
A Titania-Supported Polyoxometalate and Au Cocatalyst for Efficient Photocatalytic Environmental Remediation



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
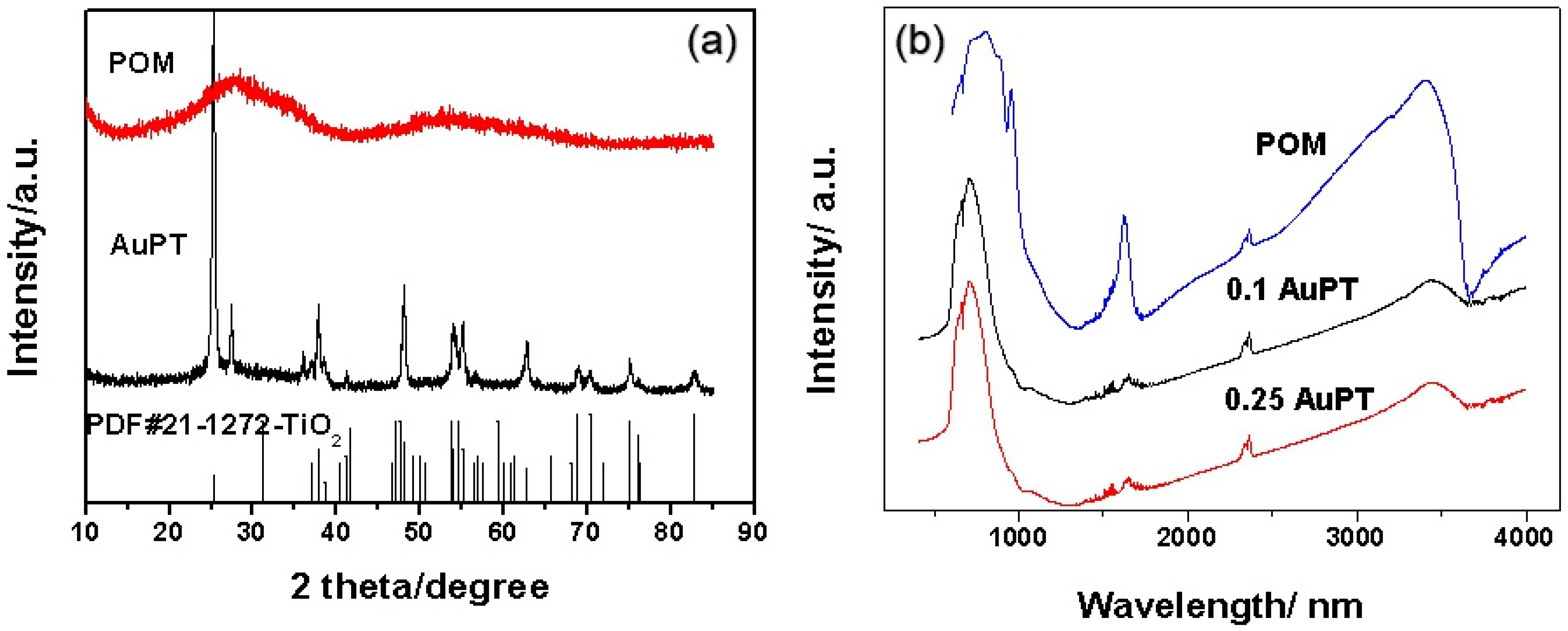
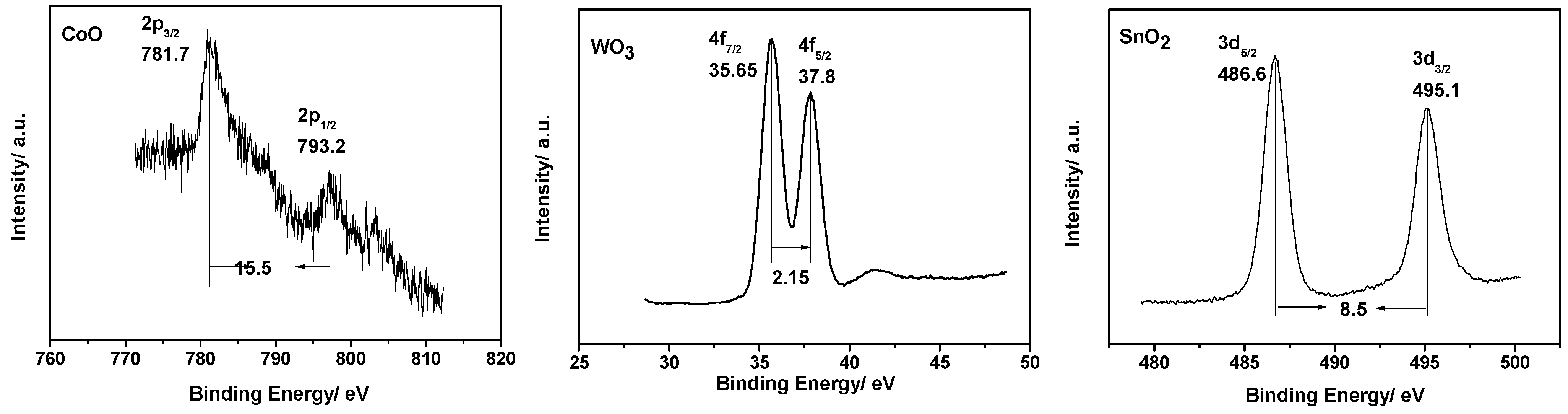
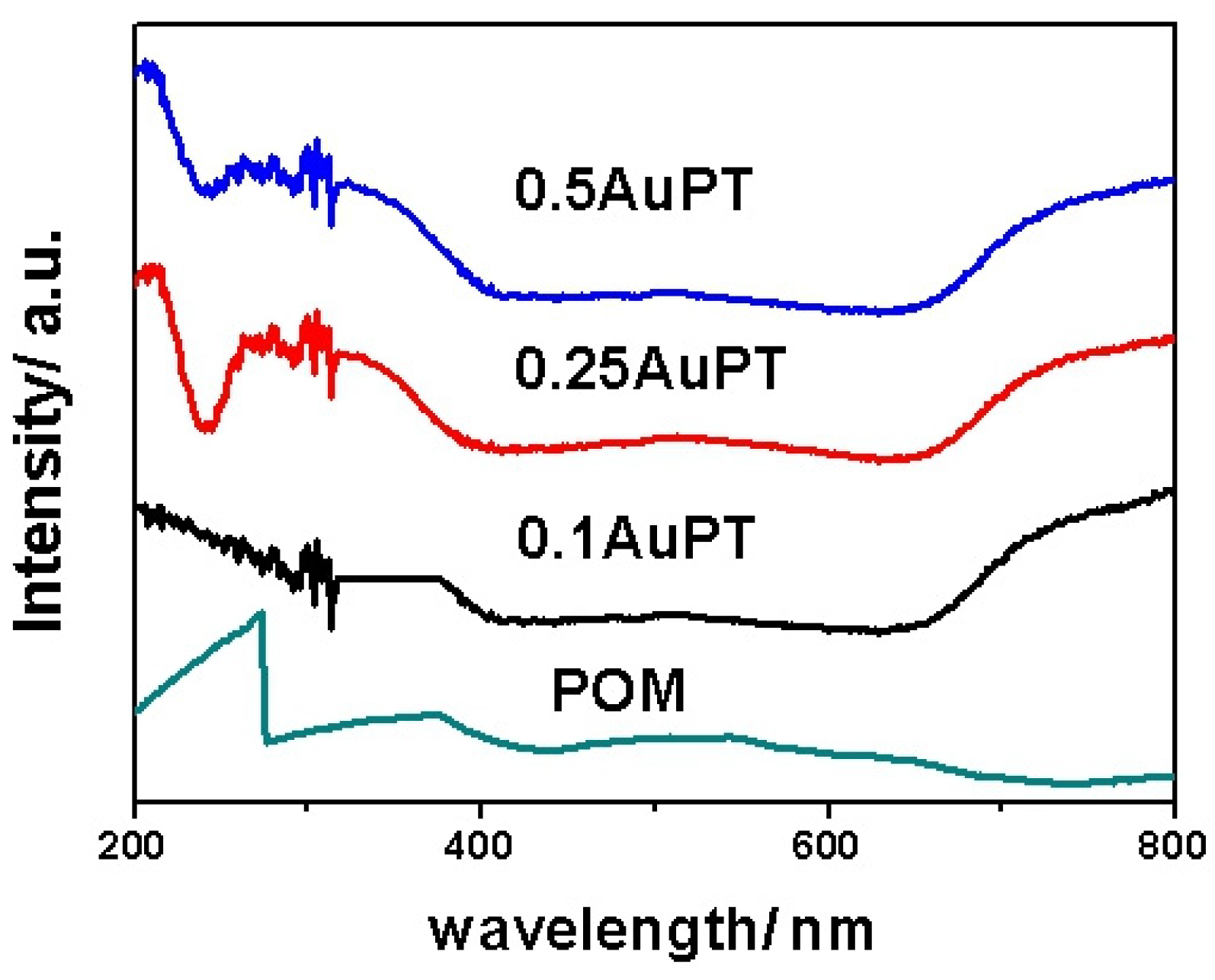
2.1. Characterization of Photocatalysts
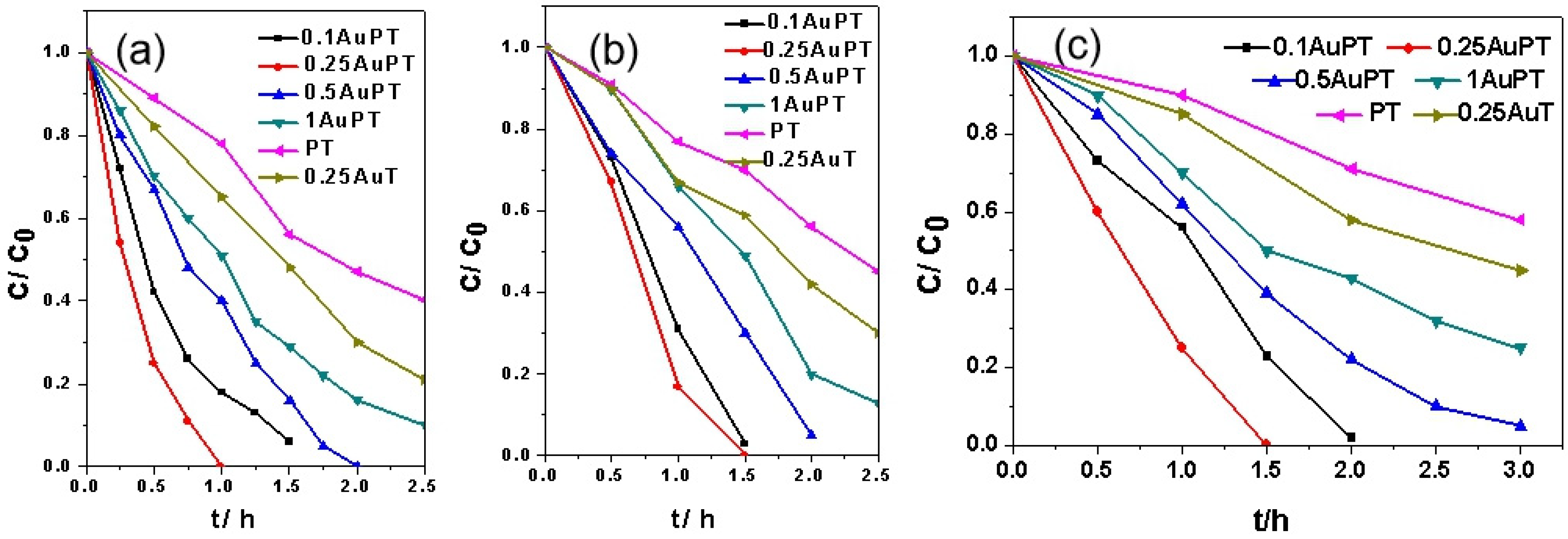
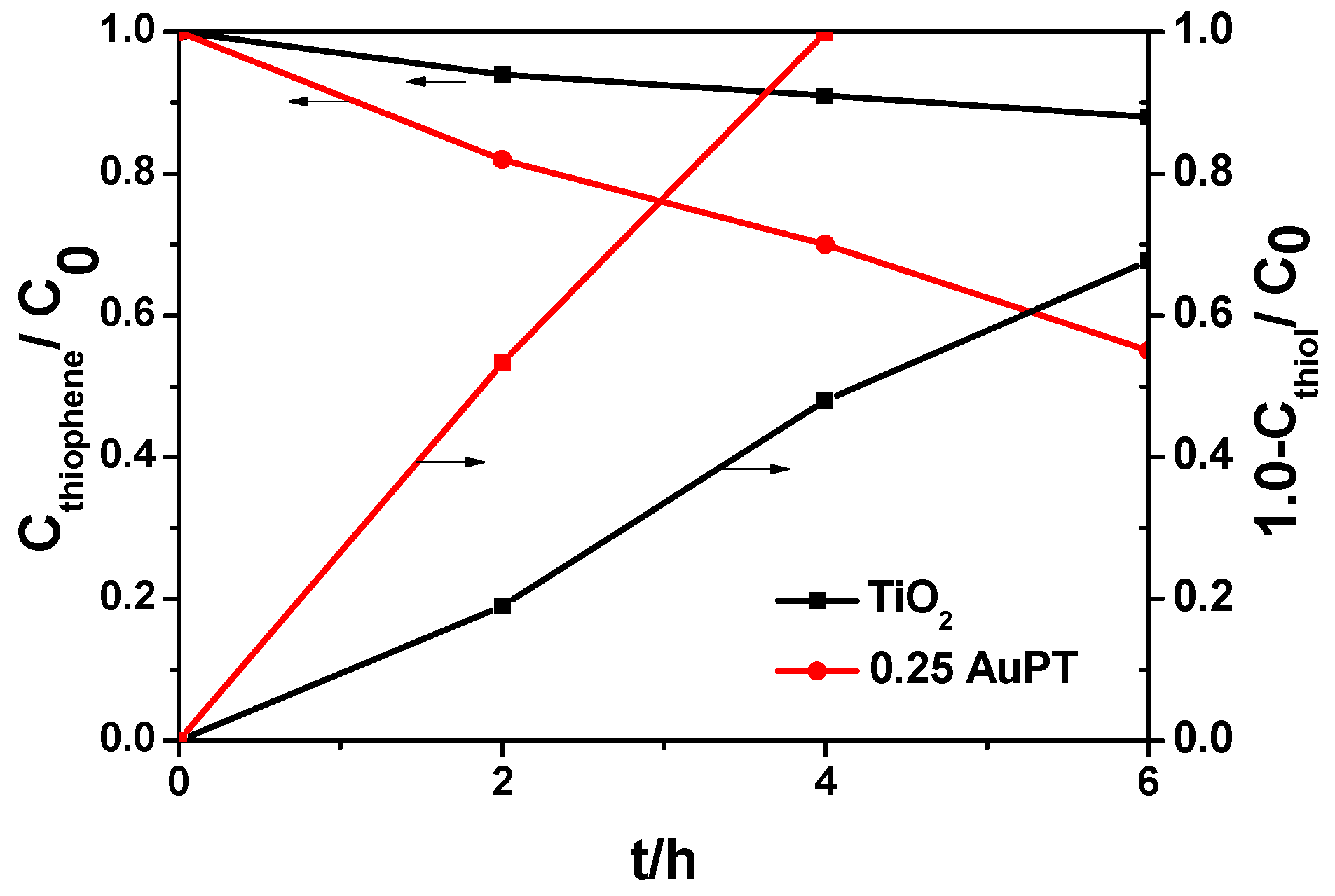
2.2. Effect of Au SPR on the Catalytic Activities of Photocatalysts
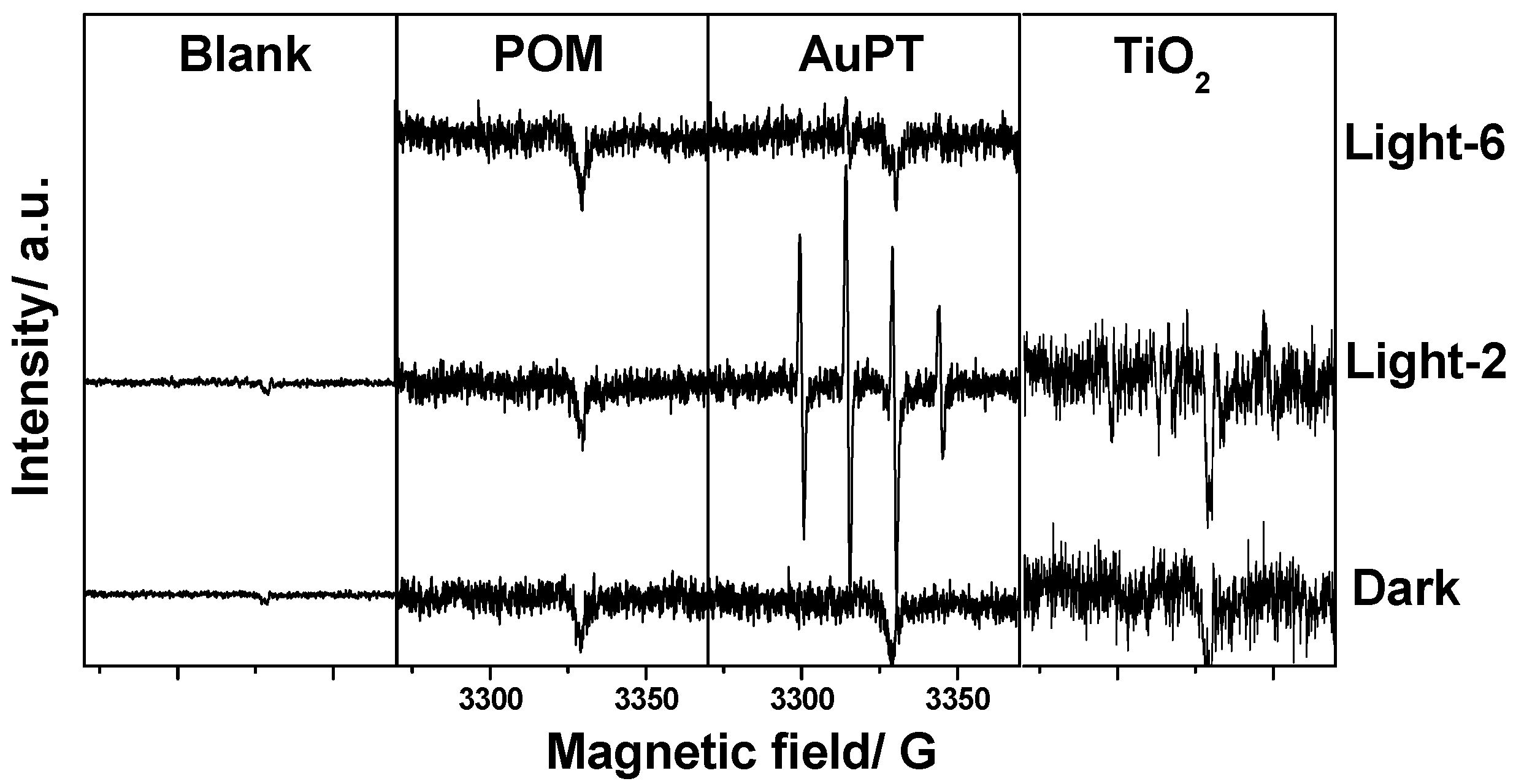
2.3. Proposed Reaction Mechanism
3. Materials and Methods
3.1. Fabrication of AuPT Catalysts
3.2. Characterization
3.3. Photocatalytic Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tian, J.; Zhao, Z.; Kumar, A.; Boughton, R.; Liu, H. Recent progress in design, synthesis, and applications of one-dimensional TiO2 nanostructured surface heterostructures: A review. Chem. Sci. Rev. 2014, 43, 6920–6937. [Google Scholar] [CrossRef]
- Li, W.; Elzatahry, A.; Aldhayan, D.; Zhao, D. Core–shell structured titanium dioxide nanomaterials for solar energy utilization. Chem. Sci. Rev. 2018, 47, 8203–8237. [Google Scholar] [CrossRef]
- Tseng, E.; Hsiao, Y.; Chen, Y.; Chen, S.; Gloter, A.; Song, J. Magnetism and plasmonic performance of mesoscopic hollow ceria spheres decorated with silver nanoparticles. Nanoscale 2019, 11, 3574–3582. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Shao, B.; Li, Z.; Zhang, J.; Wang, H.; Zhang, S.; Haruta, M.; Huang, J. Visible light photocatalysis over solid acid: Enhanced by gold plasmonic effect. Appl. Catal. B Environ. 2017, 218, 480–487. [Google Scholar] [CrossRef]
- Umakoshi, T.; Saitoa, Y.; Verma, P. Highly efficient plasmonic tip design for plasmon nanofocusing in near-field optical microscopy. Nanoscale 2016, 8, 5634–5640. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.X.; Dzade, N.Y.; Zhang, Y.; Hofmann, J.P.; Leeuw, N.H.; Weckhuysen, B.M. Identification of Photoexcited Electron Relaxation in a Cobalt Phosphide Modified Carbon Nitride Photocatalyst. ChemPhotoChem 2021. [Google Scholar] [CrossRef]
- Thomann, I.; Pinaud, B.A.; Chen, Z.; Clemens, B.M.; Jaramillo, T.F.; Brongersma, M.L. Plasmon Enhanced Solar-to-Fuel Energy Conversion. Nano Lett. 2011, 11, 3440–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadakane, M.; Steckhan, E. Electrochemical Properties of Polyoxometalates as Electrocatalysts. Chem. Rev. 1998, 98, 219–238. [Google Scholar] [CrossRef]
- Maradur, S.P.; Halligudi, S.B.; Gokavi, G. Oxidation of Aliphatic and Benzylic Alcohols by Oxone®, Catalysed by 12-Tungstocobaltate (II). Catal. Lett. 2004, 96, 3–4. [Google Scholar] [CrossRef]
- Wu, Q.; Sang, X.; Shao, F.; Pang, W. Synthesis and conductivity of undecatungstocobaltoindic heteropoly acid. Mater. Chem. Phys. 2004, 92, 16–20. [Google Scholar] [CrossRef]
- Yang, Y.; He, J.; Wang, X.; Li, B.; Liu, J. Preparation, characterization and in vitro antitumoral activity of a nanosize liposome complex encapsulated polyoxotungstate K6H2[CoW11TiO40]. Transit. Met. Chem. 2004, 29, 96–99. [Google Scholar] [CrossRef]
- Ji, Y.; Shen, L.; Wang, A.; Wu, M.; Tang, Y.; Chen, Y.; Lu, T. Electrocatalytic performance of carbon supported Pd catalyst modified with Keggin type of Sn-substituted polyoxometalatate for formic acid oxidization. J. Power Sources 2014, 260, 82–88. [Google Scholar] [CrossRef]
- Liu, X.; Iocozzia, J.; Wang, Y.; Cui, X.; Chen, Y.; Zhao, S.; Li, Z.; Lin, Z. Noble metal–metal oxide nanohybrids with tailored nanostructures for efficient solar energy conversion, photocatalysis and environmental remediation. Energy Environ. Sci. 2017, 10, 402–434. [Google Scholar] [CrossRef]
- Qi, J.; Chen, J.; Li, G.; Li, S.; Gao, Y.; Tang, Z. Facile synthesis of core–shell Au@CeO2 nanocomposites with remarkably enhanced catalytic activity for CO oxidation. Energy Environ. Sci. 2012, 5, 8937–8941. [Google Scholar] [CrossRef]
- Bawaked, S.; Dummer, N.; Dimitratos, N.; Bethell, D.; He, Q.; Kielyb, C.; Hutchings, G. Solvent-free selective epoxidation of cyclooctene using supported gold catalysts. Green Chem. 2009, 11, 1037–1044. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S.; Dutta, S.; Zboril, R.; Gawande, M. Silica-nanosphere-based organic–inorganic hybrid nanomaterials: Synthesis, functionalization and applications in catalysis. Green Chem. 2015, 17, 3207–3230. [Google Scholar] [CrossRef]
- Eustis, S.; El-Sayed, M.A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217. [Google Scholar] [CrossRef]
- Cushing, S.K.; Li, J.T.; Meng, F.K.; Senty, T.R.; Suri, S.; Zhi, M.J.; Li, M.; Bristow, A.D.; Wu, N.Q. Photocatalytic Activity Enhanced by Plasmonic Resonant Energy Transfer from Metal to Semiconductor. J. Am. Chem. Soc. 2012, 134, 15033–15041. [Google Scholar] [CrossRef]
- Ide, Y.; Matsuoka, M.; Ogawa, M. Efficient Visible-Light-Induced Photocatalytic Activity on Gold-Nanoparticle-Supported Layered Titanate. J. Am. Chem. Soc. 2010, 132, 16762–16764. [Google Scholar] [CrossRef]
- Zhang, Q.; Lima, D.Q.; Lee, I.; Zaera, F.; Chi, M.; Yin, Y. A Highly Active Titanium Dioxide Based Visible-Light Photocatalyst with Nonmetal Doping and Plasmonic Metal Decoration. Angew. Chim. 2011, 123, 7226–7230. [Google Scholar] [CrossRef]
- Song, J.; Luo, Z.; Zhu, H.; Huang, Z.; Lian, T.; Kaledin, A.L.; Musaev, D.G.; Lense, S.; Hardcastle, K.I.; Hill, C.L. Synthesis, structure, and characterization of two polyoxometalate–photosensitizer hybrid materials. Inorg. Chim. Acta 2010, 363, 4381–4386. [Google Scholar] [CrossRef]
- Niu, J.; Wang, Z.; Wang, J. Hydrothermal synthesis and structure characterization of a Keggin tungstocobaltate [Co(2,2′-bipy)3]2H2[CoW12O40] · 9.5H2O. Polyhedron 2004, 23, 773–777. [Google Scholar] [CrossRef]
- Nomiya, K.; Murasaki, H.; Miwa, M. Catalysis by heteropolyacids—VIII. Immobilization of keggin-type heteropolyacids on poly(4-vinylpyridine). Polyhedron 1986, 5, 1031–1033. [Google Scholar] [CrossRef]
- Guo, Z.; Han, D.; Wexler, D.; Zeng, R.; Liu, H. Polyoxometallate-stabilized platinum catalysts on multi-walled carbon nanotubes for fuel cell applications. Electrochim. Acta 2008, 53, 6410–6416. [Google Scholar] [CrossRef]
- Pan, D.; Tao, W.; Nie, L.; Yao, S. Polyoxometalate-Modified Carbon Nanotubes: New Catalyst Support for Methanol Electro-oxidation. Langmuir 2006, 22, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.R.; Hair, M.L. Detection of superoxide ions in nonaqueous media. Generation by photolysis of pigment dispersions. J. Phys. Chem. 1978, 82, 1397–1399. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Ma, W.; Cheng, M.; Zhao, J.; Yu, J.C. Efficient H2O2 Oxidation of Organic Pollutants Catalyzed by Supported Iron Sulfophenylporphyrin under Visible Light Irradiation. J. Phys. Chem. B 2004, 108, 7263–7270. [Google Scholar] [CrossRef]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/Gold Nanocomposites. Effect of Metal Particle Size on the Fermi Level Equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Jiang, Z.; Tang, N.; Zhang, C.; Chen, Z.; Liu, T.; Dong, B. Photocatalytic oxidation of thiophene on RuO2/SO42−-TiO2: Insights for cocatalyst and solid-acid. Appl. Catal. B Environ. 2016, 188, 253–258. [Google Scholar] [CrossRef]
- Sandulescu, A.; Anastasescu, C.; Papa, F.; Raciulete, M.; Vasile, A.; Spataru, T.; Scarisoreanu, M.; Fleaca, C.; Mihailescu, C.N.; Teodorescu, V.S.; et al. Advancements on Basic Working Principles of Photo-Driven Oxidative Degradation of Organic Substrates over Pristine and Noble Metal-Modified TiO2. Model Case of Phenol Photo Oxidation. Catalysts 2021, 11, 487. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, F.; Yang, Y.; Zhang, Z.; Tang, N.; Zhu, G. A Titania-Supported Polyoxometalate and Au Cocatalyst for Efficient Photocatalytic Environmental Remediation. Catalysts 2021, 11, 1045. https://doi.org/10.3390/catal11091045
Lin F, Yang Y, Zhang Z, Tang N, Zhu G. A Titania-Supported Polyoxometalate and Au Cocatalyst for Efficient Photocatalytic Environmental Remediation. Catalysts. 2021; 11(9):1045. https://doi.org/10.3390/catal11091045
Chicago/Turabian StyleLin, Feng, Yun Yang, Zhen Zhang, Nanfang Tang, and Guangqi Zhu. 2021. "A Titania-Supported Polyoxometalate and Au Cocatalyst for Efficient Photocatalytic Environmental Remediation" Catalysts 11, no. 9: 1045. https://doi.org/10.3390/catal11091045
APA StyleLin, F., Yang, Y., Zhang, Z., Tang, N., & Zhu, G. (2021). A Titania-Supported Polyoxometalate and Au Cocatalyst for Efficient Photocatalytic Environmental Remediation. Catalysts, 11(9), 1045. https://doi.org/10.3390/catal11091045