New Bromo- and Iodo-Hydroxylactones with Two Methyl Groups Obtained by Biotransformation of Bicyclic Halolactones



Abstract
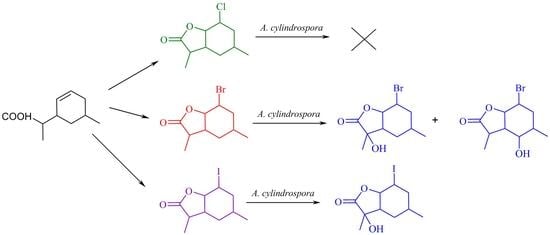
:
1. Introduction
2. Results and Discussion
2.1. Obtaining of Substrates for Biotransformation and Analysis of Their Structures
2.2. Screening Biotransformations of Halolactones 4–6
2.3. Preparative Biotransformation of Halolactones 5 and 6. Analysis of the Structures of Obtained Derivatives
3. Materials and Methods
3.1. General Methods
3.2. Organic Synthesis
Ethyl ester of (4,7-dimethylcyclohex-2-en-1-yl)acetic acid 2A + 2B
(4,7-dimethylcyclohex-2-en-1-yl)acetic acid 3A + 3B
2-Chloro-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 4A +4B
2-Bromo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 5A + 5B
2-Iodo-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 6A + 6B
3.3. Biotransformation
3.3.1. Microorganisms
3.3.2. Screening Biotransformation
3.3.3. Preparative Biotransformation
2-bromo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 7
2-bromo-5-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 8
2-iodo-7-hydroxy-4,7-dimethyl-9-oxabicyclo[4.3.0]nonan-8-one 9
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Faber, K. Biotransformations in Organic Chemistry, 7th ed.; Springer International Publishing: Graz, Austria, 2018. [Google Scholar]
- Braga, A.; Guerreiro, C.; Belo, I. Generation of Flavors and Fragrances through Biotransformation and De Novo Synthesis. Food. Bioprocess Technol. 2018, 11, 2217–2228. [Google Scholar] [CrossRef] [Green Version]
- Mangas-Sánchez, J.; Busto, E.; Gotor-Fernández, V.; Gotor, V. Straightforward Synthesis of Enantiopure 2,3-Dihydrobenzofurans by a Sequential Stereoselective Biotransformation and Chemical Intramolecular Cyclization. Org. Lett. 2010, 12, 3498–3501. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Ahmed, R.; Khan, R. Biotransformation: A green and efficient way of antioxidant synthesis. Free Radic. Res. 2016, 50, 939–948. [Google Scholar] [CrossRef] [PubMed]
- de Souza, G.G.; Oliveira, T.S.; Takahashi, J.A.; Collado, I.G.; Macías-Sánchez, A.J.; Hernández-Galán, R. Biotransformation of clovane derivatives. Whole cell fungi mediated domino synthesis of rumphellclovane A. Org. Biomol. Chem. 2012, 10, 3315–3320. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z. Cascade Biotransformations via Enantioselective Reduction, Oxidation, and Hydrolysis: Preparation of (R)-δ-Lactones from 2-Alkylidenecyclopentanones. ACS Catal. 2013, 3, 908–911. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Sutherland, J.B. The use of Aspergillus niger cultures for biotransformation of terpenoids. Process Biochem. 2014, 49, 2086–2100. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Yang, X.W. Four new eudesmane-type sesquiterpenoid lactones from atractylenolide II by biotransformation of rat hepatic microsomes. J. Asian Nat. Prod. Res. 2013, 15, 344–356. [Google Scholar] [CrossRef]
- Cano-Flores, A.; Delgado, G. Transformations of Some Sesquiterpene Lactones by Filamentous Fungi and Cytotoxic Evaluations. Chem. Biodivers. 2017, 14, e1700211. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Wawrzeńczyk, C. Microbial Asymmetric Functionalization of β-Cyclocitral-Derived Tetramethyl-Substituted γ-Lactone. Molecules 2019, 24, 666. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, S.; Habibi, Z.; Mohajeri, M.; Yousefi, M. Biotransformation of two furanocoumarins by the fungi species Aspergillus sp. PTCC 5266 and Aspergillus niger PTCC 5010. Nat. Prod. Res. 2019, 3, 835–842. [Google Scholar] [CrossRef]
- Lv, X.; Liu, D.; Hou, J.; Dong, P.; Zhan, L.; Wang, L.; Deng, S.; Wang, C.; Yao, J.; Shu, X.; et al. Biotransformation of imperatorin by Penicillium janthinellum. Anti-osteoporosis activities of its metabolites. Food Chem. 2013, 138, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Ahmed, M.; Khan, T.A.; Akhter, Y. Fungal P 450 monooxygenases-the diversity in catalysis and their promising roles in biocontrol activity. Appl. Microbiol. Biotechnol. 2020, 104, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R.; Urlacher, V.B. Cytochromes P450 as promising catalysts for biotechnological application: Chances and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, J.E.; Lee, Y.W.; Son, H. Fungal cytochrome P450s and the P450 complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, C.; Municoy, M.; Guallar, V.; Kiebist, J.; Scheibner, K.; Ullrich, R.; del Rio, J.C.; Hofrichter, M.; Martínez, A.T.; Gutiérrez, A. Selective synthesis of 4-hydroxyisophorone and 4-ketoisophorone by fungal peroxygenases. Catal. Sci. Technol. 2019, 9, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Hofrichter, M.; Kellner, H.; Pecyna, M.J.; Ullrich, R. Fungal unspecific peroxygenases: Heme-thiolate proteins that combine peroxidase and cytochrome P450 properties. Adv. Exp. Med. Biol. 2015, 851, 341–368. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Mączka, W.; Wińska, K.; Żarowska, B.; Anioł, M. Antimicrobial activity of hydroxylactone obtained by biotransformation of bromo- and iodolactone with gem-dimethylcyclohexane ring. J. Br. Chem. Soc. 2013, 24, 1913–1919. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Żołnierczyk, A.K.; Żarowska, B.; Anioł, M. Lactones with methylcyclohexane system obtained by chemical and microbiological methods and their antimicrobial activity. Molecules 2015, 20, 3335–3353. [Google Scholar] [CrossRef] [Green Version]
- Wińska, K.; Grabarczyk, M.; Mączka, W.; Żarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Szumny, A.M. Anioł, Biotransformation of bicyclic halolactones with a methyl group in the cyclohexane ring into hydroxylactones and their biological activity. Molecules 2016, 21, 1453. [Google Scholar] [CrossRef] [Green Version]
- Wińska, K.; Grabarczyk, M.; Mączka, W.; Żarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Anioł, M. Biotransformation of lactones with methylcyclohexane ring and their biological activity. Appl. Sci. 2017, 7, 12. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Żarowska, B.; Białońska, A.; Anioł, M. Hydroxylactones with the gem-dimethylcyclohexane system—synthesis and antimicrobial activity. Arabian J. Chem. 2019, 12, 2280–22988. [Google Scholar] [CrossRef] [Green Version]
- Gładkowski, W.; Mazur, M.; Białońska, A.; Wawrzeńczyk, C. Lactones 35 [1]. Metabolism of iodolactones with cyclohexane ring in Absidia cylindrospora culture. Enzyme Microb. Technol. 2011, 48, 326–333. [Google Scholar] [CrossRef]
- Mazur, M.; Kudrynska, A.; Pawlak, A.; Hernandez-Suarez, B.; Obmińska-Mrukowicz, B.; Gładkowski, W. Biotechnological Approach for the Production of Enantiomeric Hydroxylactones Derived from Benzaldehyde and Evaluation of Their Cytotoxic Activity. Catalysts 2020, 10, 1313. [Google Scholar] [CrossRef]
- Mazur, M.; Gładkowski, W.; Srček, V.G.; Radošević, K.; Maciejewska, G.; Wawrzeńczyk, C. Regio- and enantioselective microbial hydroxylation and evaluation of cytotoxic activity of β-cyclocitral-derived halolactones. PLoS ONE 2017, 12, e0183429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabarczyk, M.; Mączka, W.; Wińska, K.; Żarowska, B.; Maciejewska, G.; Gębarowska, E.; Pietr, S.J. Antimicrobial chloro-hydroxylactones derived from the biotransformations of bicyclic halolactones by cultures of Pleurotus ostreatus. Bioorg. Chem. 2020, 104, 104250. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Szumny, A.; Gładkowski, W.; Białońska, A.; Ciunik, Z.; Wawrzeńczyk, C. Lactones 18. Synthesis of bicyclic lactones with methyl-, di- and trimethyl substituted cyclohexane system. Polish J. Chem. 2005, 79, 1763–1771. [Google Scholar] [CrossRef]
- Loupy, A.; Seyden-Penne, J. The influence of lithium complexing agents on the regioselectivity of reductions of substituted 2-cyclohexenones by LiAlH4 and LiBH4. Tetrahedron 1980, 36, 1937–1942. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Białońska, A. Biotransformations of chloro-, bromo- and iodolactone with trimethylcyclohexane system using fungal strains. Biocatal. Biotransform. 2010, 28, 408–414. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Lactone 4 | Lactone 5 | Lactone 7 | Lactone 8 | Lactone 6 | Lactone 9 |
|---|---|---|---|---|---|---|
| Fusarium culmorum AM10 | 100 | 100 | 0 | 0 | 100 | 0 |
| Fusarium avenaceum AM12 | 100 | 86.1 | 0 | 13.9 | 100 | 0 |
| Fusarium semitectum AM20 | 100 | 100 | 0 | 0 | 100 | 0 |
| Fusarium solani AM203 | 100 | 85.8 | 0 | 14.2 | 100 | 0 |
| Absidia coerulea AM93 | 100 | 100 | 0 | 0 | 100 | 0 |
| Absidia cylindrospora AM336 | 100 | 64.7 | 23.2 | 12.1 | 66.0 | 34.0 |
| Penicillium chermesinum AM113 | 100 | 100 | 0 | 0 | 100 | 0 |
| Penicillium frequentans AM351 | 100 | 100 | 0 | 0 | 100 | 0 |
| Substrate | Unreacted Substrate (%) | Lactone 7 (%) | Isolated Yield (g/%) | Lactone 8 (%) | Isolated Yield (g/%) | Lactone 9 (%) | Isolated Yield (g/%) |
|---|---|---|---|---|---|---|---|
| Lactone 5 | 61.6 | 27.7 | 0.008/8.5 | 10.7 | 0.003/3.2 | 0 | 0 |
| Lactone 6 | 67.5 | 0 | 0 | 0 | 0 | 32.5 | 0.009/8.5 |
| Compound | ee (%) | |
|---|---|---|
| Lactone 7 | 32.4 | −7.25 (C = 0.35, CHCl3) |
| Lactone 8 | 84.7 | −15.08 (C = 0.13, CHCl3) |
| Lactone 9 | 9.5 | −10.28 (C = 0.40, CHCl3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Mączka, W.; Maciejewska, G.; Wińska, K. New Bromo- and Iodo-Hydroxylactones with Two Methyl Groups Obtained by Biotransformation of Bicyclic Halolactones. Catalysts 2021, 11, 73. https://doi.org/10.3390/catal11010073
Grabarczyk M, Mączka W, Maciejewska G, Wińska K. New Bromo- and Iodo-Hydroxylactones with Two Methyl Groups Obtained by Biotransformation of Bicyclic Halolactones. Catalysts. 2021; 11(1):73. https://doi.org/10.3390/catal11010073
Chicago/Turabian StyleGrabarczyk, Małgorzata, Wanda Mączka, Gabriela Maciejewska, and Katarzyna Wińska. 2021. "New Bromo- and Iodo-Hydroxylactones with Two Methyl Groups Obtained by Biotransformation of Bicyclic Halolactones" Catalysts 11, no. 1: 73. https://doi.org/10.3390/catal11010073
APA StyleGrabarczyk, M., Mączka, W., Maciejewska, G., & Wińska, K. (2021). New Bromo- and Iodo-Hydroxylactones with Two Methyl Groups Obtained by Biotransformation of Bicyclic Halolactones. Catalysts, 11(1), 73. https://doi.org/10.3390/catal11010073