
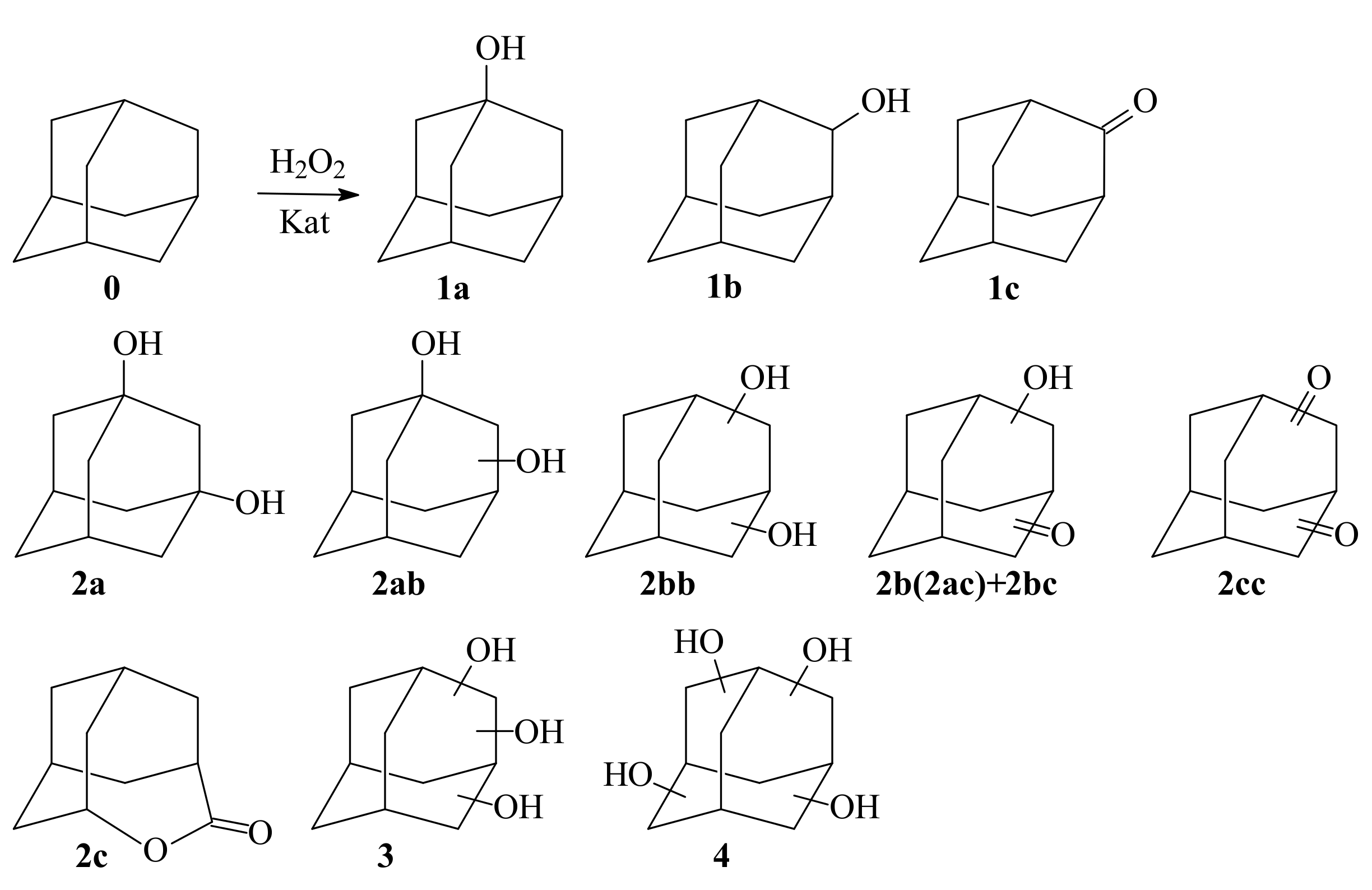
One-Stage Catalytic Oxidation of Adamantane to Tri-, Tetra-, and Penta-Ols



Abstract
:1. Introduction
2. Results
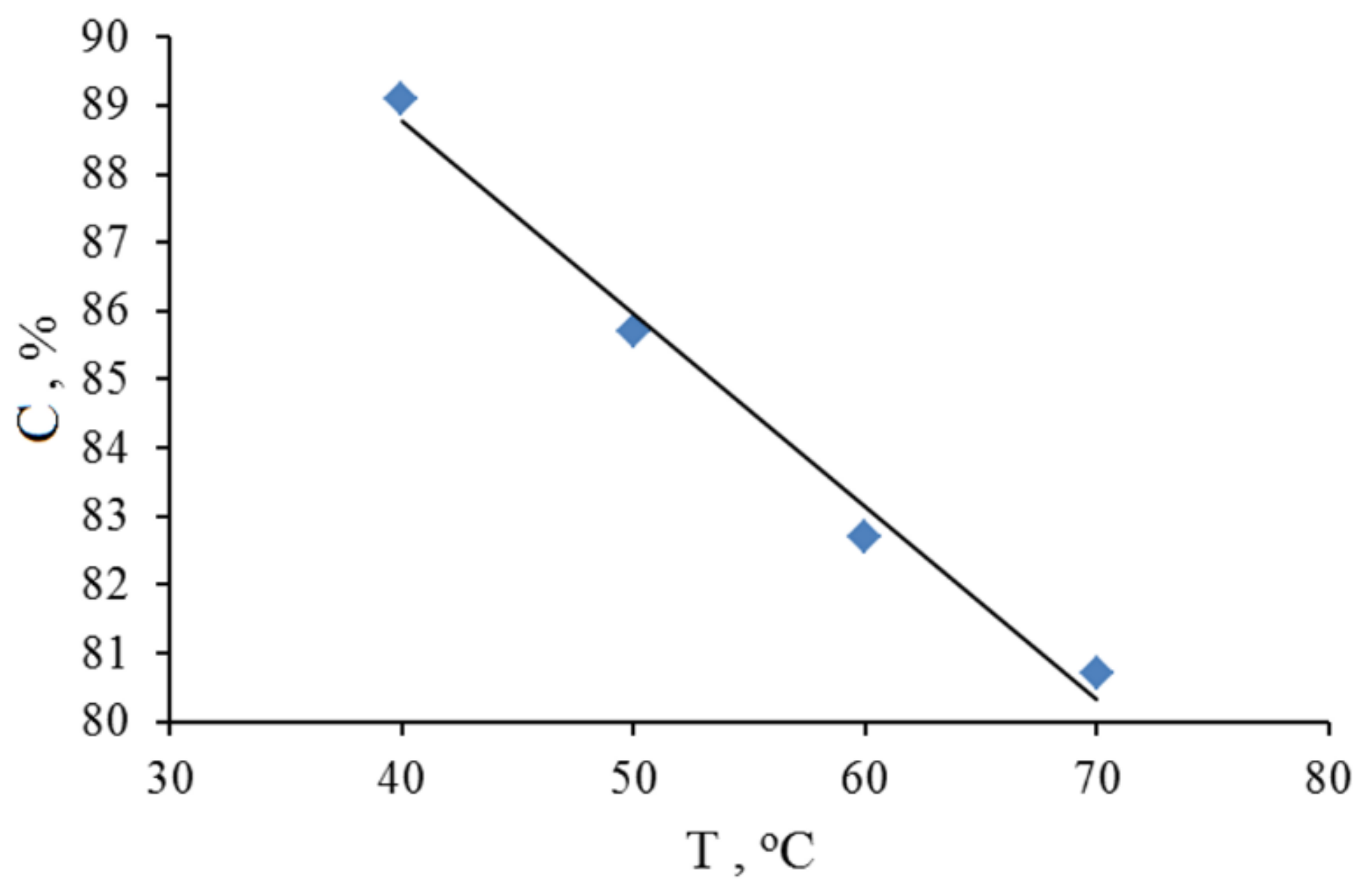
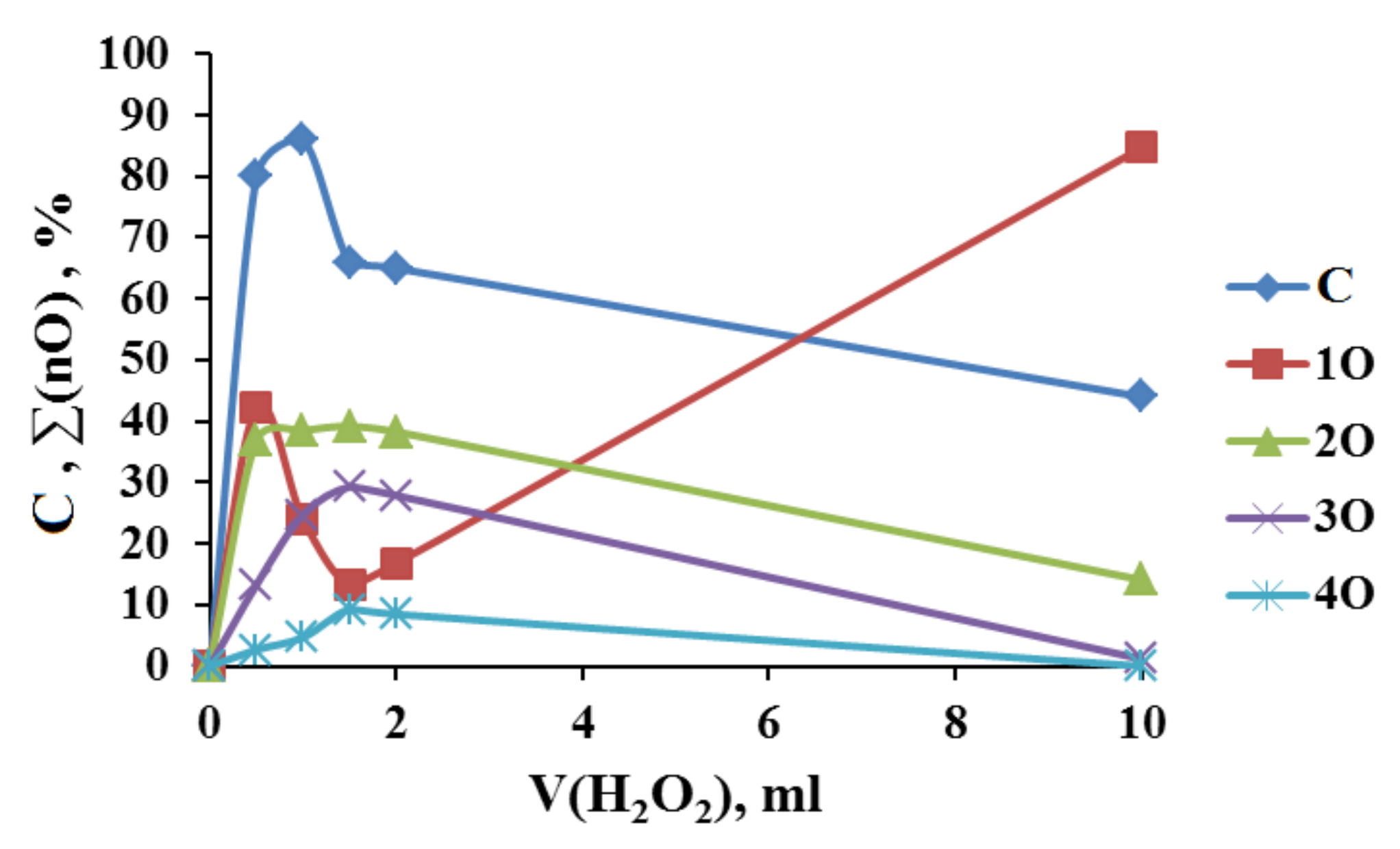
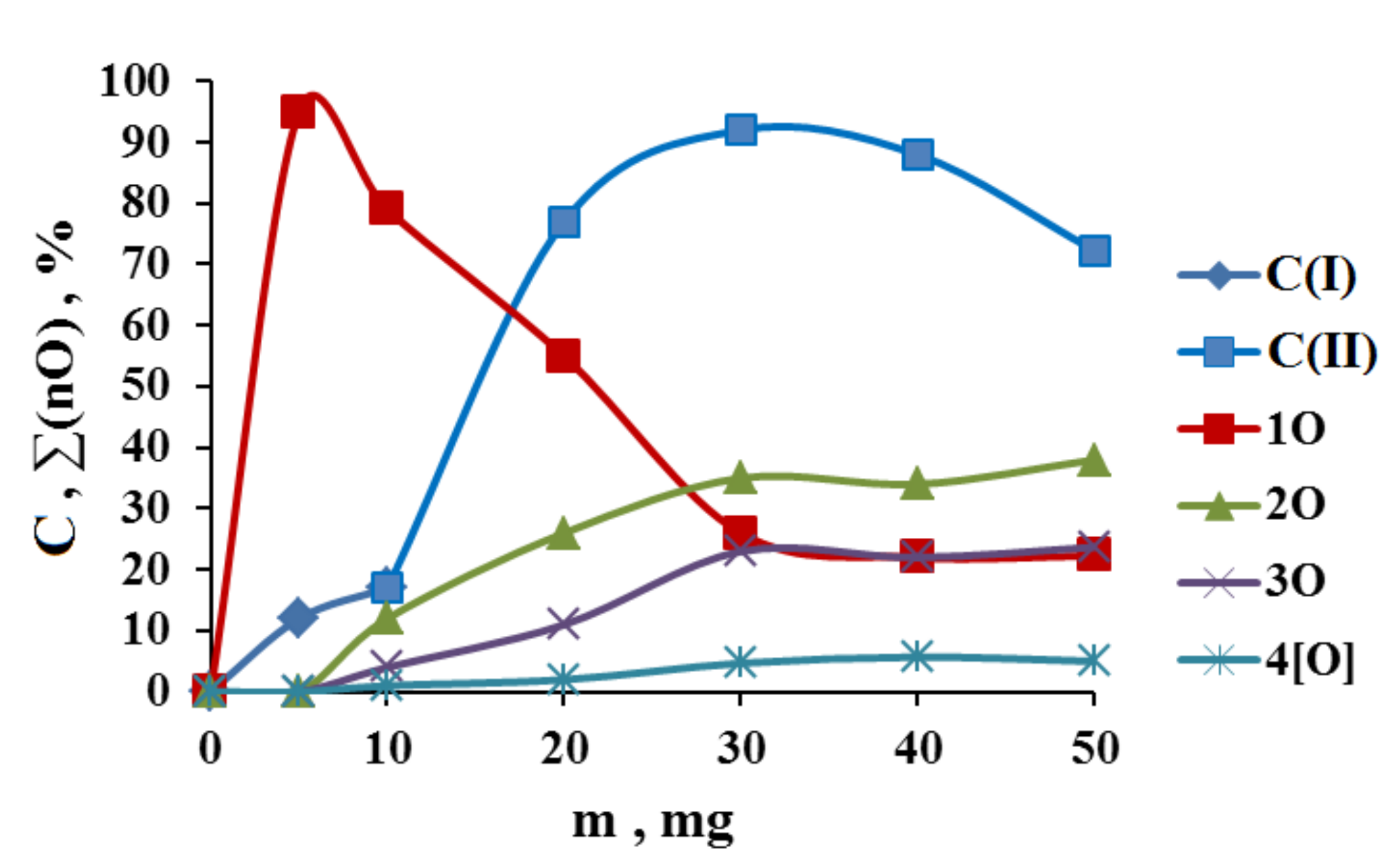
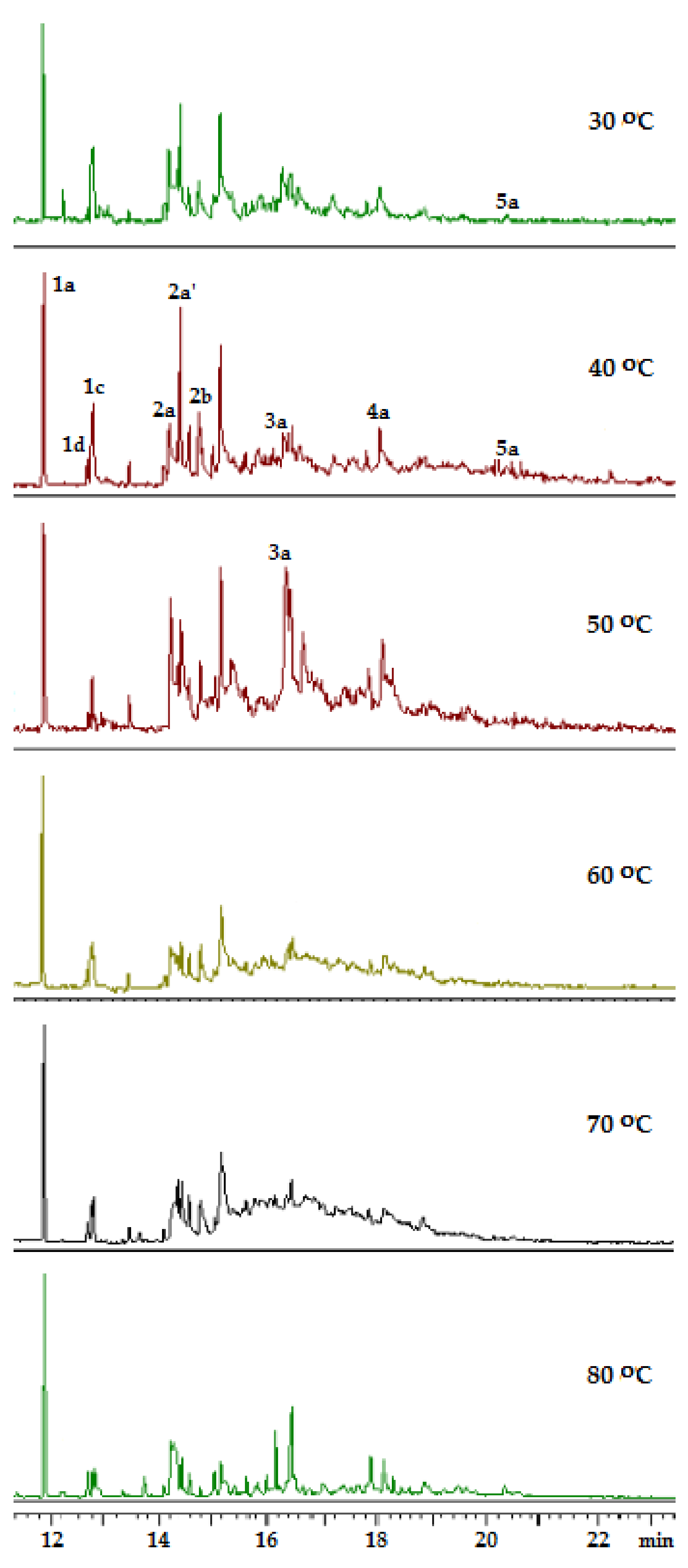
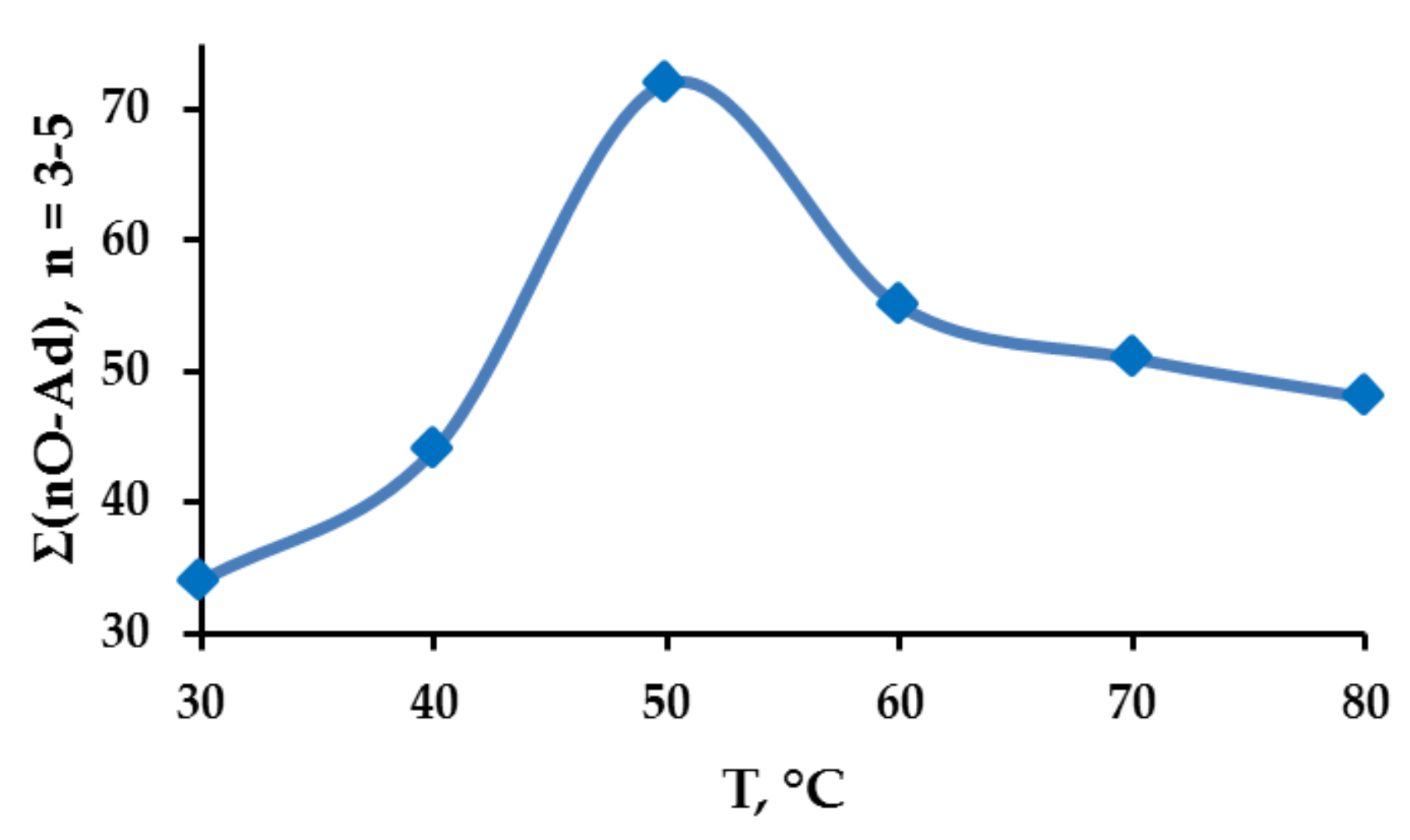
2.1. Search for Optimal 0 Oxidation Conditions upon the Fast (~3 s) Method of Oxidizer Solution Introduction
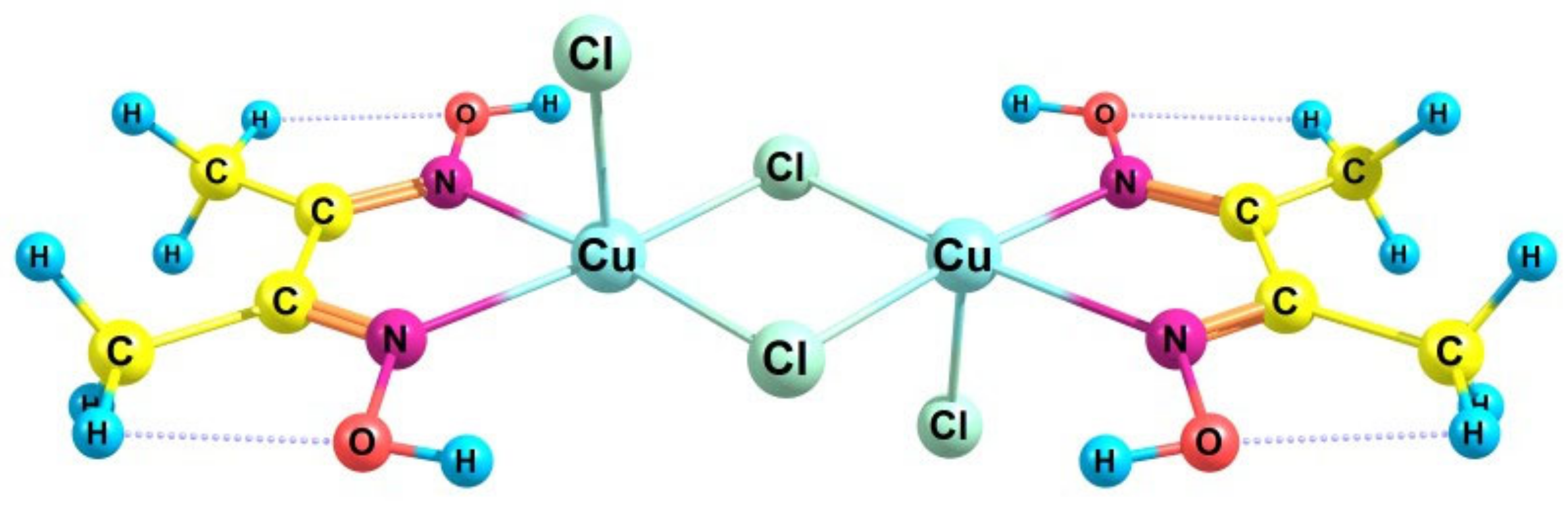
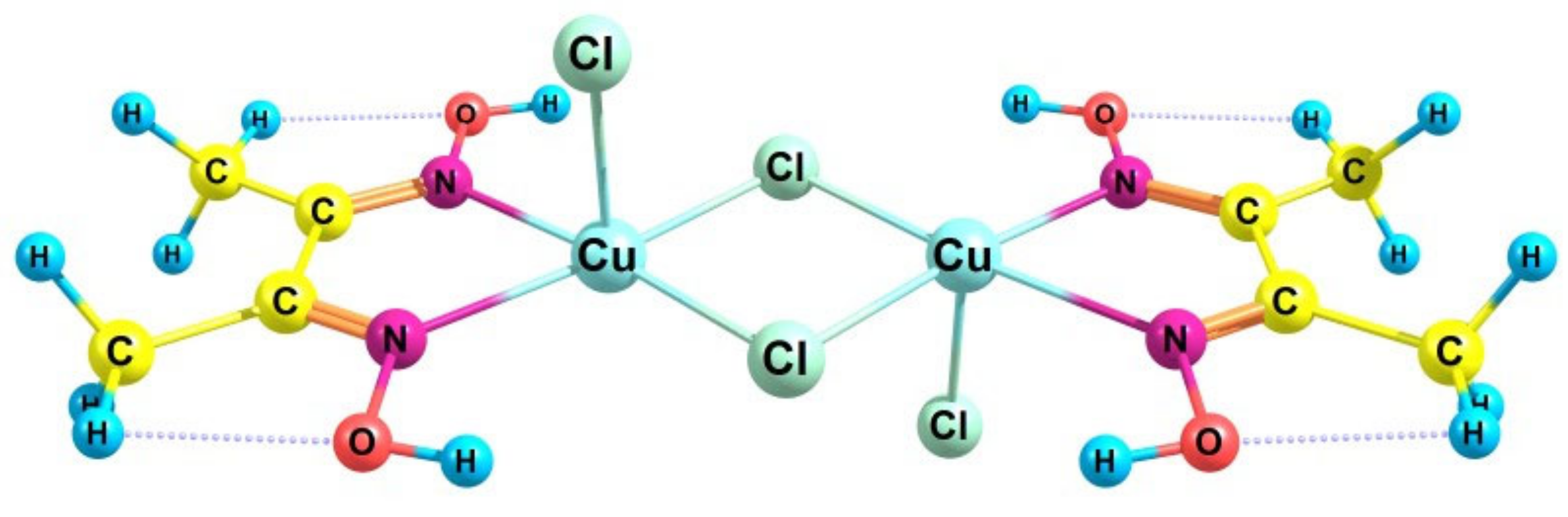
2.2. Catalytic Activity of the Cu2Cl4·2DMG Complex and the Release of Molecular Oxygen upon the Fast (~3 s) Method of Oxidizer Solution Introduction
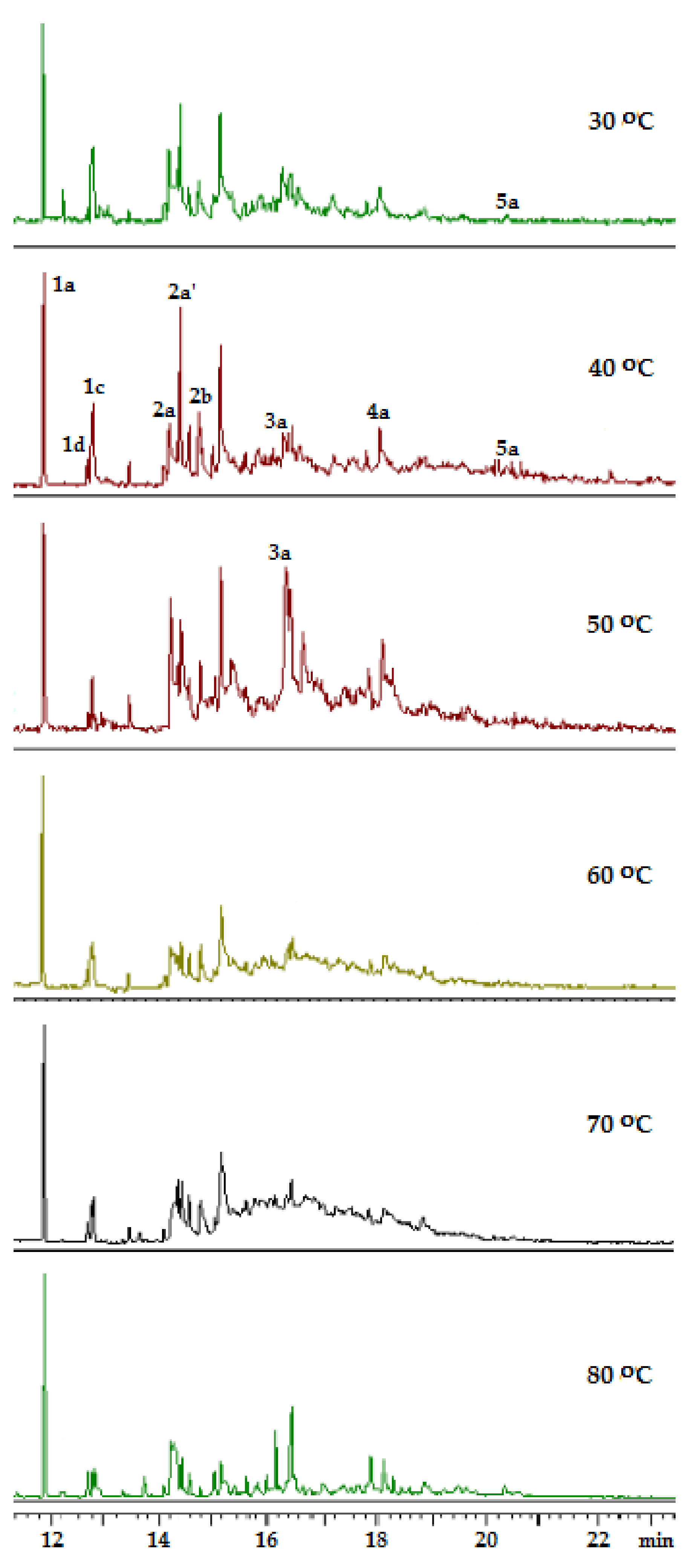
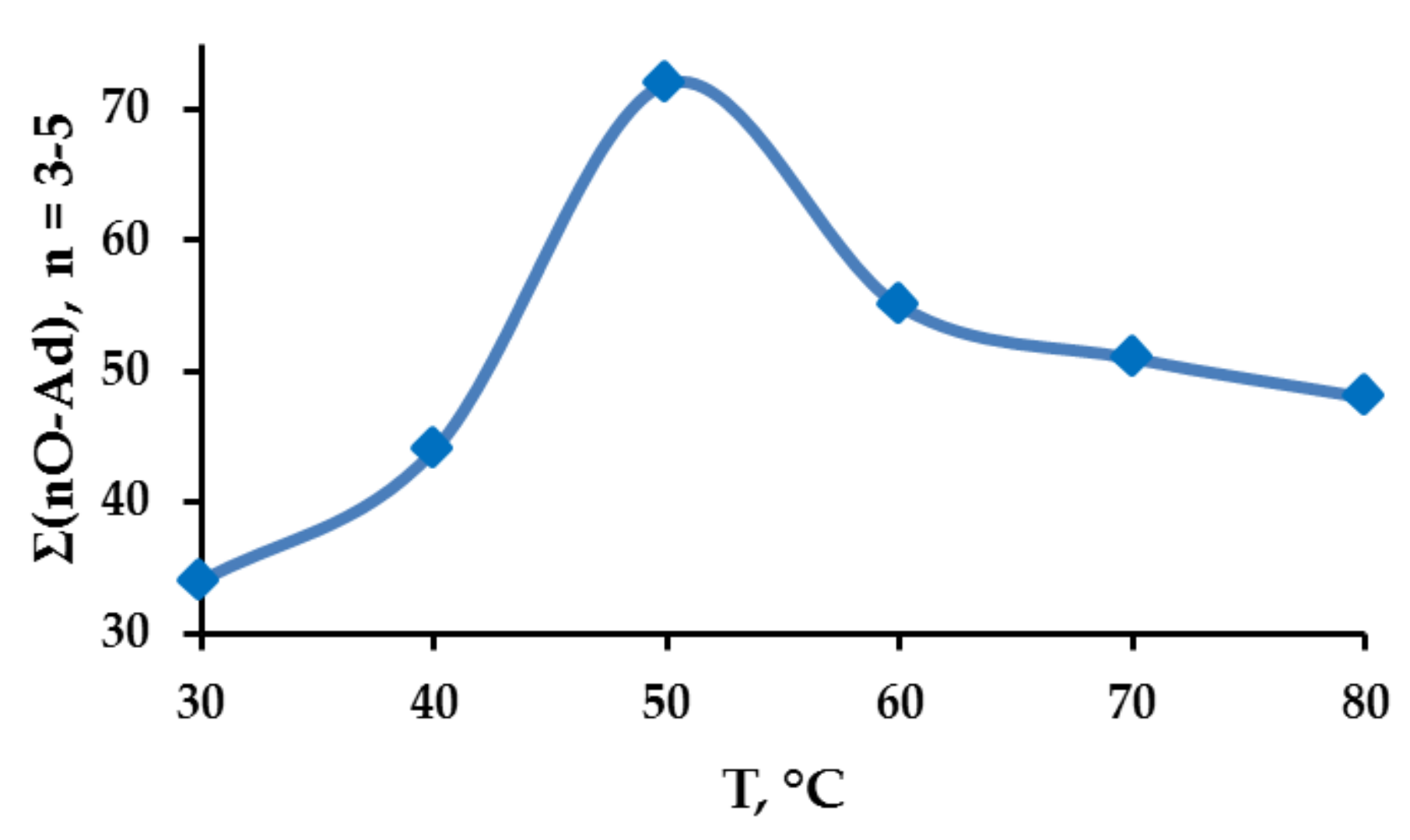
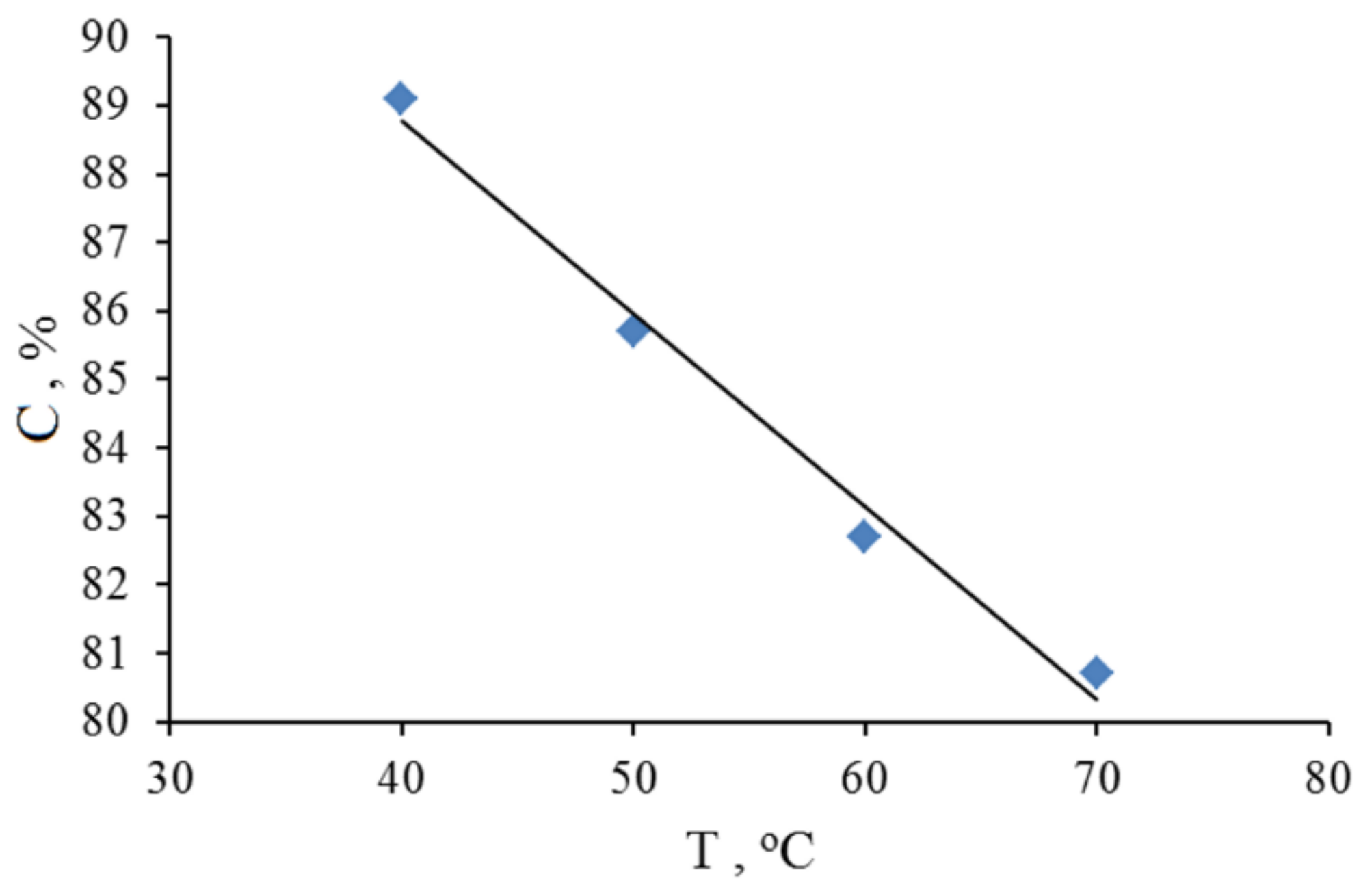
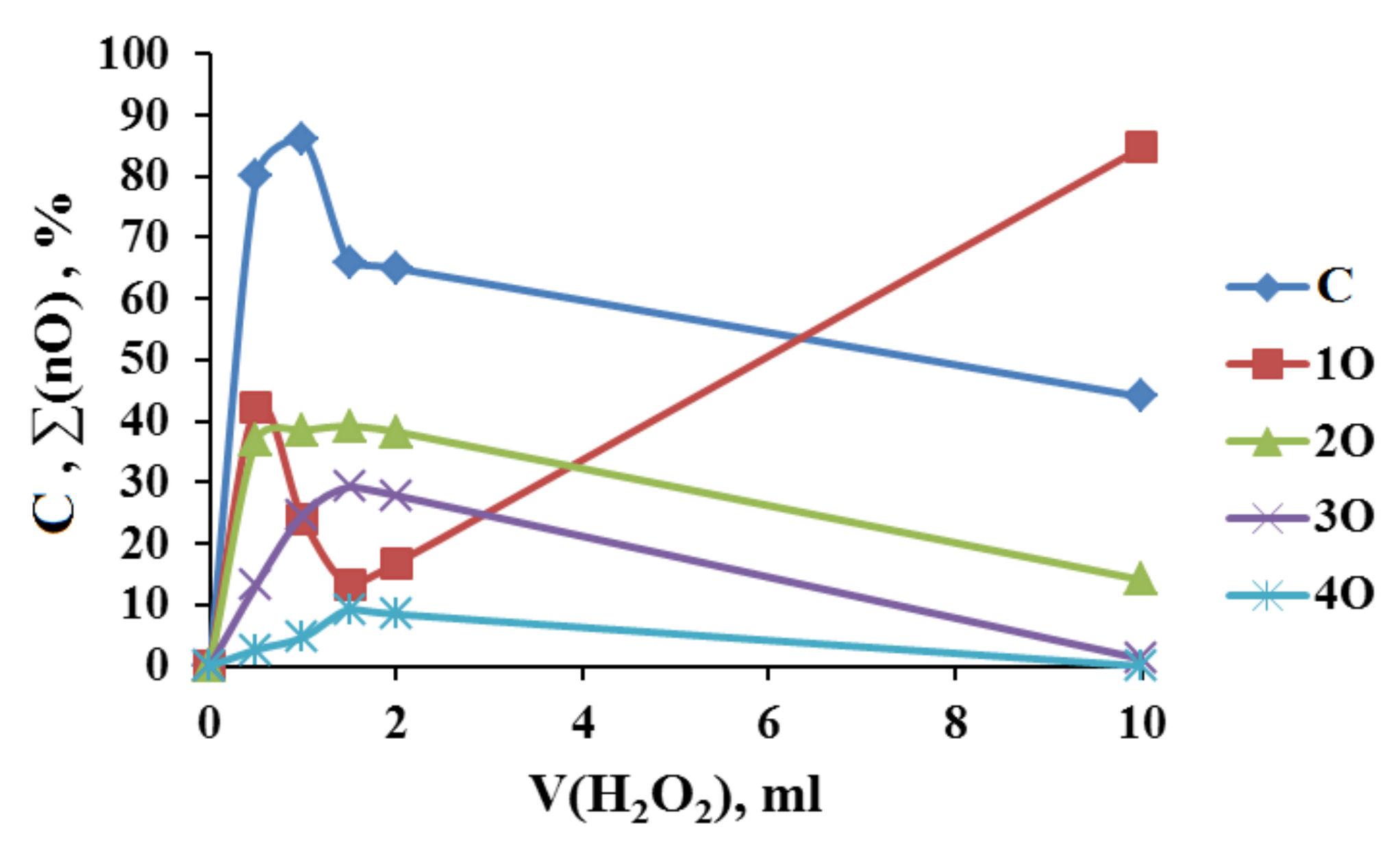
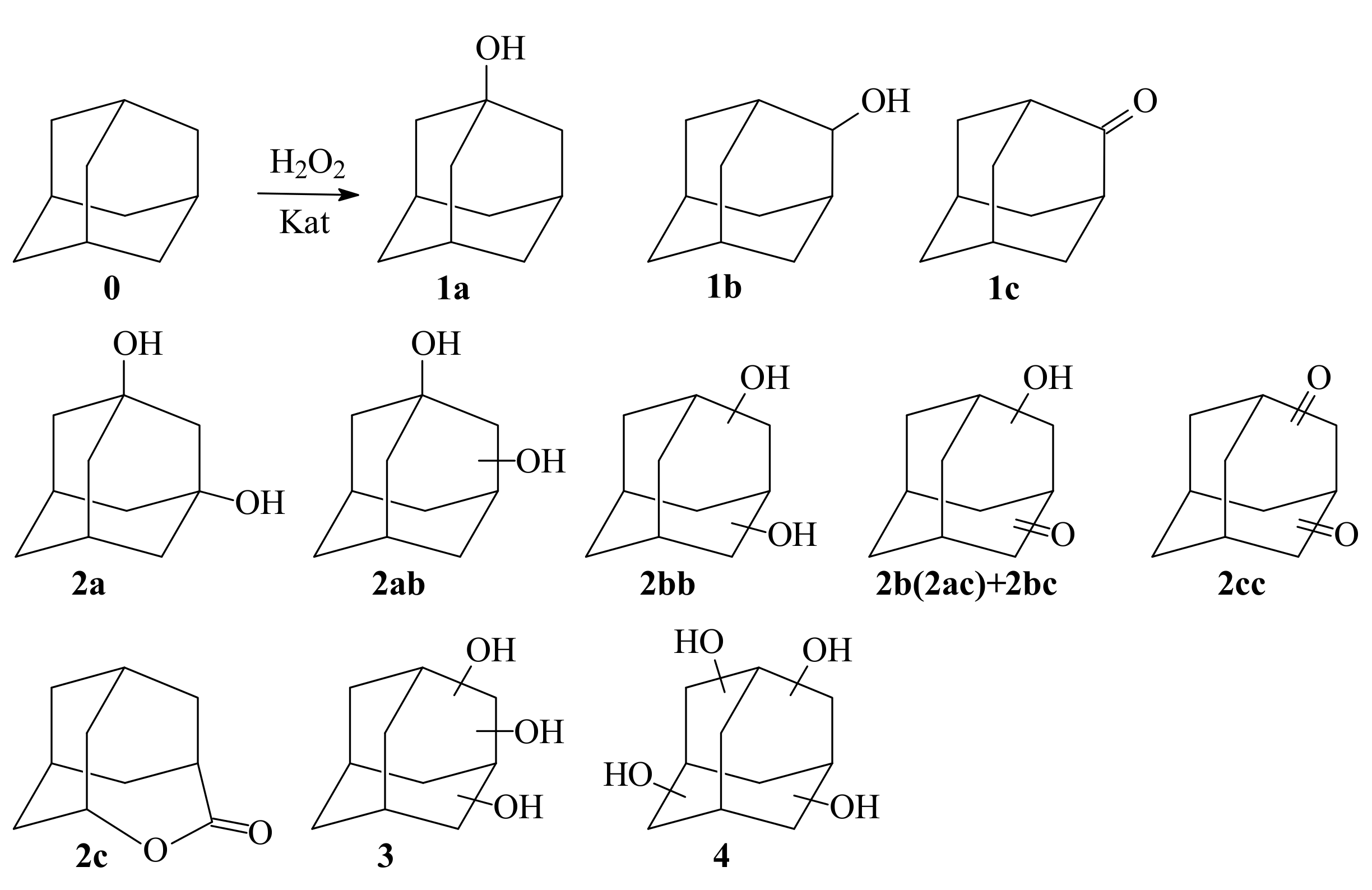
2.3. Adamantane (0) Oxidation upon the Slow (60 min) Method of Oxidizer Solution Introduction
3. Discussion
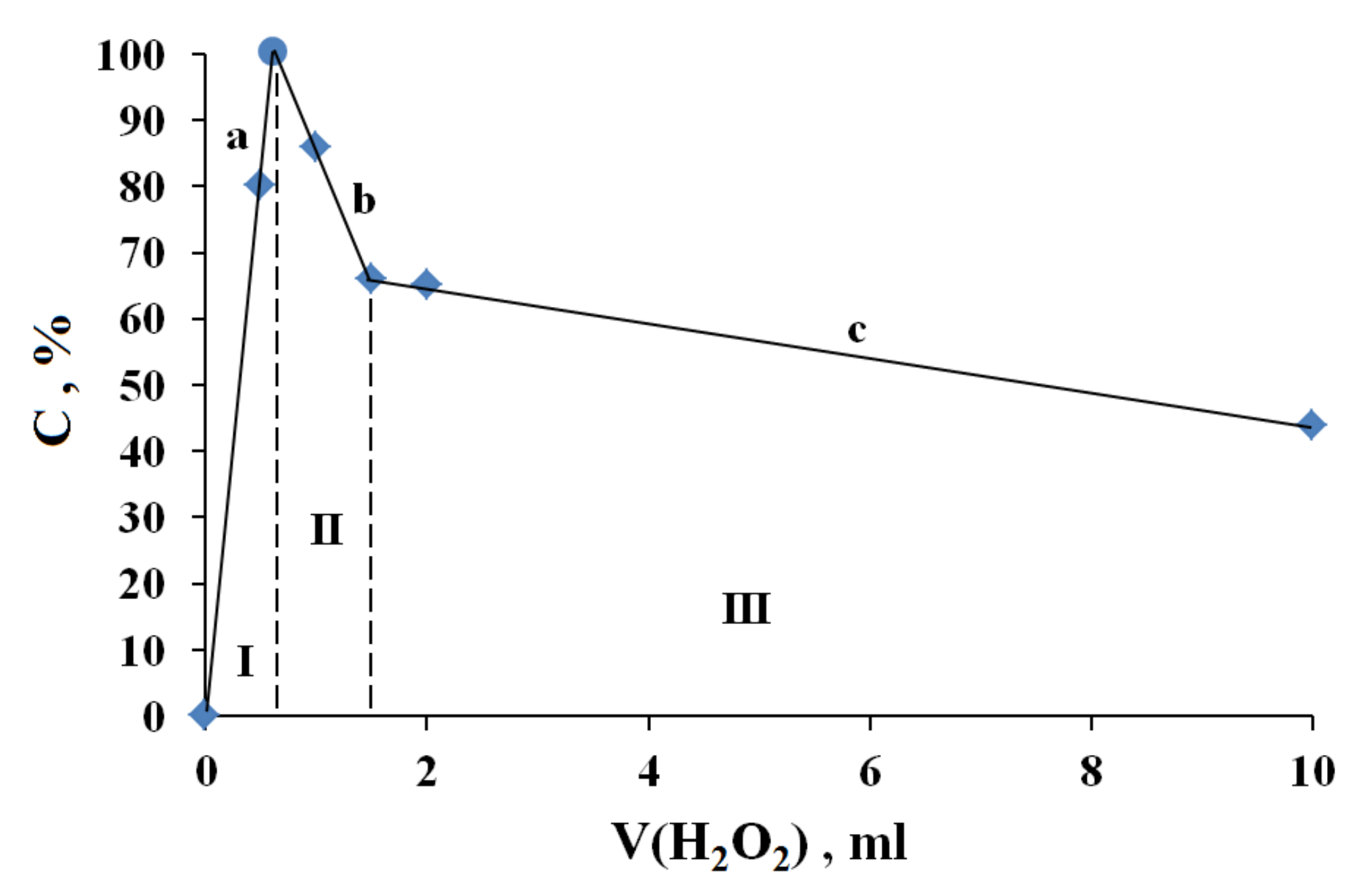
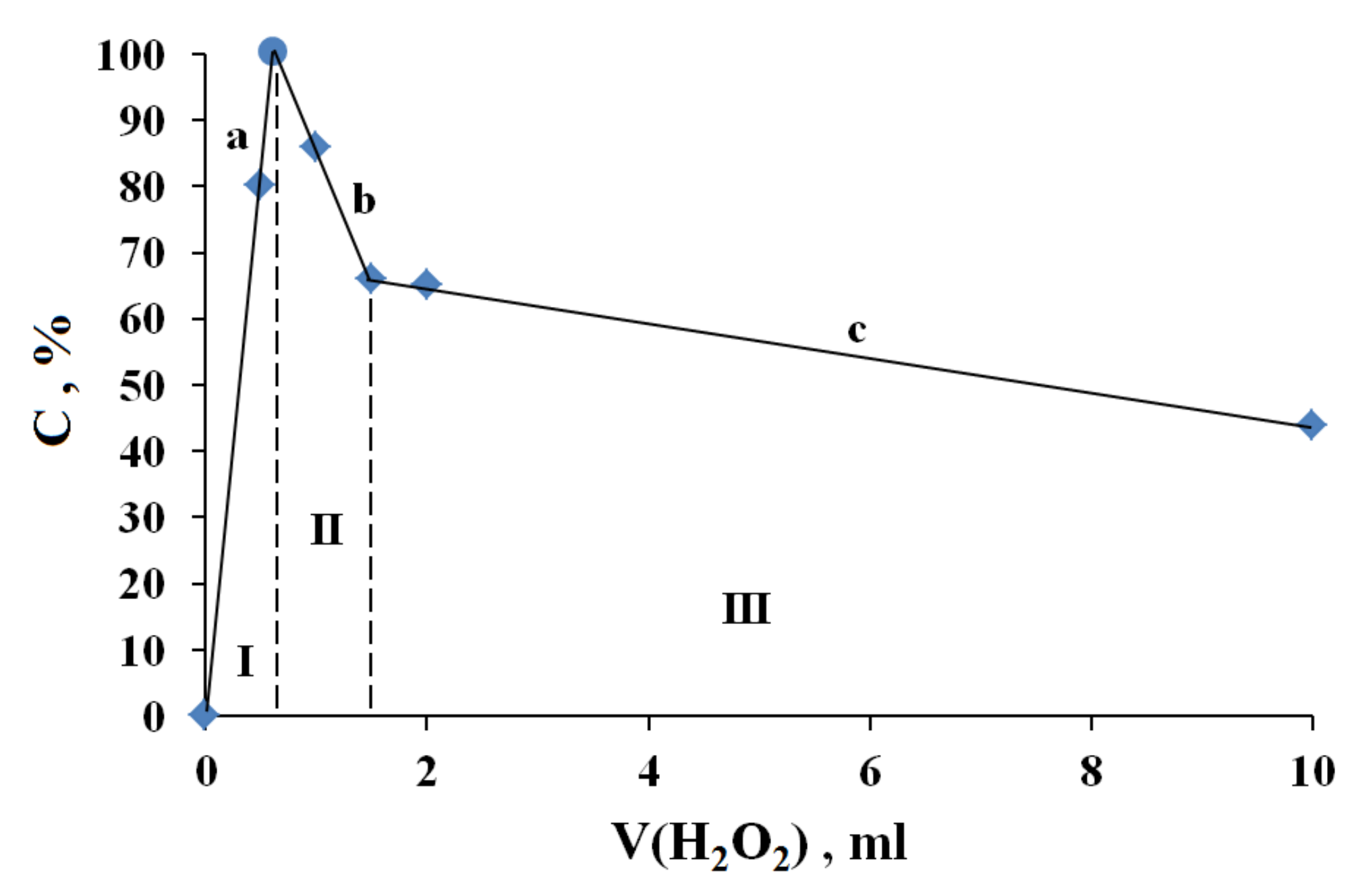
3.1. The Dependence of the Conversion (C) 0 on the Volume V(H2O2) of the Fast Introduced Oxidizer Solution: Three Regions with Different Catalytic Activity and the Structure of the Solvent
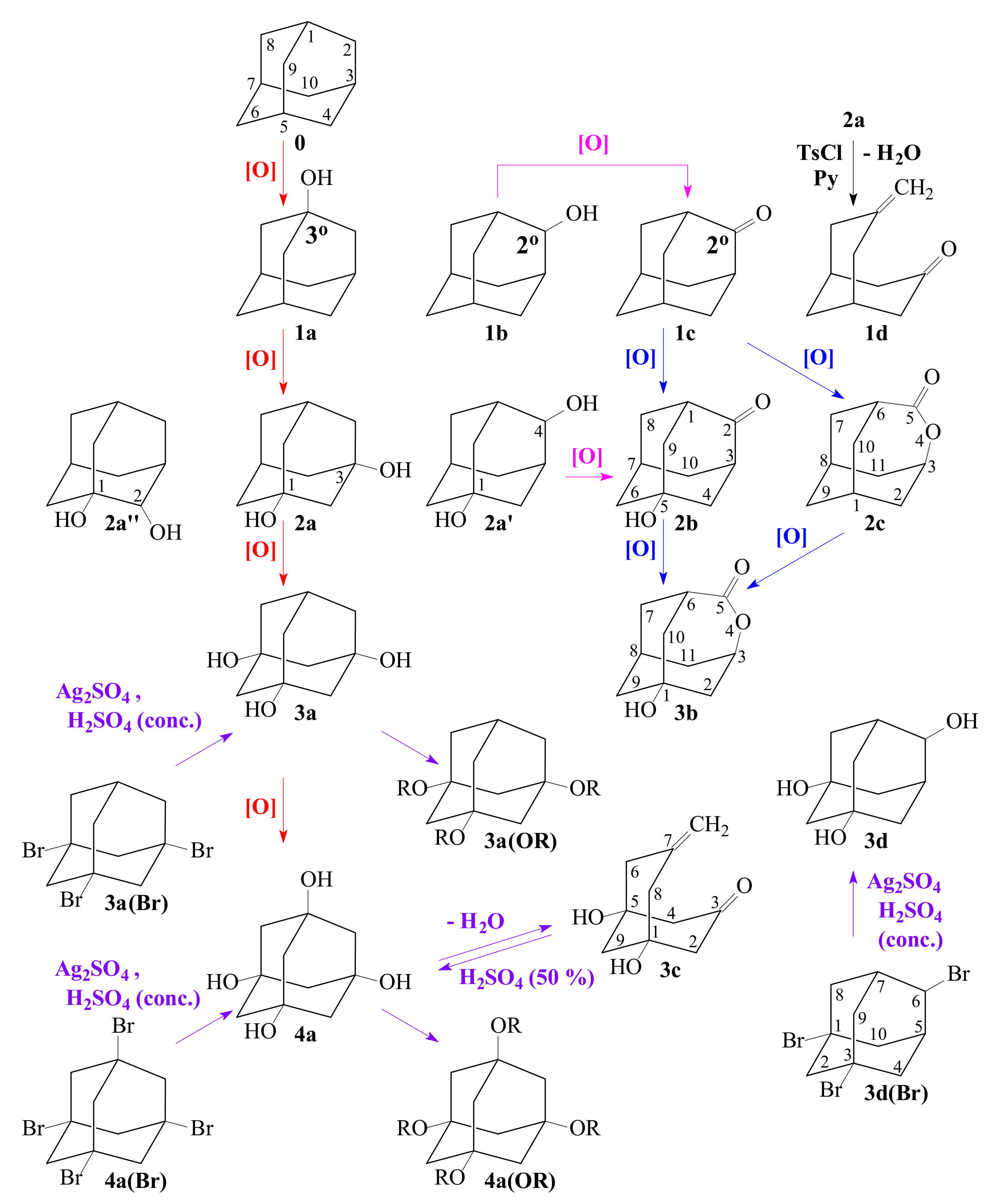
3.2. Comparison of the Results of the Authors’ and Other Methods of 0 Oxidation Described in the Literature
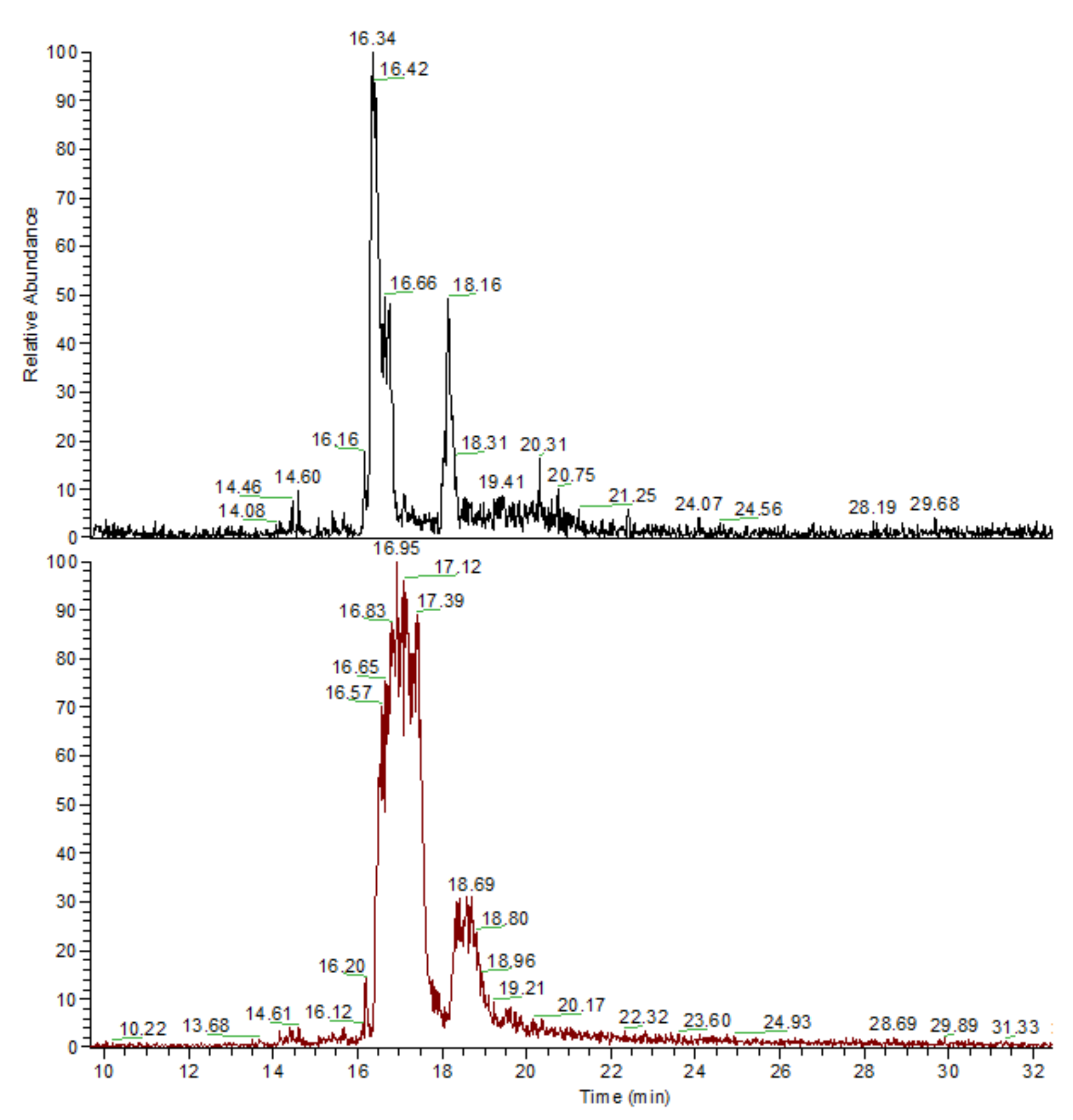
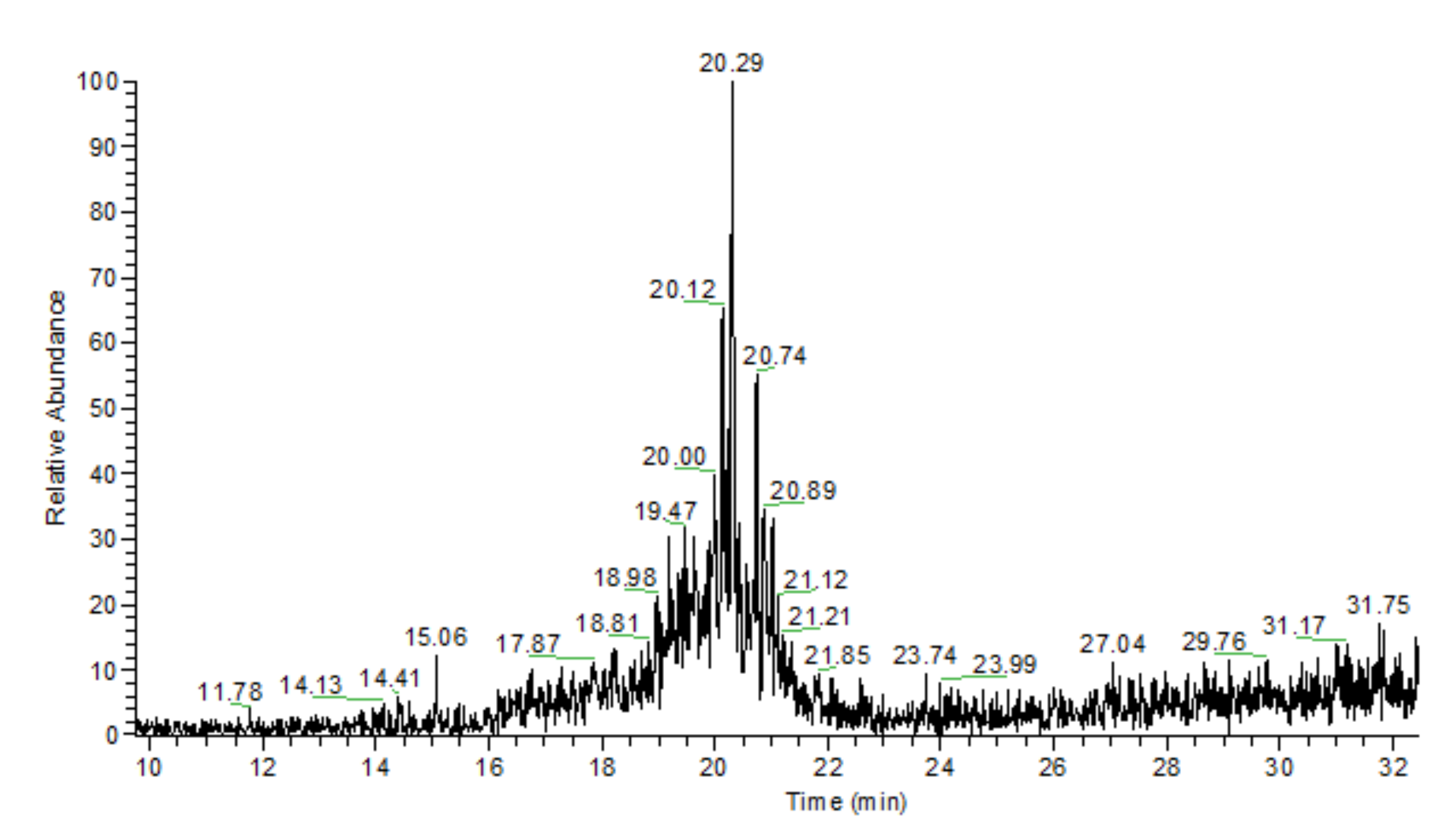
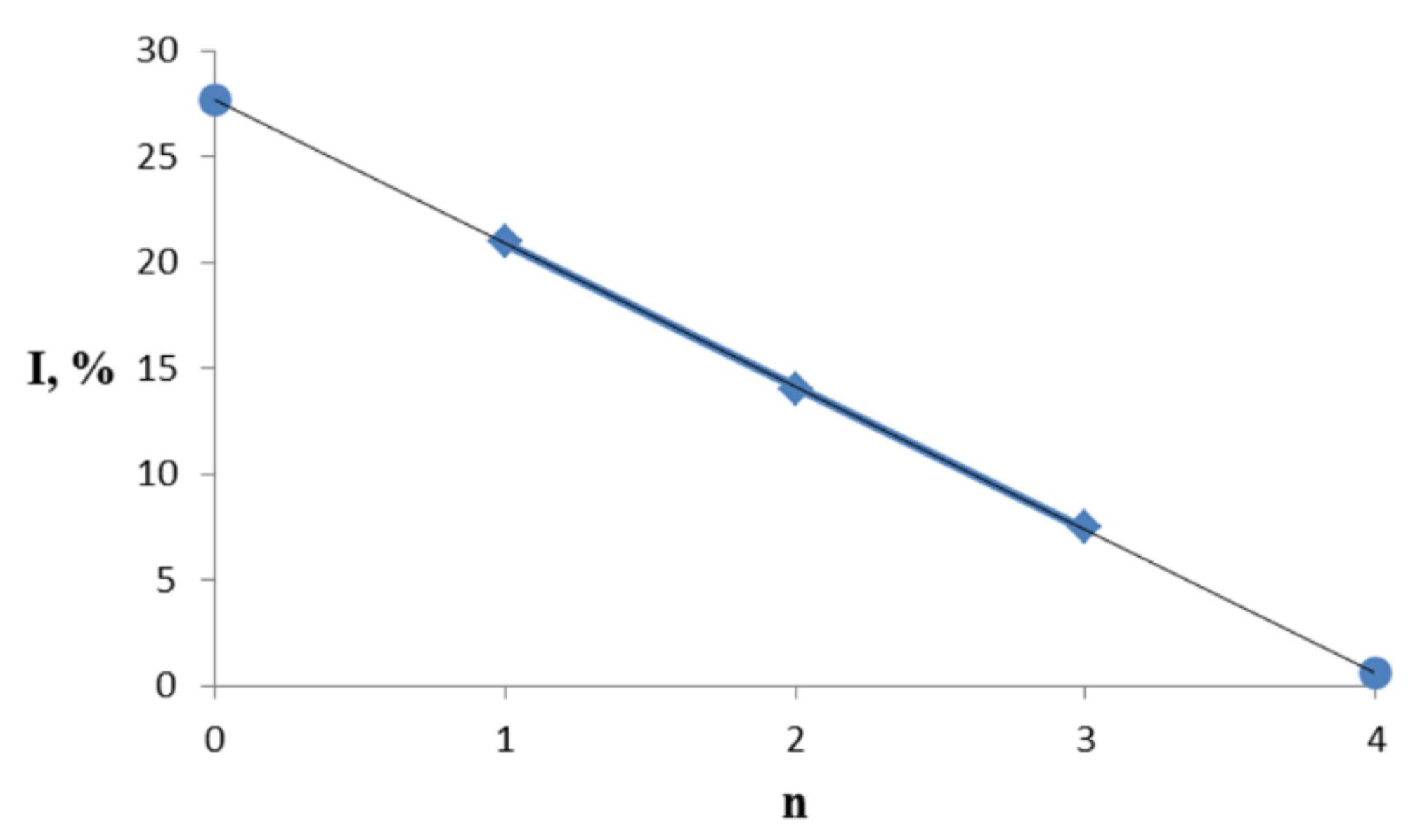
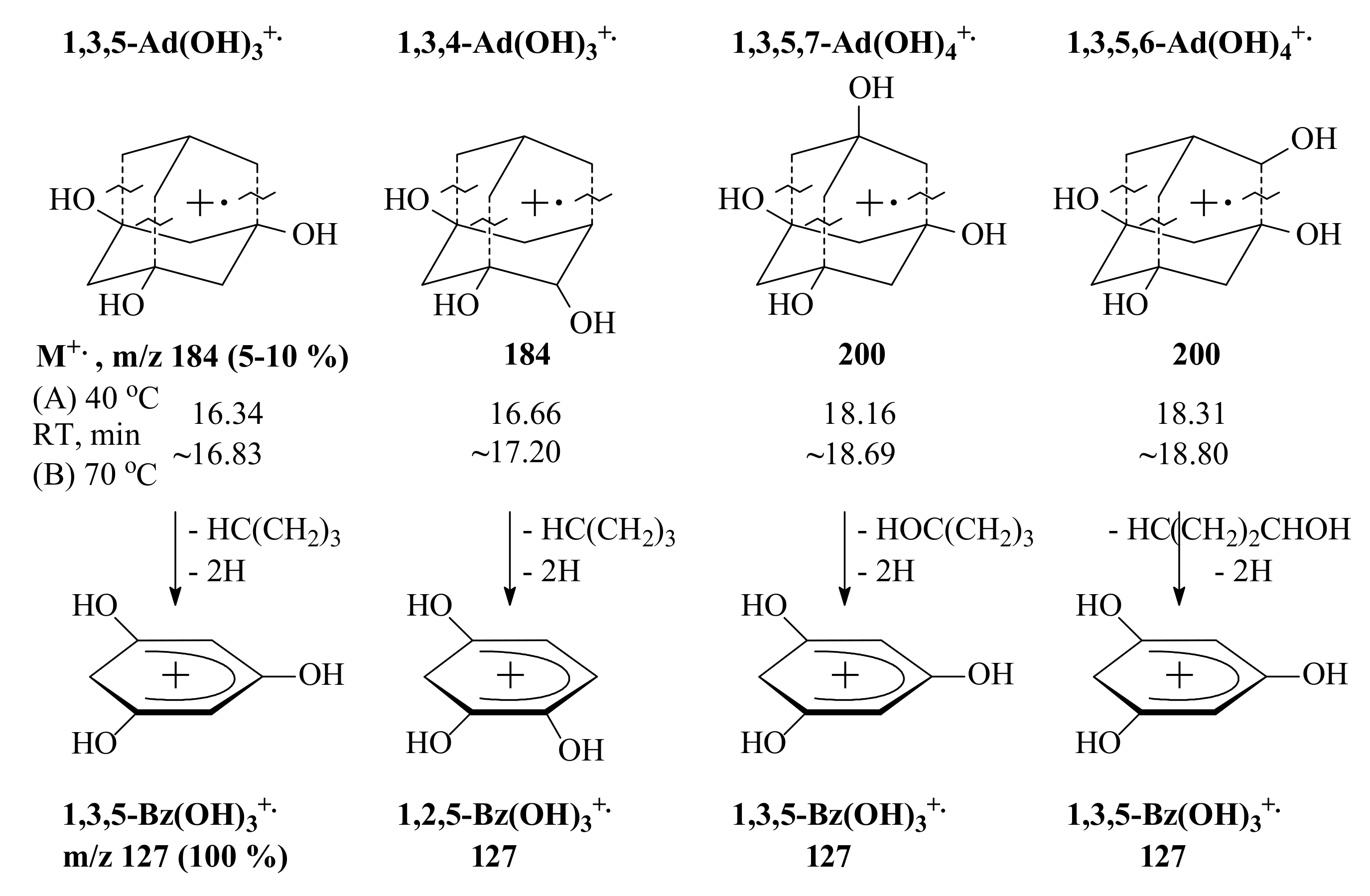
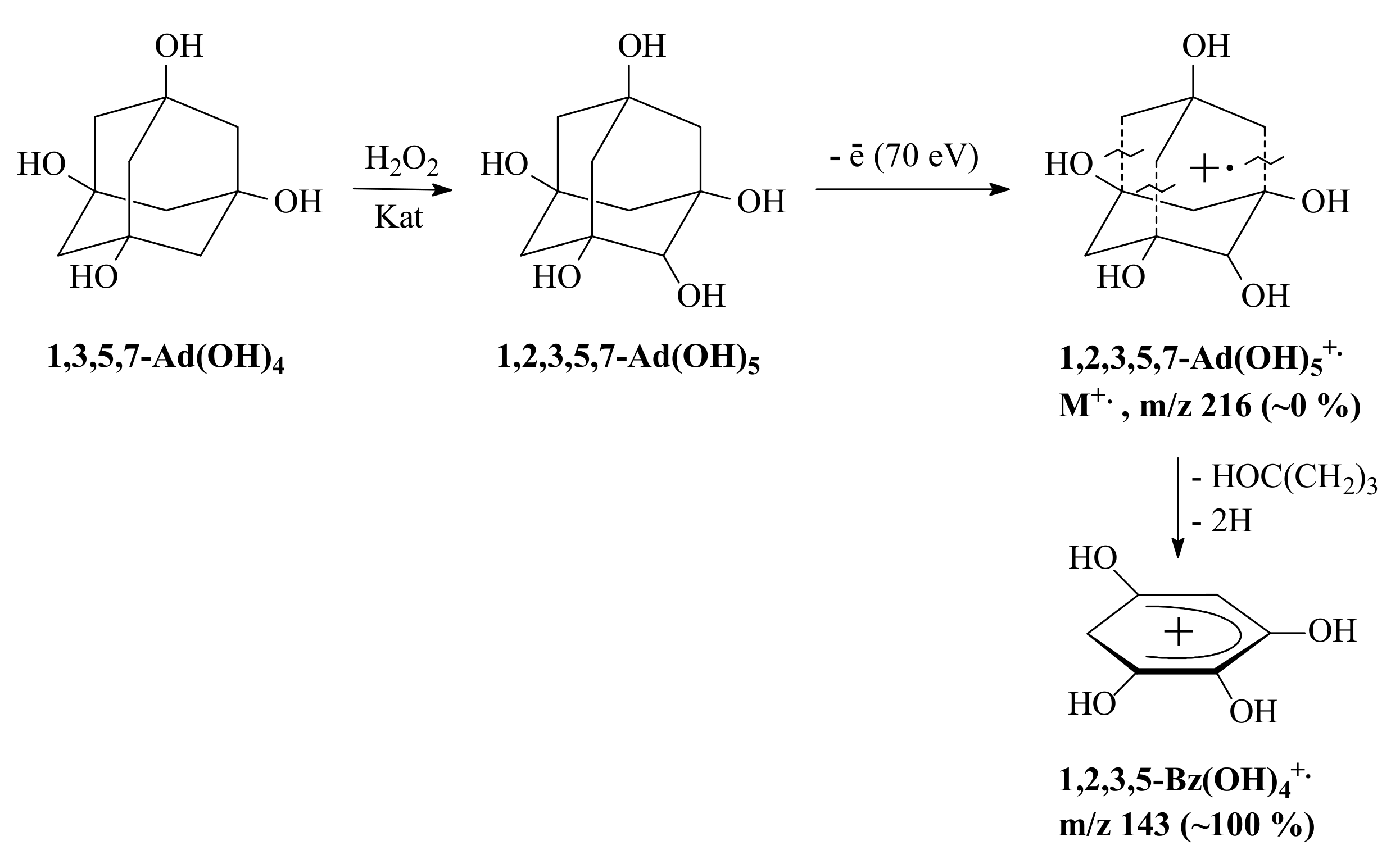
3.3. General Regularity in the Fragmentation of Molecular Ions Ad(OH)n+•, n = 0–4
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Spasov, A.A.; Khamidova, T.V.; Bugaeva, L.I.; Morozov, I.S. Adamantane Derivatives: Pharmacological and Toxicological Properties (Review). Pharm. Chem. J. 2000, 34, 1–7. [Google Scholar] [CrossRef]
- Lamoureux, G.; Artavia, G. Use of the Adamantane Structure in Medicinal Chemistry. Curr. Med. Chem. 2010, 17, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Klimochkin, Y.N.; Shiryaev, V.A.; Leonova, M.V. Antiviral properties of cage compounds. New prospects. Russ. Chem. Bull. 2015, 64, 1473–1496. [Google Scholar] [CrossRef]
- Štimac, A.; Šekutor, M.; Mlinarić-Majerski, K.; Frkanec, L.; Frkanec, R. Adamantane in Drug Delivery Systems and Surface Recognition. Molecules 2017, 22, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomaston, J.L.; Polizzi, N.F.; Konstantinidi, A.; Wang, J.; Kolocouris, A.; DeGrado, W.F. Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters. J. Am. Chem. Soc. 2018, 140, 15219–15226. [Google Scholar] [CrossRef]
- Bagrii, E.I.; Nekhaev, A.I.; Maksimov, A.L. Oxidative Functionalization of Adamantanes (Review). Petrol. Chem. 2017, 57, 183–197. [Google Scholar] [CrossRef]
- Shibata, S.; Sugahara, K.; Kamata, M.; Hara, K. Liquid-phase oxidation of alkanes with molecular oxygen catalyzed by high valent iron-based perovskite. Chem. Commun. 2018, 50, 6772–6775. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. On the nature of the active intermediates in iron-catalyzed oxidation of cycloalkanes with hydrogen peroxide and peracids. Mol. Catal. 2018, 455, 6–13. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kasyanova, K.V.; Makhankova, V.G.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Pombeiro, A.J. Stereospecific sp3 C–H oxidation with m-CPBA: A CoIII Schiff base complex as pre-catalyst vs. its CoIIICdII heterometallic derivative. Appl. Catal. A 2018, 560, 171–184. [Google Scholar] [CrossRef]
- Kislitsina, K.S.; Shchadneva, N.A.; Khusnutdinov, R.I. Oxidation of Adamantane with H2O2–CF3COCF3·1.5H2O in the Presence of VO(acac)2. Russ. J. Org. Chem. 2018, 54, 992–995. [Google Scholar] [CrossRef]
- Lesieur, M.; Battilocchio, C.; Labes, R.; Jacq, J.; Genicot, C.; Ley, S.V.; Pasau, P. Direct Oxidation of Csp3-H bonds using in Situ Generated Trifluoromethylated Dioxirane in Flow. Chem. Eur. J. 2019, 25, 1203–1207. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Du, R.; Yuan, H.; Wang, Y.; Yao, J.; Li, H. Selective One-Step Aerobic Oxidation of Cyclohexane to ε-Caprolactone Mediated by N-Hydroxyphthalimide (NHPI). ChemCatChem 2019, 11, 2260–2264. [Google Scholar] [CrossRef]
- Tabushi, I.; Nakajima, T.; Seto, K. The oxidation of adamantane with an iron salen complex modelling ω-hydroxylase. Tetrahedron Lett. 1980, 21, 2565–2568. [Google Scholar] [CrossRef]
- Song, S.J.; Kim, K.S.; Kim, K.H.; Kim, J.B.; Kim, J.H.; Kim, K.S.; Shin, H.; Cho, D.L. Highly Selective Production of 2-Adamantanone by Photocatalytic Oxidation of Adamantane. Chem. Lett. 2008, 37, 1052–1053. [Google Scholar] [CrossRef]
- Vasil’eva, V.V.; Nekhaev, A.I.; Shchapin, I.Yu.; Bagrii, E.I. On the selectivity of the biomimetic oxidation of the saturated hydrocarbon 1,3-dimethyladamantane in a Gif-type system containing a Fe2+ salt, picolinic acid, and pyridine. Kinet. Catal. 2006, 47, 610–623. [Google Scholar] [CrossRef]
- Shchapin, I.Yu.; Vasil’eva, V.V.; Nekhaev, A.I.; Bagrii, E.I. On a possible radical-cation mechanism of the biomimetic oxidation of the saturated hydrocarbon 1,3-dimethyladamantane in a Gif-type system containing a Fe2+ salt, picolinic acid, and pyridine. Kinet. Catal. 2006, 47, 624–637. [Google Scholar] [CrossRef]
- Geluk, H.W.; Schlatmann, J.L.M.A. Hydride transfer reactions of the adamantyl cation (IV): Synthesis of 1,4- and 2,6-substituted adamantanes by oxidation with sulfuric acid. Recueil Travaux Chim. Pays-Bas 1971, 90, 516–520. [Google Scholar] [CrossRef]
- Curci, R.; D’Accolti, L.; Detomaso, A.; Fusco, C.; Takeuchi, K.I.; Ohga, Y.; Eaton, P.E.; Yip, Y.C. Selective oxidation of tertiary-secondary vic-diols to α-hydroxy ketones by dioxiranes. Tetrahedron Lett. 1993, 34, 4559–4562. [Google Scholar] [CrossRef]
- Labes, R.; Battilocchio, C.; Mateos, C.; Cumming, G.R.; de Frutos, O.; Rincón, J.A.; Binder, K.; Ley, S.V. Chemoselective continuous Ru-catalyzed hydrogen-transfer Oppenauer-type oxidation of secondary alcohols. Org. Process Res. Dev. 2017, 21, 1419–1422. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xu, Z.; Zhou, L.; Hua, L.; Zhang, S.; Wang, J. Temperature responsive polymer-supported TEMPO: An efficient and recoverable catalyst for the selective oxidation of alcohols. Tetrahedron Lett. 2019, 60, 419–422. [Google Scholar] [CrossRef]
- Zhang, W.; Carpenter, K.L.; Lin, S. Electrochemistry broadens the scope of flavin photocatalysis: Photoelectrocatalytic oxidation of unactivated alcohols. Angew. Chem. Int. Ed. 2020, 132, 417–425. [Google Scholar] [CrossRef]
- De Araujo, P.L.B.; Mansoori, G.A.; De Araujo, E.S. Diamondoids: Occurrence in fossil fuels, applications in petroleum exploration and fouling in petroleum production. A review paper. Int. J. Oil Gas Coal Technol. 2012, 5, 316–368. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Alcántara, A.R. Multienzymatic Processes Involving Baeyer–Villiger Monooxygenases. Catalysts 2021, 11, 605. [Google Scholar] [CrossRef]
- Muraoka, O.; Wang, Y.; Okumura, M.; Nishiura, S.; Tanabe, G.; Momose, T. A Facile Synthesis of 7-Methylenebicyclo-[3.3.1]nonan-3-one and its Transformation Leading to the Novel Tricyclic System, Protoadamantane. Synth. Commun. 1996, 26, 1555–1562. [Google Scholar] [CrossRef]
- Ho, T.C. The total synthesis of c1’-azacycloalkyl hexahydrocannabinoids; the total synthesis of 3-oxaadamantyl hexahydrocannabinoids; the synthesis of bicyclic 3-adamantyl cannabinoids. Doctoral dissertation, University of Hawaii at Manoa, Honolulu, HI, USA, 2014. Available online: http://hdl.handle.net/10125/101085 (accessed on 18 August 2021).
- Itoh, H.; Kato, I.; Unoura, K.; Senda, Y. Regioselectivity on Electroreductive Transannular Reaction of 7-Methylenebicyclo[3.3.1]nonan-3-one. Bull. Chem. Soc. Jpn. 2001, 74, 339–345. [Google Scholar] [CrossRef]
- White, R.E.; McCarthy, M.B.; Egeberg, K.D.; Sligar, S.G. Regioselectivity in the cytochromes P-450: Control by protein constraints and by chemical reactivities. Arch. Biochem. Biophys. 1984, 228, 493–502. [Google Scholar] [CrossRef]
- Mitsukura, K.; Kondo, Y.; Yoshida, T.; Nagasawa, T. Regioselective hydroxylation of adamantane by Streptomyces griseoplanus cells. Appl. Microbiol. Biotechnol. 2006, 71, 502–504. [Google Scholar] [CrossRef]
- Mitsukura, K.; Sakamoto, H.; Kubo, H.; Yoshida, T.; Nagasawa, T. Bioconversion of 1-adamantanol to 1,3-adamantanediol using Streptomyces sp. SA8 oxidation system. J. Biosci. Bioeng. 2010, 109, 550–553. [Google Scholar] [CrossRef]
- Mitsukura, K.; Yamanaka, N.; Yoshida, T.; Nagasawa, T. Regioselective synthesis of 1,3,5-adamantanetriol from 1,3-adamantanediol using Kitasatospora cells. Biotechnol. Lett. 2012, 34, 1741–1744. [Google Scholar] [CrossRef]
- Bell, S.G.; Yang, W.; Dale, A.; Zhou, W.; Wong, L.L. Improving the affinity and activity of CYP101D2 for hydrophobic substrates. Appl. Microbiol. Biotechnol. 2013, 97, 3979–3990. [Google Scholar] [CrossRef]
- Md Sarkar, R.; Hall, E.A.; Dasgupta, S.; Bell, S.G. The use of directing groups enables the selective and efficient biocatalytic oxidation of unactivated adamantyl C-H bonds. ChemistrySelect 2016, 1, 6700–6707. [Google Scholar] [CrossRef] [Green Version]
- Alcántara, C.M.; Alcántara, A.R. Biocatalyzed synthesis of antidiabetic drugs: A review. Biocatal. Biotransform. 2018, 36, 12–46. [Google Scholar] [CrossRef]
- Sarkar, M.R. Application of the Monooxygenase Enzymes CYP101B1 and CYP101C1 from Novosphingobium aromaticivorans for Selective and Efficient Functionalisation of Inert CH bonds. Doctoral dissertation, School of Physical Sciences, University of Adelaide, Adelaide, Australia, 2019. Available online: http://hdl.handle.net/2440/119892 (accessed on 18 August 2021).
- Furuya, T.; Kanno, T.; Yamamoto, H.; Kimoto, N.; Matsuyama, A.; Kino, K. Biocatalytic production of 5-hydroxy-2-adamantanone by P450cam coupled with NADH regeneration. J. Mol. Catal. B Enzym. 2013, 94, 111–118. [Google Scholar] [CrossRef]
- Selifonov, S.A. Microbial oxidation of adamantanone by Pseudomonas putida carrying the camphor catabolic plasmid. Biochem. Biophys. Res. Commun. 1992, 186, 1429–1436. [Google Scholar] [CrossRef]
- Wei, Z.; Moldowan, J.M.; Peters, K.E.; Wang, Y.; Xiang, W. The abundance and distribution of diamondoids in biodegraded oils from the San Joaquin Valley: Implications for biodegradation of diamondoids in petroleum reservoirs. Org. Geochem. 2007, 38, 1910–1926. [Google Scholar] [CrossRef]
- Ohshita, J.; Hino, K.; Inata, K.; Kunai, A.; Maehara, T. Palladium-catalyzed silation of adamantanedi- and triol, leading to adamantane–siloxane alternating polymers with high heat resistance. Polymer 2007, 48, 4301–4304. [Google Scholar] [CrossRef]
- Sayed, S.M.; Lin, B.P.; Yang, H. Generation of liquid crystallinity from a Td-symmetry central unit. Soft Matter 2016, 12, 6148–6156. [Google Scholar] [CrossRef]
- Stetter, H.; Krause, M. Zur kenntnis der 1.3.5.7-tetrasubstituierten adamantane. Tetrahedron Lett. 1967, 8, 1841–1843. [Google Scholar] [CrossRef]
- Stetter, H.; Krause, M. Über Verbindungen mit Urotropin-Struktur, XLII. Über 1.3.5.7-tetrasubstituierte Adamantane. Justus Liebigs Ann. Chem. 1968, 717, 60–63. [Google Scholar] [CrossRef]
- Moiseev, I.K.; Belyaev, P.G.; Tartakovskii, V.A. Fragmentation reaction of 1,3,5,7-tetrabromoadamantane. Russ. Chem. Bull. 1973, 22, 245. [Google Scholar] [CrossRef]
- Sollott, G.P.; Gilbert, E.E. A facile route to 1,3,5,7-tetraaminoadamantane. Synthesis of 1,3,5,7-tetranitroadamantane. J. Org. Chem. 1980, 45, 5405–5408. [Google Scholar] [CrossRef]
- Menger, F.M.; Migulin, V.A. Synthesis and Properties of Multiarmed Geminis. J. Org. Chem. 1999, 64, 8916–8921. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Lee, H.-F.; Kuo, S.-W.; Xu, H.; Chang, F.-C. Star polymers via atom transfer radical polymerization from adamantane-based cores. Polymer 2004, 45, 2261–2269. [Google Scholar] [CrossRef]
- Grillaud, M.; Bianco, A. Multifunctional adamantane derivatives as new scaffolds for the multipresentation of bioactive peptides. J. Pept. Sci. 2015, 21, 330–345. [Google Scholar] [CrossRef]
- Fu, S.-Q.; Guo, J.-W.; Zhu, D.-Y.; Yang, Z.; Yang, C.-F.; Xian, J.-X. Li, X. Novel halogen-free flame retardants based on adamantane for polycarbonate. RSC Adv. 2015, 5, 67054–67065. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Y.; Ezell, E.L.; Garrison, J.C.; Wood, J.K.; Hagen, J.P.; Vennerstrom, J.L. Semipinacol and protoadamantane-adamantane rearrangements of 5,6-dibromoadamantan-2-one and -2-ol. Tetrahedron 2017, 73, 2972–2976. [Google Scholar] [CrossRef]
- Schwenger, A.; Birchall, N.; Richert, C. Solution-Phase Synthesis of Branched Oligonucleotides with up to 32 Nucleotides and the Reversible Formation of Materials. Eur. J. Org. Chem. 2017, 39, 5852–5864. [Google Scholar] [CrossRef]
- Nasrallah, H.; Hierso, J.C. Porous Materials Based on 3-Dimensional Td-Directing Functionalized Adamantane Scaffolds and Applied as Recyclable Catalysts. Chem. Mater. 2018, 31, 619–642. [Google Scholar] [CrossRef]
- Klapoetke, T.M.; Krumm, B.; Widera, A. Synthesis and Properties of Tetranitro-Substituted Adamantane Derivatives. ChemPlusChem 2018, 83, 61–69. [Google Scholar] [CrossRef]
- Fu, S.; Zhu, J.; Chen, S. Tunable Shape Memory Polyurethane Networks Cross-Linked by 1,3,5,7-Tetrahydroxyadamantane. Macromol. Res. 2018, 26, 1035–1041. [Google Scholar] [CrossRef]
- Li, M.; Guo, J.-W.; Wen, W.-Q.; Chen, J.K. Biodegradable Redox-Sensitive Star Polymer Nanomicelles for Enhancing Doxorubicin Delivery. Nanomaterials 2019, 9, 547. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, M.; Katagiri, K.; Azumaya, I. Hydrogen-bonded networks formed from tri- and tetra-substituted adamantanes bearing dimethoxyphenol moieties and their 1,3,5-trinitrobenzene complexes via charge-transfer interactions. CrystEngComm. 2010, 12, 1164–1170. [Google Scholar] [CrossRef]
- Alexandre, P.E.; Schwenger, A.; Frey, W.; Richert, C. High-Loading Crystals of Tetraaryladamantanes and the Uptake and Release of Aromatic Hydrocarbons from the Gas Phase. Chem. Eur. J. 2017, 23, 9018–9021. [Google Scholar] [CrossRef]
- Reddy, D.S.; Craig, D.C.; Desiraju, G.R. Topological Equivalences between Organic and Inorganic Crystal Structures: 1,3,5,7-Tetrahydroxyadamantane and Caesium Chloride. J. Chem. Soc. Chem. Commun. 1995, 3, 339–340. [Google Scholar] [CrossRef]
- Mello, R.; Cassidei, L.; Fiorentino, M.; Fusco, C.; Curci, R. Oxidations by methyl(trifluoromethyl)dioxirane. 3. Selective polyoxyfunctionalization of adamantane. Tetrahedron Lett. 1990, 31, 3067–3070. [Google Scholar] [CrossRef]
- D’Accolti, L.; Annese, C.; Fusco, C. Continued Progress towards Efficient Functionalization of Natural and Non-natural Targets under Mild Conditions: Oxygenation by C–H Bond Activation with Dioxirane. Chem. Eur. J. 2019, 25, 12003–12017. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Arakawa, K.; Kodera, M. Electronic Tuning of Iron–Oxo-Mediated C–H Activation: Effect of Electron-Donating Ligand on Selectivity. Chem. Eur. J. 2013, 19, 14697–14701. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Kato, S.; Iwahama, T.; Sakaguchi, S. Hydroxylation of polycyclic alkanes with molecular oxygen catalyzed by N-hydroxyphthalimide (NHPI) combined with transition metal salts. Tetrahedron Lett. 1996, 37, 4993–4996. [Google Scholar] [CrossRef]
- Ivleva, E.A.; Platonov, I.A.; Klimochkin, Y.N. Improved approach towards synthesis of adamantane-1,3,5-triol. Russ. J. Gen. Chem. 2015, 85, 1830–1833. [Google Scholar] [CrossRef]
- Fu, C.A.; Guo, J.; Liu, S.; Cui, Y.; Yang, C.; Peng, J.; Cui, Y. A novel synthesis of adamantane polyhydric alcohols by direct oxidation of adamantine. CIESC J. 2011, 62, 3428. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, J.; Yang, C.; Liu, S.; Cui, Y.; Zhong, X. Synthesis of Adamantane-Based Trimeric Cationic Surfactants. Synth. Commun. 2013, 43, 1161–1167. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Krogul-Sobczak, A.; da Silva, M.F.C.G.; Pombeiro, A.J. Synthesis, crystal structures and catalytic activity of Cu (II) and Mn (III) Schiff base complexes: Influence of additives on the oxidation catalysis of cyclohexane and 1-phenylehanol. J. Mol. Cat. A Chem. 2017, 426, 506–515. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Shul’pina, L.S. Oxidation of Organic Compounds with Peroxides Catalyzed by Polynuclear Metal Compounds. Catalysts 2021, 11, 186. [Google Scholar] [CrossRef]
- Stavropoulos, P.; Çelenligil-Çetin, R.; Tapper, A.E. The Gif paradox. Acc. Chem. Res. 2001, 34, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Butov, G.M.; Mokhov, V.M.; Ledenev, S.M.; Terent’ev, A.O. Method of producing 1-adamantylhydroperoxide. Patent RU 2471780 C1, 25 November 2011. (In Russian). [Google Scholar]
- Terent’ev, A.O.; Platonov, M.M.; Ogibin, Y.N.; Nikishin, G.I. Convenient synthesis of geminal bishydroperoxides by the reaction of ketones with hydrogen peroxide. Synth. Commun. 2007, 37, 1281–1287. [Google Scholar] [CrossRef]
- Sharma, C.; Joy, J.; Nethaji, M.; Jemmis, E.D.; Awasthi, S.K. Synthetic, Crystallographic, and Computational Studies of Extensively Hydrogen Bonded Bilayers in Thermally Stable Adamantane Hydroperoxides. Asian J. Org. Chem. 2016, 5, 1398–1405. [Google Scholar] [CrossRef]
- Ramazanov, D.N.; Kluev, M.V. Oxidation of cyclohexane to adipinic acid by hydrogen peroxide in the presence of copper complexes. Russ. J. Chem. And Chem. Tech. 2009, 52, 44–46. (In Russian) [Google Scholar]
- Svedung, D.H. The crystal structure of copper dimethylglyoxime dichloride. Acta Chem. Scand. 1969, 23, 2865–2878. [Google Scholar] [CrossRef]
- Gawlig, C.; Schindler, S.; Becker, S. One-Pot Conversion of Cyclohexane to Adipic Acid Using a µ4-Oxido-Copper Cluster as Catalyst Together with Hydrogen Peroxide. Eur. J. Inorg. Chem. 2020, 2020, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.P.C.; Spada, E.; Bertani, R.; Martins, L.M.D.R.S. Adipic Acid Route: Oxidation of Cyclohexene vs. Cyclohexane. Catalysts 2020, 10, 1443. [Google Scholar] [CrossRef]
- Stein, S.E. NIST/EPA/NIH Mass Spectral Database (NIST 11,17) and NIST Mass Spectral Search Program, version 2.0g; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2011. [Google Scholar]
- Takamuku, T.; Tabata, M.; Yamaguchi, A.; Nishimoto, J.; Kumamoto, M.; Wakita, H.; Yamaguchi, T. Liquid Structure of Acetonitrile−Water Mixtures by X-ray Diffraction and Infrared Spectroscopy. J. Phys. Chem. B 1998, 102, 8880–8888. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Mushtakova, S.P.; Kolesnikova, S.S.; Gribov, L.A. Spectral-chemometric and quantum-chemical study of the water-acetonitrile system. J. Anal. Chem. 2011, 66, 53–59. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Mushtakova, S.P. Methanol and acetonitrile associates in aqueous and chloroform solutions according to 1H NMR spectroscopic data. Russ. J. Phys. Chem. A 2014, 88, 798–802. [Google Scholar] [CrossRef]
- Stepakova, L.V.; Skripkin, M.Yu.; Korneeva, V.V.; Grigoriev, Ya.M.; Burkov, K.A. Organic solvent effect on the solution-solid phase equilibria in the systems CuCl2-L-H2O (L = DMSO, DMF, acetonitrile) at 25°C. Russ. J. Gen. Chem. 2009, 79, 1053–1056. [Google Scholar] [CrossRef]
- Bouwman, J.; Horst, S.; Oomens, J. Spectroscopic Characterization of the Product Ions Formed by Electron Ionization of Adamantane. Chem. Phys. Chem. 2018, 19, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Candian, A.; Bouwman, J.; Hemberger, P.; Bodi, A.; Tielens, A.G. Dissociative ionisation of adamantane: A combined theoretical and experimental study. Phys. Chem. Chem. Phys. 2018, 20, 5399–5406. [Google Scholar] [CrossRef] [Green Version]
- Solcà, N.; Dopfer, O. Protonated Benzene: IR Spectrum and Structure of C6H7+. Angew. Chem. Int. Ed. 2002, 41, 3628–3631. [Google Scholar] [CrossRef]
- Rode, M.F.; Sobolewski, A.L.; Dedonder, C.; Jouvet, C.; Dopfer, O. Computational Study on the Photophysics of Protonated Benzene. J. Phys. Chem. A 2009, 113, 5865–5873. [Google Scholar] [CrossRef] [PubMed]
- Garkusha, I.; Fulara, J.; Nagy, A.; Maier, J.P. Electronic Transitions of Protonated Benzene and Fulvene, and of C6H7 Isomers in Neon Matrices. J. Am. Chem. Soc. 2010, 132, 14979–14985. [Google Scholar] [CrossRef]
- Chen, H.-F.; Wu, Y.-J. Structure, stability, and vibrational fundamentals of low-lying isomers of C6H7+. Comput. Theor. Chem. 2013, 1006, 100–104. [Google Scholar] [CrossRef]




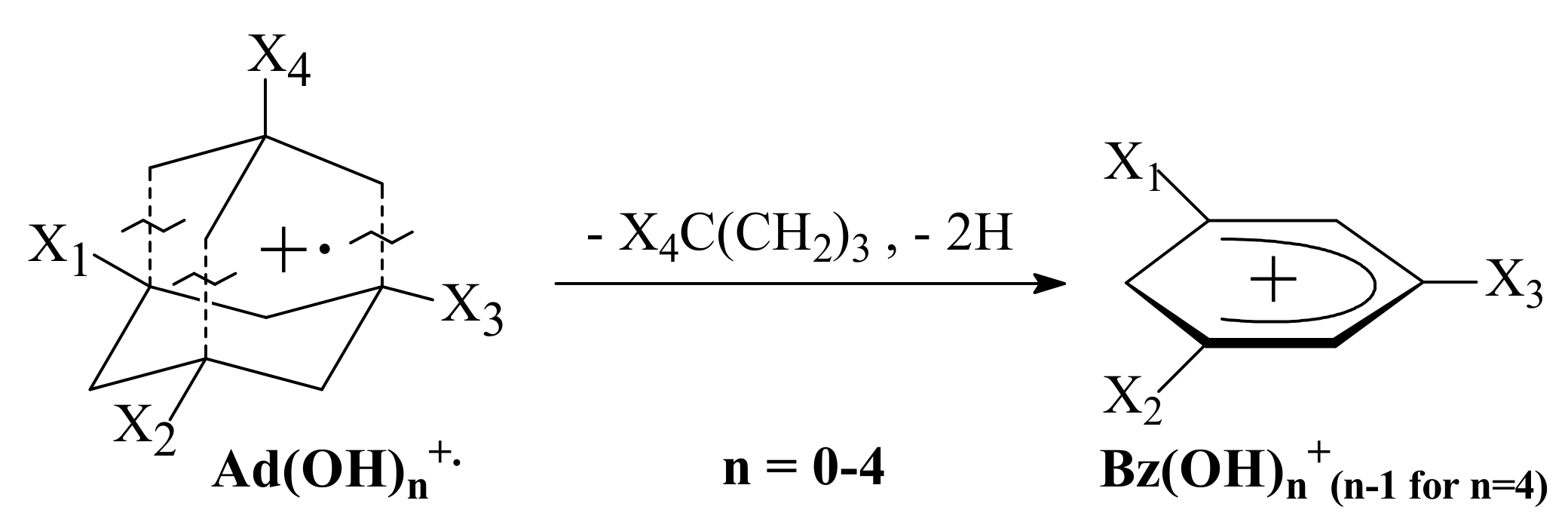


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
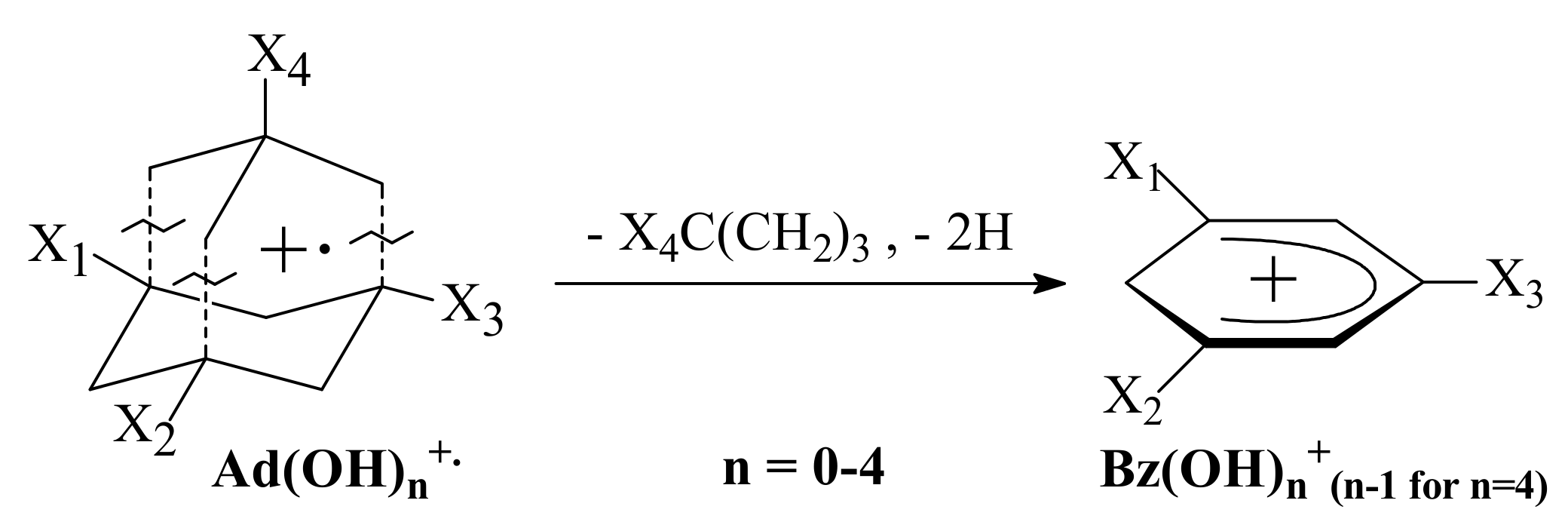{kind=link}
| n | T, °C | |||||
|---|---|---|---|---|---|---|
| 30 | 40 | 50 | 60 | 70 | 80 | |
| % | ||||||
| 1 | 20 | 14 | 10 | 9 | 9 | 22 |
| 2 | 46 | 40 | 18 | 36 | 40 | 30 |
| 3 | 26 | 27 | 45 | 35 | 34 | 31 |
| 4 | 8 | 12 | 25 | 18 | 16 | 15 |
| 5 | 0 | 7 | 2 | 2 | 1 | 2 |
| V, mL | X(AN), wt% | Y(AN), mol Fraction | N(AN): N(Ox), AN Molecules: Ox Molecules | |
|---|---|---|---|---|
| Ox * | AN ** | |||
| 0.625 | 8 | 91 | 0.81 | 4.4:1 |
| 1.5 | 8 | 80 | 0.65 | 1.8:1 |
| 10 | 10 | 43 | 0.25 | 1:2.9 |
| Sub | Products, % | C, % | Ox | Cat | Sol | T, °C | t, h | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 2a | 3a | 4a | ||||||||
| 0 | 10 | 53 | >99 | O2 | NHPI/ Co(acac)2 | AcOH | 75 | 6 | [60] | ||
| 1a | 76 | 18 | 95 | 15 | |||||||
| 2a | 85 | 46 | 24 | ||||||||
| 2a | 42 | 56 | 100 | +MnO2 | 60 | 30 | [61] | ||||
| 0 | 42 | n.r. | CrO3 | +H2SO4 | 80 | 3 | [62] | ||||
| 0 | 50 * | n.r. | AcOH | 100 | >1 | [63] | |||||
| 0 | 90 | 98 | O2C(CF3)CH3 | CH2Cl2/TFP (2:1) | −20 | 2 | [57] | ||||
| 78 | n.r. | 3 | |||||||||
| 0 | 2 | 12 | 32 | 46 | H2O2 | Fe(III) ** | CH3CN | 25 | 5 | [59] | |
| 0 | 5 | 4 | 10 | 3 | 100 | Cu2Cl4 2DMG | 50 | 1 | *** | ||
| Ad(OH)n | X1 | X2 | X3 | X4 | M+ | Bz(OH)n+ | |||
|---|---|---|---|---|---|---|---|---|---|
| No. | n | m/z | % | m/z | % | ||||
| 0 | 0 | H | H | H | H | 136 | 100 | 79 | 74 |
| 28 * | 100 * | ||||||||
| 1a | 1 | OH | H | H | H | 152 | 21 | 95 | 100 |
| 2a | 2 | OH | OH | H | H | 168 | 14 | 111 | 100 |
| 3a | 3 | OH | OH | OH | H | 184 | 7.5 | 127 | 100 |
| 4a | 4 | OH | OH | OH | OH | 200 | ~1 ** | 127 *** | 100 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchapin, I.Y.; Ramazanov, D.N.; Nekhaev, A.I.; Borisov, R.S.; Buravlev, E.A.; Maximov, A.L. One-Stage Catalytic Oxidation of Adamantane to Tri-, Tetra-, and Penta-Ols. Catalysts 2021, 11, 1017. https://doi.org/10.3390/catal11081017
Shchapin IY, Ramazanov DN, Nekhaev AI, Borisov RS, Buravlev EA, Maximov AL. One-Stage Catalytic Oxidation of Adamantane to Tri-, Tetra-, and Penta-Ols. Catalysts. 2021; 11(8):1017. https://doi.org/10.3390/catal11081017
Chicago/Turabian StyleShchapin, Igor Yu., Dzhamalutdin N. Ramazanov, Andrey I. Nekhaev, Roman S. Borisov, Evgeny A. Buravlev, and Anton L. Maximov. 2021. "One-Stage Catalytic Oxidation of Adamantane to Tri-, Tetra-, and Penta-Ols" Catalysts 11, no. 8: 1017. https://doi.org/10.3390/catal11081017
APA StyleShchapin, I. Y., Ramazanov, D. N., Nekhaev, A. I., Borisov, R. S., Buravlev, E. A., & Maximov, A. L. (2021). One-Stage Catalytic Oxidation of Adamantane to Tri-, Tetra-, and Penta-Ols. Catalysts, 11(8), 1017. https://doi.org/10.3390/catal11081017