–842. [Google Scholar] [CrossRef]
- Lv, X.; Liu, D.; Hou, J.; Dong, P.; Zhan, L.; Wang, L.; Deng, S.; Wang, C.; Yao, J.; Shu, X.; et al. Biotransformation of imperatorin by Penicillium janthinellum. Anti-osteoporosis activities of its metabolites. Food Chem. 2013, 138, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Ahmed, M.; Khan, T.A.; Akhter, Y. Fungal P 450 monooxygenases-the diversity in catalysis and their promising roles in biocontrol activity. Appl. Microbiol. Biotechnol. 2020, 104, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R.; Urlacher, V.B. Cytochromes P450 as promising catalysts for biotechnological application: Chances and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, J.E.; Lee, Y.W.; Son, H. Fungal cytochrome P450s and the P450 complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Aranda, C.; Municoy, M.; Guallar, V.; Kiebist, J.; Scheibner, K.; Ullrich, R.; del Rio, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of 4-hydroxyisophorone and 4-ketoisophorone by fungal peroxygenases. Catal. Sci. Technol. 2019, 9, 1398–1405. [Google Scholar] [CrossRef]
- Hofrichter, M.; Kellner, H.; Pecyna, M.J.; Ullrich, R. Fungal unspecific peroxygenases: Heme-thiolate proteins that combine peroxidase and cytochrome P450 properties. Adv. Exp. Med. Biol. 2015, 851, 341–368. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Mączka, W.; Wińska, K.; Żarowska, B.; Anioł, M. Antimicrobial activity of hydroxylactone obtained by biotransformation of bromo- and iodolactone with gem-dimethylcyclohexane ring. J. Br. Chem. Soc. 2013, 24, 1913–1919. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Żołnierczyk, A.K.; Żarowska, B.; Anioł, M. Lactones with methylcyclohexane system obtained by chemical and microbiological methods and their antimicrobial activity. Molecules 2015, 20, 3335–3353. [Google Scholar] [CrossRef]
- Wińska, K.; Grabarczyk, M.; Mączka, W.; Żarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Szumny, A.M. Anioł, Biotransformation of bicyclic halolactones with a methyl group in the cyclohexane ring into hydroxylactones and their biological activity. Molecules 2016, 21, 1453. [Google Scholar] [CrossRef]
- Wińska, K.; Grabarczyk, M.; Mączka, W.; Żarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Anioł, M. Biotransformation of lactones with methylcyclohexane ring and their biological activity. Appl. Sci. 2017, 7, 12. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Żarowska, B.; Białońska, A.; Anioł, M. Hydroxylactones with the gem-dimethylcyclohexane system—synthesis and antimicrobial activity. Arabian J. Chem. 2019, 12, 2280–22988. [Google Scholar] [CrossRef]
- Gładkowski, W.; Mazur, M.; Białońska, A.; Wawrzeńczyk, C. Lactones 35 [1]. Metabolism of iodolactones with cyclohexane ring in Absidia cylindrospora culture. Enzyme Microb. Technol. 2011, 48, 326–333. [Google Scholar] [CrossRef]
- Mazur, M.; Kudrynska, A.; Pawlak, A.; Hernandez-Suarez, B.; Obmińska-Mrukowicz, B.; Gładkowski, W. Biotechnological Approach for the Production of Enantiomeric Hydroxylactones Derived from Benzaldehyde and Evaluation of Their Cytotoxic Activity. Catalysts 2020, 10, 1313. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Srček, V.G.; Radošević, K.; Maciejewska, G.; Wawrzeńczyk, C. Regio- and enantioselective microbial hydroxylation and evaluation of cytotoxic activity of β-cyclocitral-derived halolactones. PLoS ONE 2017, 12, e0183429. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, M.; Mączka, W.; Wińska, K.; Żarowska, B.; Maciejewska, G.; Gębarowska, E.; Pietr, S.J. Antimicrobial chloro-hydroxylactones derived from the biotransformations of bicyclic halolactones by cultures of Pleurotus ostreatus. Bioorg. Chem. 2020, 104, 104250. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Szumny, A.; Gładkowski, W.; Białońska, A.; Ciunik, Z.; Wawrzeńczyk, C. Lactones 18. Synthesis of bicyclic lactones with methyl-, di- and trimethyl substituted cyclohexane system. Polish J. Chem. 2005, 79, 1763–1771. [Google Scholar] [CrossRef]
- Loupy, A.; Seyden-Penne, J. The influence of lithium complexing agents on the regioselectivity of reductions of substituted 2-cyclohexenones by LiAlH4 and LiBH4. Tetrahedron 1980, 36, 1937–1942. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Białońska, A. Biotransformations of chloro-, bromo- and iodolactone with trimethylcyclohexane system using fungal strains. Biocatal. Biotransform. 2010, 28, 408–414. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).