
Highly Active Rutile TiO2 for Photocatalysis under Violet Light Irradiation at 405 nm


Abstract
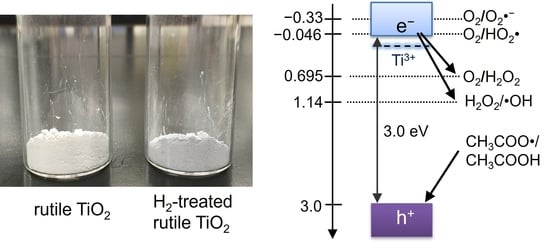
:
1. Introduction
2. Results
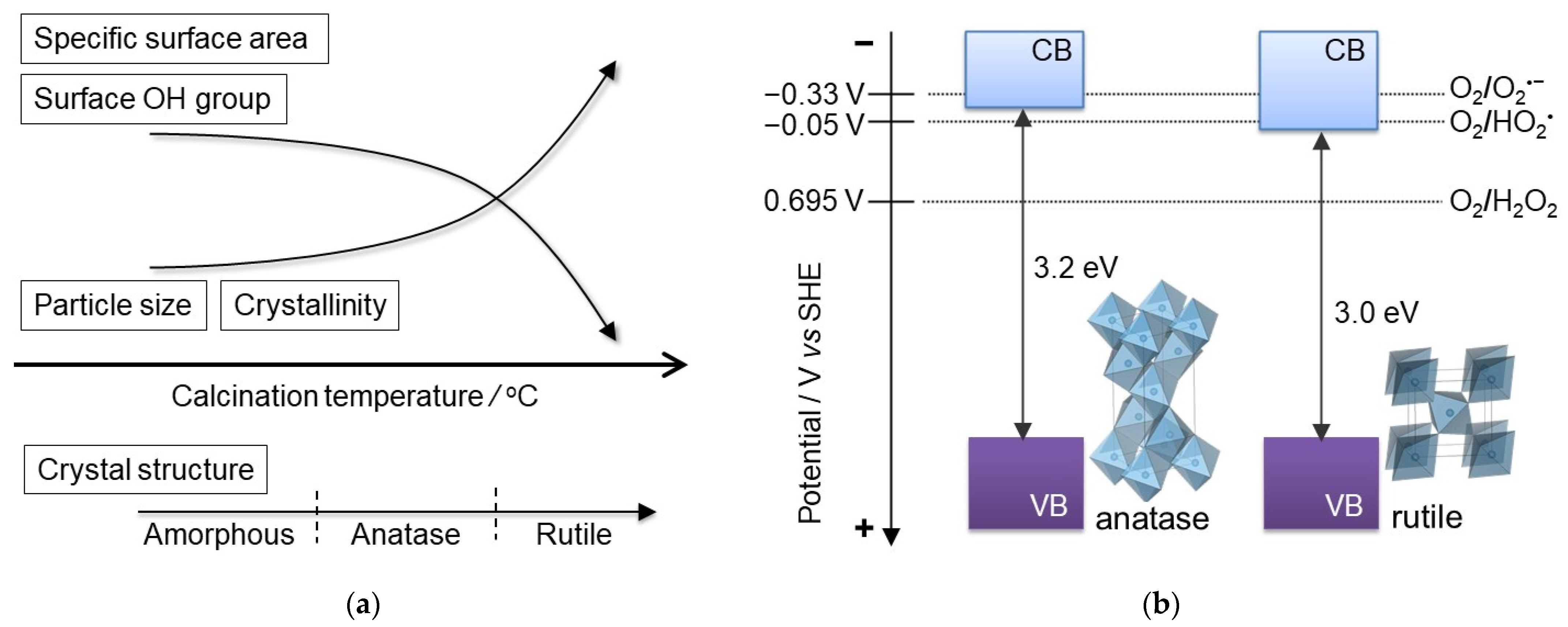
2.1. Preparation and Characterization
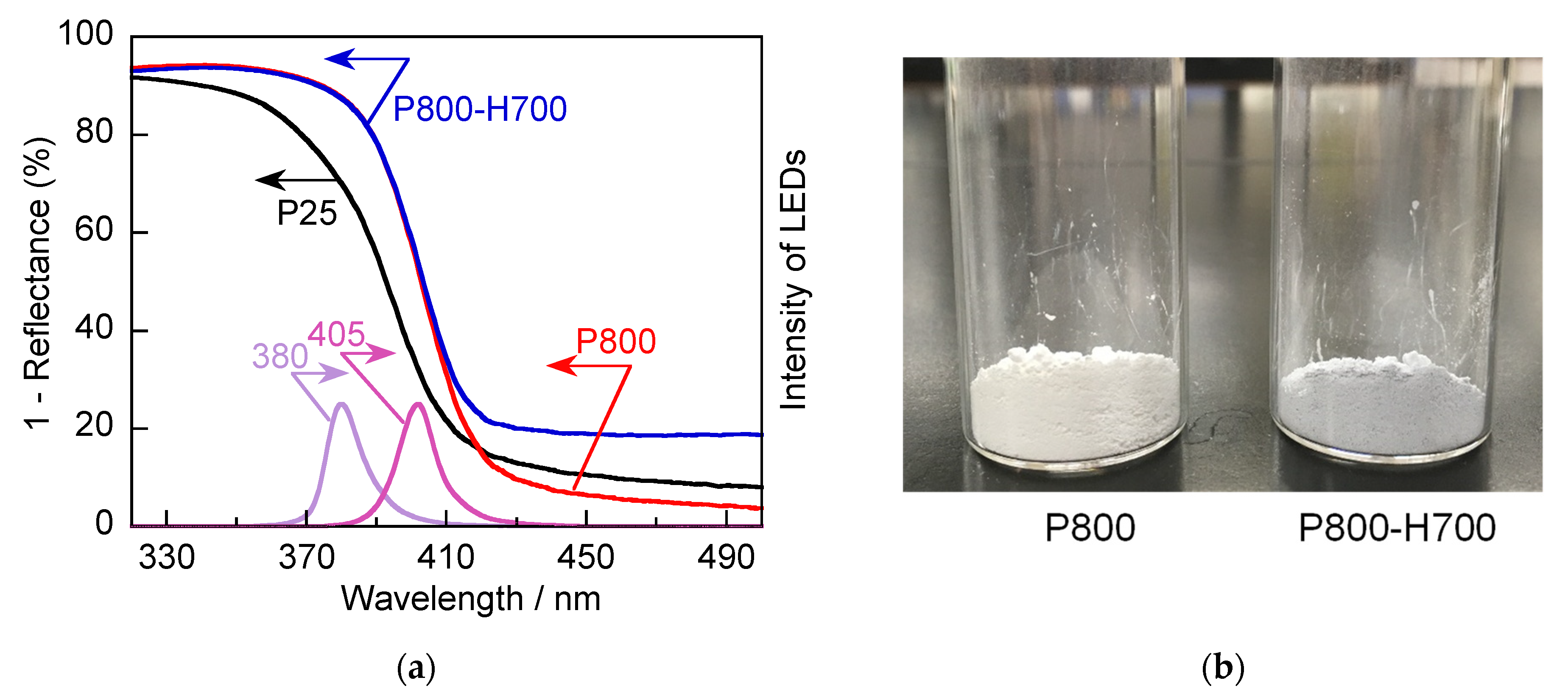
2.2. Optical Properties of TiO2 Particles
2.3. Photocatalytic Activity Test
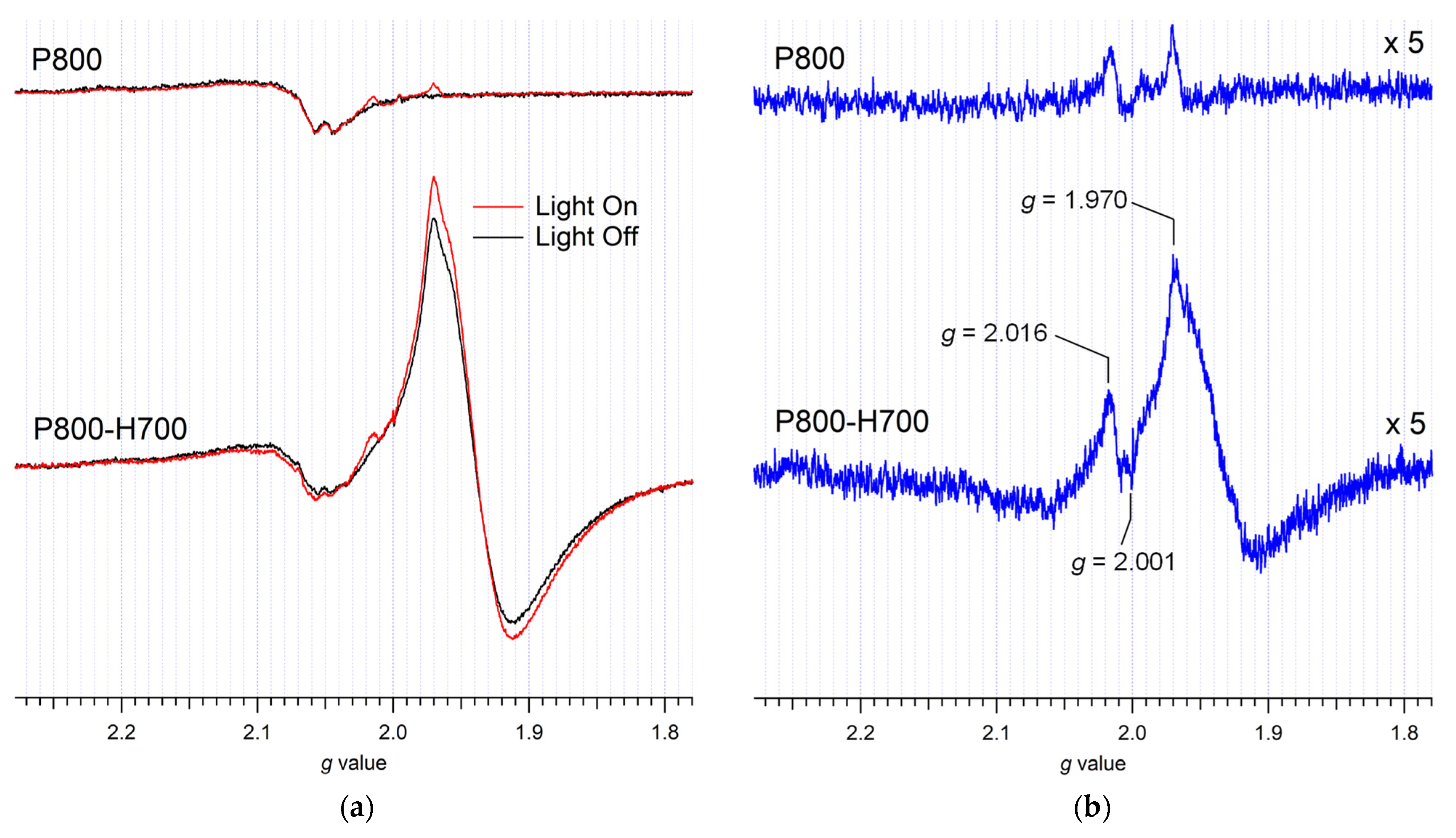
2.4. ESR Study
2.5. Effect of H2O2 Addition on the Photocatalytic Activity
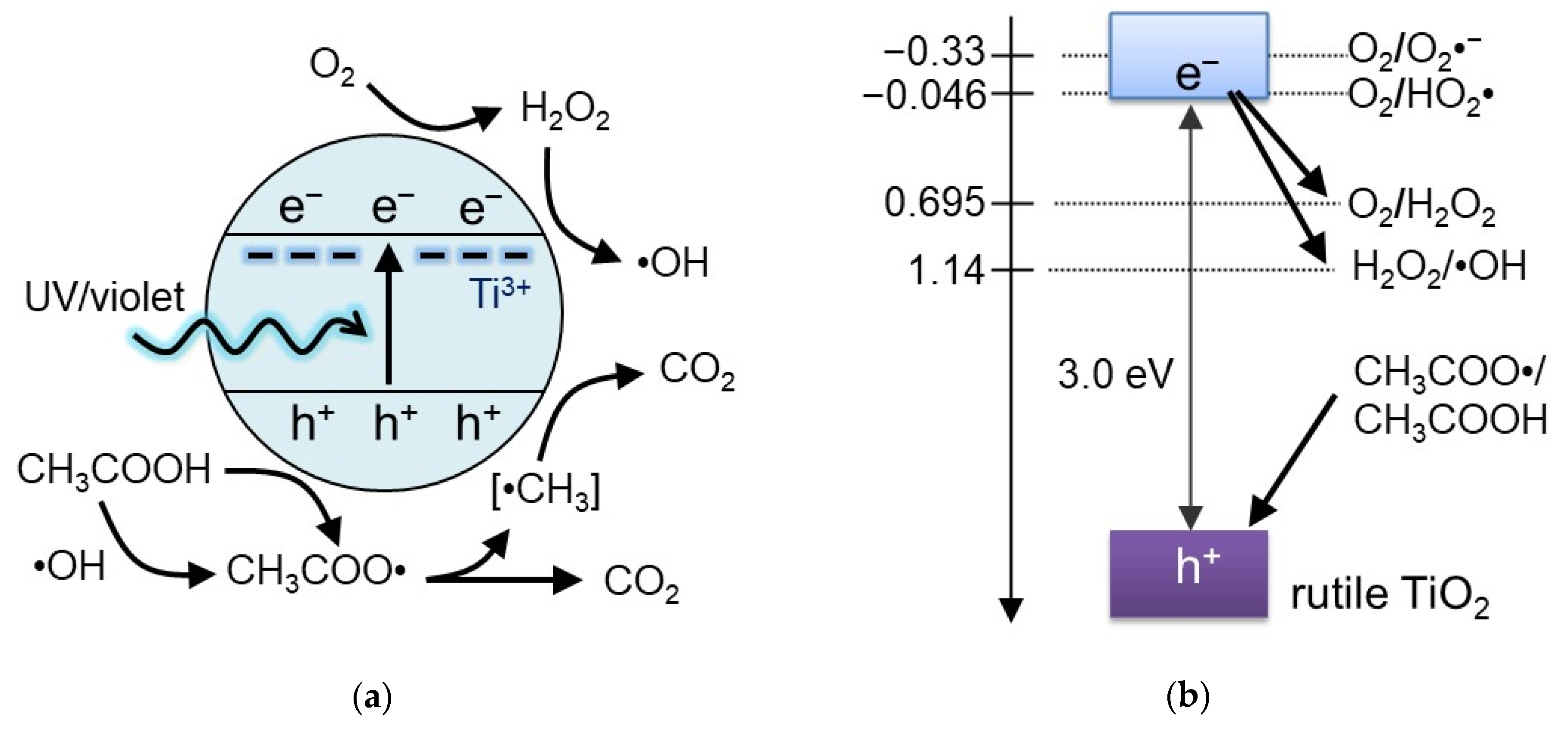
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Linsebigler, A.L.; Lu, G.; Yates, J.T. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.L.; Reller, A. Photoinduced reactivity of titanium dioxide. Prog. Solid. State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Sclafani, A.; Herrmann, J.M. Comparison of the photoelectronic and photocatalytic activities of various anatase and rutile forms of titania in pure liquid organic phases and in aqueous solutions. J. Phys. Chem. 1996, 100, 13655–13661. [Google Scholar] [CrossRef]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 photocatalysis: A historical overview and future prospects. Jpn. J. Appl. Phys 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Ohno, T.; Sarukawa, K.; Matsumura, M. Photocatalytic activities of pure rutile particles isolated from TiO2 powder by dissolving the anatase component in HF solution. J. Phys. Chem. B 2001, 105, 2417–2420. [Google Scholar] [CrossRef]
- Kavan, L.; Grätzel, M.; Gilbert, S.E.; Klemenz, C.; Scheel, H.J. Electrochemical and photoelectrochemical investigation of single-crystal anatase. J. Am. Chem. Soc. 1996, 118, 6716–6723. [Google Scholar] [CrossRef]
- Amano, F.; Yasumoto, T.; Prieto-Mahaney, O.O.; Uchida, S.; Shibayama, T.; Ohtani, B. Photocatalytic activity of octahedral single-crystalline mesoparticles of anatase titanium(IV) oxide. Chem. Commun. 2009, 45, 2311–2313. [Google Scholar] [CrossRef]
- Turchi, C.S.; Ollis, D.F. Photocatalytic degradation of organic water contaminants: Mechanisms involving hydroxyl radical attack. J. Catal. 1990, 122, 178–192. [Google Scholar] [CrossRef]
- Du, P.; Bueno-López, A.; Verbaas, M.; Almeida, A.R.; Makkee, M.; Moulijn, J.A.; Mul, G. The effect of surface OH-population on the photocatalytic activity of rare earth-doped P25-TiO2 in methylene blue degradation. J. Catal. 2008, 260, 75–80. [Google Scholar] [CrossRef]
- Gerischer, H.; Heller, A. The role of oxygen in photooxidation of organic molecules on semiconductor particles. J. Phys. Chem. 1991, 95, 5261–5267. [Google Scholar] [CrossRef]
- Gerischer, H.; Heller, A. Photocatalytic oxidation of organic molecules at TiO2 particles by sunlight in aerated water. J. Electrochem. Soc. 1992, 139, 113–118. [Google Scholar] [CrossRef]
- Wang, C.M.; Heller, A.; Gerischer, H. Palladium catalysis of O2 reduction by electrons accumulated on TiO2 particles during photoassisted oxidation of organic compounds. J. Am. Chem. Soc. 1992, 114, 5230–5234. [Google Scholar] [CrossRef]
- Yamakata, A.; Vequizo, J.J.M.; Matsunaga, H. Distinctive behavior of photogenerated electrons and holes in anatase and rutile TiO2 powders. J. Phys. Chem. C 2015, 119, 24538–24545. [Google Scholar] [CrossRef]
- Yamada, Y.; Kanemitsu, Y. Determination of electron and hole lifetimes of rutile and anatase TiO2 single crystals. Appl. Phys. Lett. 2012, 101, 133907. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Noda, K.; Nishi, T.; Kobayashi, K.; Yamada, H.; Matsushige, K. Investigation of electrical transport in anodized single TiO2 nanotubes. Appl. Phys. Lett. 2013, 102, 043105. [Google Scholar] [CrossRef]
- Luttrell, T.; Halpegamage, S.; Tao, J.; Kramer, A.; Sutter, E.; Batzill, M. Why is anatase a better photocatalyst than rutile?—Model studies on epitaxial TiO2 films. Sci. Rep. 2014, 4, 4043. [Google Scholar] [CrossRef] [PubMed]
- Amano, F.; Nakata, M.; Asami, K.; Yamakata, A. Photocatalytic activity of titania particles calcined at high temperature: Investigating deactivation. Chem. Phys. Lett. 2013, 579, 111–113. [Google Scholar] [CrossRef]
- Zhen, C.; Wang, L.; Liu, L.; Liu, G.; Lu, G.Q.; Cheng, H.-M. Nonstoichiometric rutile TiO2 photoelectrodes for improved photoelectrochemical water splitting. Chem. Commun. 2013, 49, 6191–6193. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M. High-temperature calcination and hydrogen reduction of rutile TiO2: A method to improve the photocatalytic activity for water oxidation. Appl. Catal. B 2014, 158, 202–208. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Ishinaga, E. Photocatalytic activity of rutile titania for hydrogen evolution. Chem. Lett. 2014, 43, 509–511. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Yamamoto, A.; Tanaka, T. Rutile titanium dioxide prepared by hydrogen reduction of Degussa P25 for highly efficient photocatalytic hydrogen evolution. Catal. Sci. Technol. 2016, 6, 5693–5699. [Google Scholar] [CrossRef]
- Amano, F.; Mukohara, H.; Shintani, A. Rutile titania particulate photoelectrodes fabricated by two-step annealing of titania nanotube arrays. J. Electrochem. Soc. 2018, 165, H3164–H3169. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Vequizo, J.J.M.; Yamakata, A. Enhanced visible light response of TiO2 codoped with Cr and Ta photocatalysts by electron doping. ACS Appl. Energy Mater. 2019, 2, 3274–3282. [Google Scholar] [CrossRef]
- Ohtani, B.; Mahaney, O.O.P.; Amano, F.; Murakami, N.; Abe, R. What are titania photocatalysts?―An exploratory correlation of photocatalytic activity with structural and physical properties. J. Adv. Oxid. Technol. 2010, 13, 247–261. [Google Scholar] [CrossRef]
- Ohtani, B.; Prieto-Mahaney, O.O.; Li, D.; Abe, R. What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test. J. Photochem. Photobiol. A 2010, 216, 179–182. [Google Scholar] [CrossRef]
- Spurr, R.A.; Myers, H. Quantitative analysis of anatase-rutile mixtures with an X-ray diffractometer. Anal. Chem. 1957, 29, 760–762. [Google Scholar] [CrossRef]
- Butler, M.A.; Ginley, D.S. Prediction of flatband potentials at semiconductor-electrolyte interfaces from atomic electronegativities. J. Electrochem. Soc. 1978, 125, 228–232. [Google Scholar] [CrossRef]
- Maruska, H.P.; Ghosh, A.K. Photocatalytic decomposition of water at semiconductor electrodes. Sol. Energy 1978, 20, 443–458. [Google Scholar] [CrossRef]
- Yamakata, A.; Ishibashi, T.; Onishi, H. Time-resolved infrared absorption spectroscopy of photogenerated electrons in platinized TiO2 particles. Chem. Phys. Lett. 2001, 333, 271–277. [Google Scholar] [CrossRef]
- Berger, T.; Anta, J.A.; Morales-Flórez, V. Electrons in the band gap: Spectroscopic characterization of anatase TiO2 nanocrystal electrodes under Fermi level control. J. Phys. Chem. C 2012, 116, 11444–11455. [Google Scholar] [CrossRef]
- Amano, F.; Nakata, M.; Yamamoto, A.; Tanaka, T. Effect of Ti3+ ions and conduction band electrons on photocatalytic and photoelectrochemical activity of rutile titania for water oxidation. J. Phys. Chem. C 2016, 120, 6467–6474. [Google Scholar] [CrossRef]
- Kumar, C.P.; Gopal, N.O.; Wang, T.C.; Wong, M.-S.; Ke, S.C. EPR investigation of TiO2 nanoparticles with temperature-dependent properties. J. Phys. Chem. B 2006, 110, 5223–5229. [Google Scholar] [CrossRef]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the enhanced photocatalytic activity of Degussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Seymour, R.C.; Wood, J.C. Paramagnetic gas adsorption. Surf. Sci. 1971, 27, 605–610. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR study of hydrated anatase under UV irradiation. J. Phys. Chem. 1987, 91, 3906–3909. [Google Scholar] [CrossRef]
- Nakaoka, Y.; Nosaka, Y. ESR investigation into the effects of heat treatment and crystal structure on radicals produced over irradiated TiO2 powder. J. Photochem. Photobiol. A 1997, 110, 299–305. [Google Scholar] [CrossRef]
- Okumura, M.; Coronado, J.M.; Soria, J.; Haruta, M.; Conesa, J.C. EPR study of CO and O2 interaction with supported Au catalysts. J. Catal. 2001, 203, 168–174. [Google Scholar] [CrossRef]
- Coronado, J.M.; Maira, A.J.; Conesa, J.C.; Yeung, K.L.; Augugliaro, V.; Soria, J. EPR study of the surface characteristics of nanostructured TiO2 under UV irradiation. Langmuir 2001, 17, 5368–5374. [Google Scholar] [CrossRef]
- Carter, E.; Carley, A.F.; Murphy, D.M. Evidence for O2– radical stabilization at surface oxygen vacancies on polycrystalline TiO2. J. Phys. Chem. C 2007, 111, 10630–10638. [Google Scholar] [CrossRef]
- Komaguchi, K.; Maruoka, T.; Nakano, H.; Imae, I.; Ooyama, Y.; Harima, Y. ESR study on the reversible electron transfer from O22− to Ti4+ on TiO2 nanoparticles induced by visible-light illumination. J. Phys. Chem. C 2009, 113, 1160–1163. [Google Scholar] [CrossRef]
- Saison, T.; Gras, P.; Chemin, N.; Chanéac, C.; Durupthy, O.; Brezová, V.; Colbeau-Justin, C.; Jolivet, J.-P. New insights into Bi2WO6 properties as a visible-light photocatalyst. J. Phys. Chem. C 2013, 117, 22656–22666. [Google Scholar] [CrossRef]
- Klassen, N.V.; Marchington, D.; McGowan, H.C.E. H2O2 determination by the I3− method and by KMnO4 titration. Anal. Chem. 1994, 66, 2921–2925. [Google Scholar] [CrossRef]
- Tomita, O.; Ohtani, B.; Abe, R. Highly selective phenol production from benzene on a platinum-loaded tungsten oxide photocatalyst with water and molecular oxygen: Selective oxidation of water by holes for generating hydroxyl radical as the predominant source of the hydroxyl group. Catal. Sci. Technol. 2014, 4, 3850–3860. [Google Scholar] [CrossRef]
- Tomita, O.; Otsubo, T.; Higashi, M.; Ohtani, B.; Abe, R. Partial oxidation of alcohols on visible-light-responsive WO3 photocatalysts loaded with palladium oxide cocatalyst. ACS Catal. 2016, 6, 1134–1144. [Google Scholar] [CrossRef]
- Hengerer, R.; Kavan, L.; Krtil, P.; Grätzel, M. Orientation dependence of charge-transfer processes on TiO2 (anatase) single crystals. J. Electrochem. Soc. 2000, 147, 1467. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution; CRC Press: Boca Raton, FL, USA, 1985. [Google Scholar]
- Sheng, J.; Li, X.; Xu, Y. Generation of H2O2 and OH radicals on Bi2WO6 for phenol degradation under visible light. ACS Catal. 2014, 4, 732–737. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Kraeutler, B.; Bard, A.J. Heterogeneous photocatalytic synthesis of methane from acetic acid—New Kolbe reaction pathway. J. Am. Chem. Soc. 1978, 100, 2239–2240. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Formed H2O2/µmol | Evolved CO2/µmol |
|---|---|---|
| P 25 | 0.17 | 1.19 |
| P800 | 0.03 | 0.88 |
| P800-H700 | 0.09 | 3.36 |
| blank | – | 0.00 |
| Photocatalyst | Residual H2O2/µmol | Evolved CO2/µmol |
|---|---|---|
| P 25 | 2.93 | 0.89 |
| P800 | 1.48 | 2.39 |
| P800-H700 | 0.03 | 8.60 |
| blank | 9.39 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amano, F.; Yamamoto, A.; Kumagai, J. Highly Active Rutile TiO2 for Photocatalysis under Violet Light Irradiation at 405 nm. Catalysts 2022, 12, 1079. https://doi.org/10.3390/catal12101079
Amano F, Yamamoto A, Kumagai J. Highly Active Rutile TiO2 for Photocatalysis under Violet Light Irradiation at 405 nm. Catalysts. 2022; 12(10):1079. https://doi.org/10.3390/catal12101079
Chicago/Turabian StyleAmano, Fumiaki, Akira Yamamoto, and Jun Kumagai. 2022. "Highly Active Rutile TiO2 for Photocatalysis under Violet Light Irradiation at 405 nm" Catalysts 12, no. 10: 1079. https://doi.org/10.3390/catal12101079