
The Activity of Ultrafine Cu Clusters Encapsulated in Nano-Zeolite for Selective Hydrogenation of CO2 to Methanol


 and
and 
Abstract
:
1. Introduction
2. Results
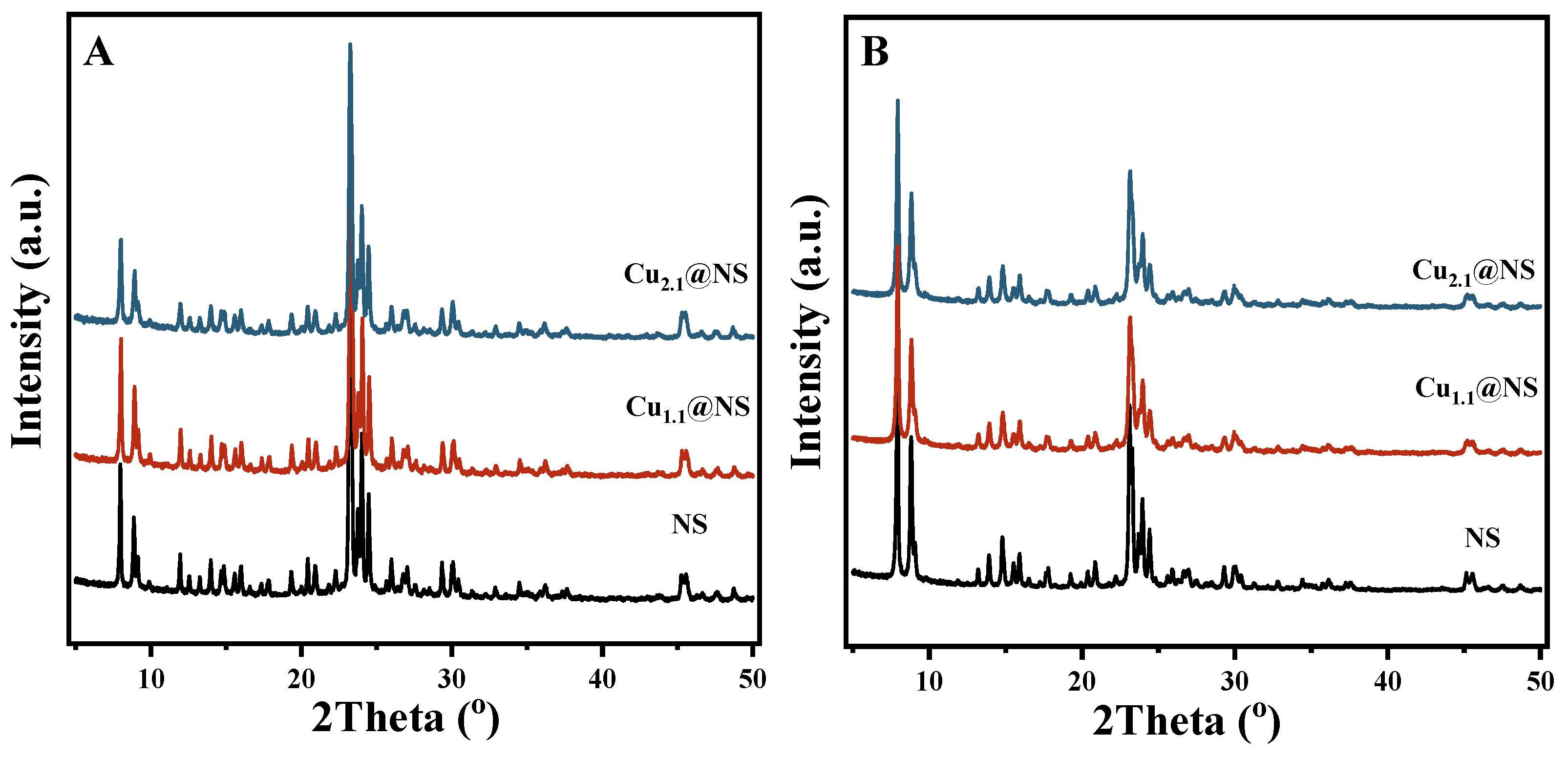
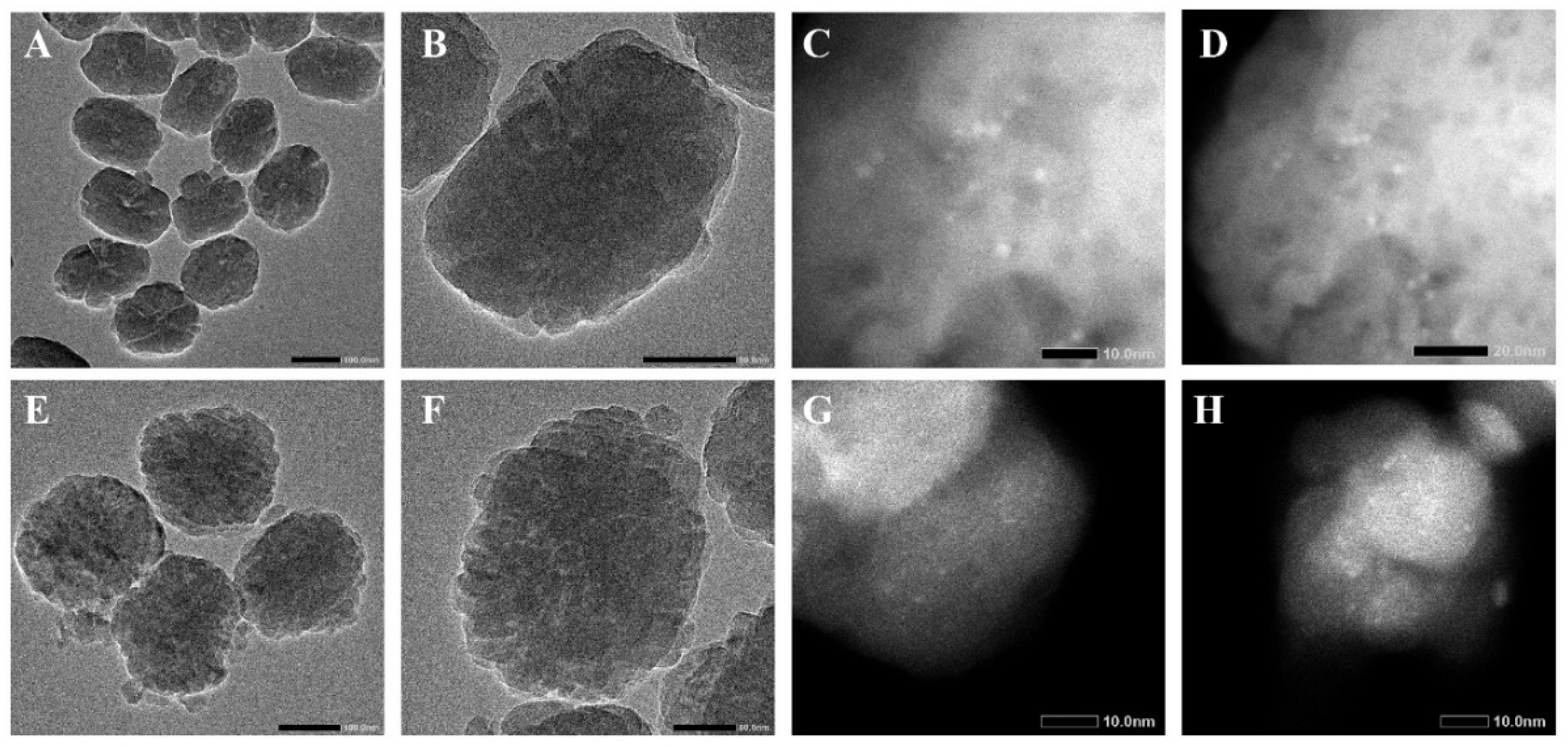
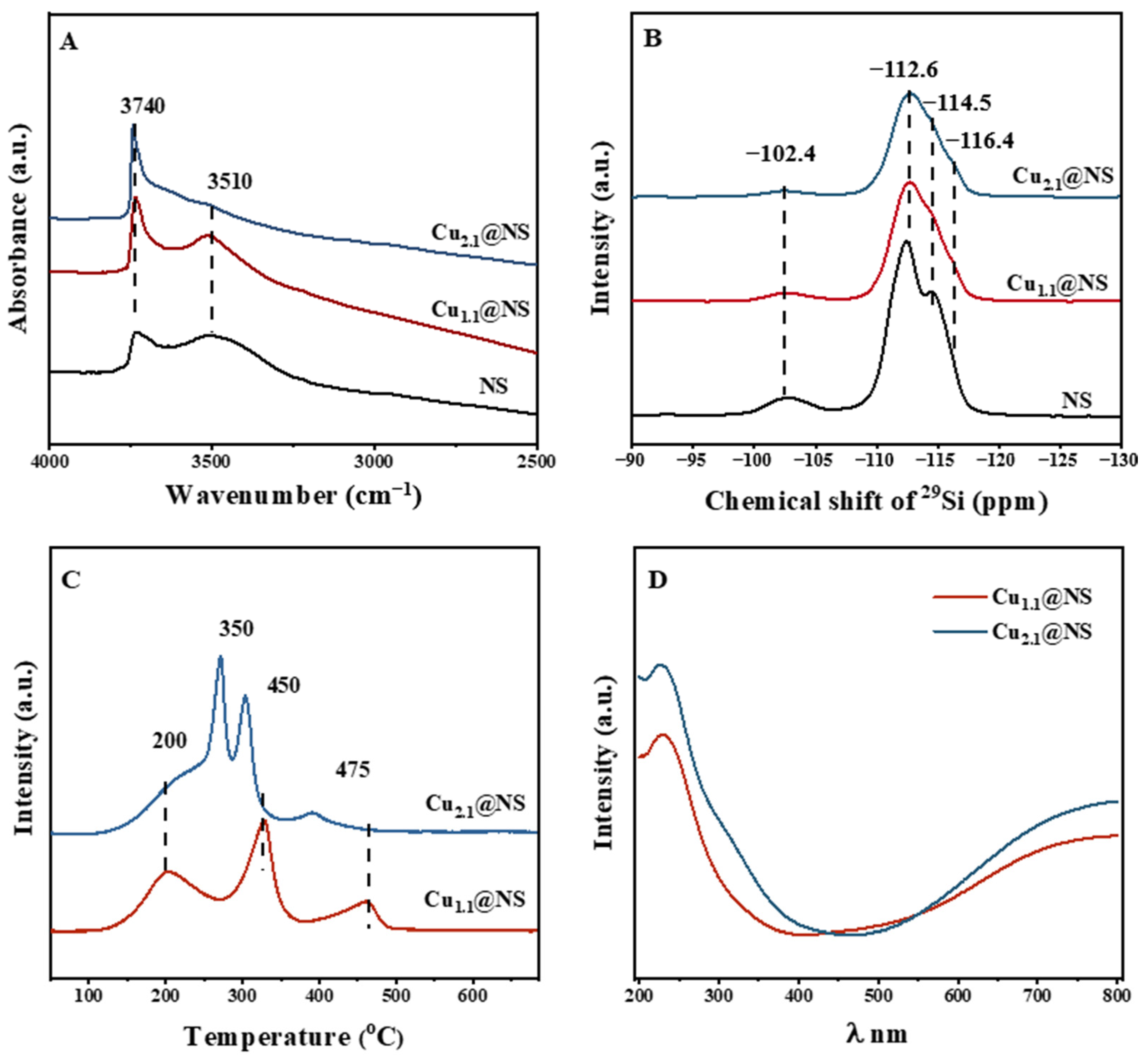
2.1. The Presence of Cu in the Synthesized Cu@NS Crystals
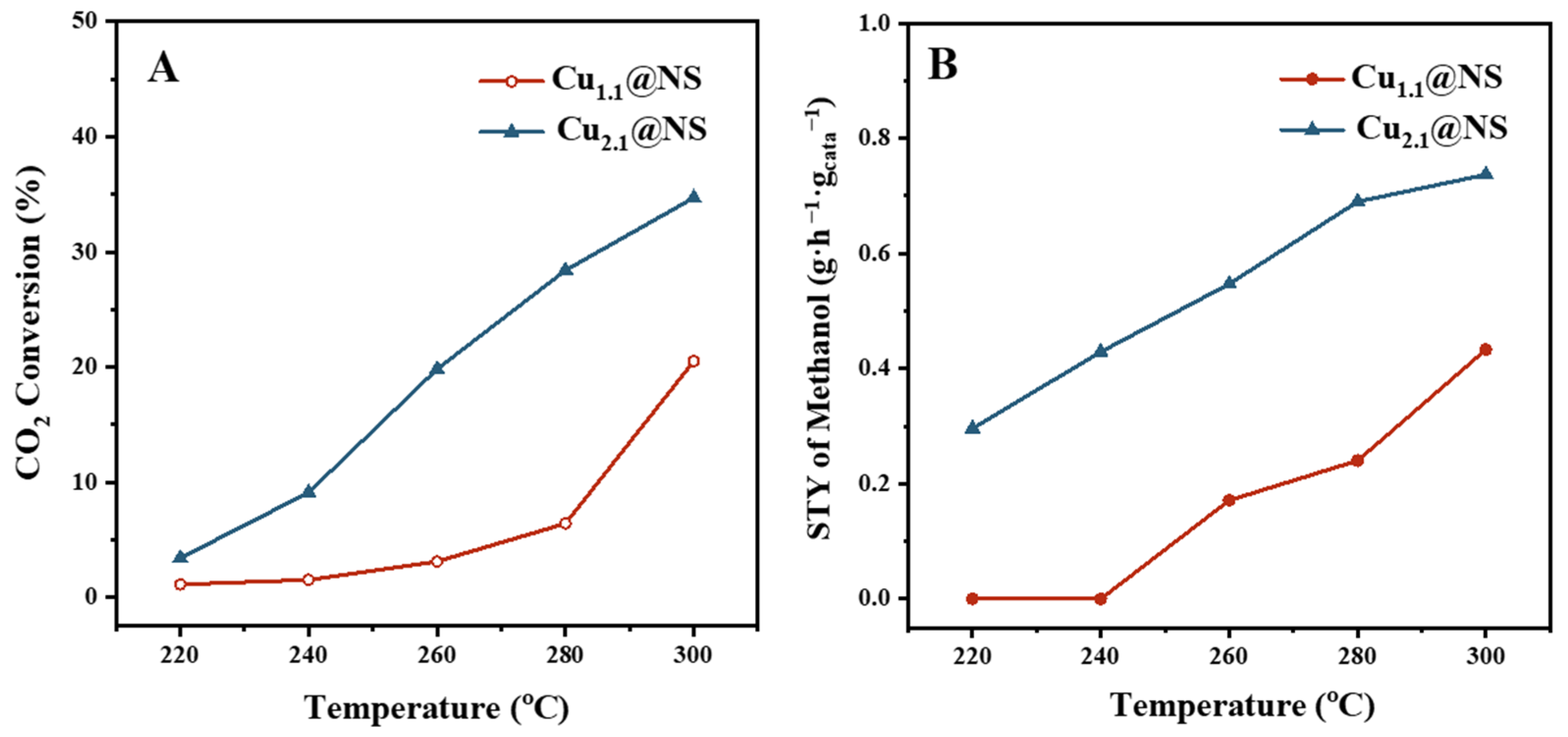
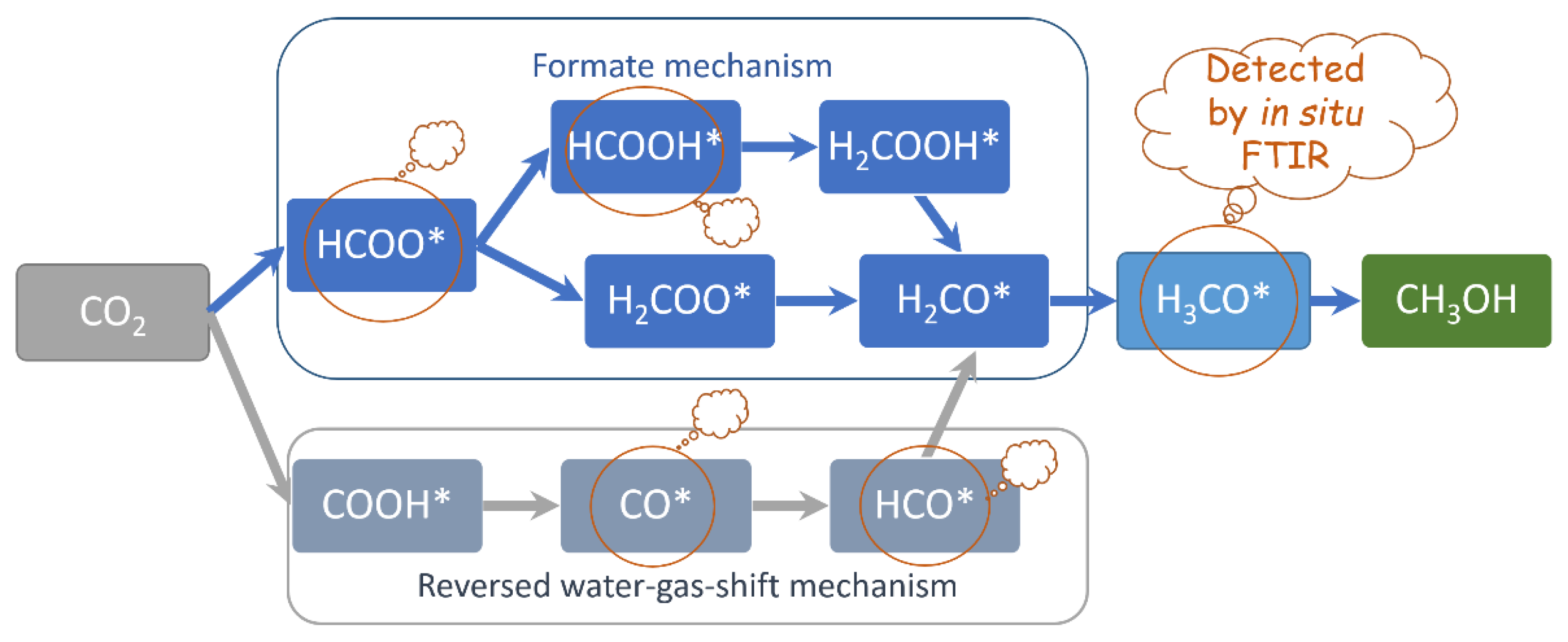
2.2. The Observed CO2 Conversion Intermediates and Their Extinguishments
3. Materials and Methods
3.1. Materials
3.2. Synthesis
3.3. Characterization
3.4. Catalytic Test
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhang, T. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.F.; Zhang, T.; Li, J.; Bai, C. Powering the Future with Liquid Sunshine. Joule 2018, 2, 1925–1949. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhang, L.; Li, S.; Zhou, Z.; Sun, Y. Novel Heterogeneous Catalysts for CO2 Hydrogenation to Liquid Fuels. ACS Cent. Sci. 2020, 6, 1657–1670. [Google Scholar] [CrossRef]
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlögl, R. CO2 Hydrogenation with Cu/ZnO/Al2O3: A Benchmark Study. ChemCatChem 2020, 12, 3216–3222. [Google Scholar] [CrossRef]
- Liang, B.; Ma, J.; Su, X.; Yang, C.; Duan, H.; Zhou, H.; Deng, S.; Li, L.; Huang, Y. Investigation on Deactivation of Cu/ZnO/Al2O3 Catalyst for CO2 Hydrogenation to Methanol. Ind. Eng. Chem. Res. 2019, 58, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wen, J.; Sun, Y.; Zhang, M.; Bao, Y.; Zhang, Y.; Liang, L.; Fu, M.; Wu, J.; Ye, D.; et al. CO2 hydrogenation to methanol over Cu/ZnO plate model catalyst: Effects of reducing gas induced Cu nanoparticle morphology. Chem. Eng. J. 2019, 374, 221–230. [Google Scholar] [CrossRef]
- Toyir, J.; Miloua, R.; Elkadri, N.E.; Nawdali, M.; Toufik, H.; Miloua, F.; Saito, M. Sustainable process for the production of methanol from CO2 and H2 using Cu/ZnO-based multicomponent catalyst. Phys. Procedia 2009, 2, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- George Olah CO2 to Renewable Methanol Plant, Reykjanes, Iceland. Available online: https://www.carbonrecycling.is/ (accessed on 2 September 2022).
- Liu, L.; Corma, A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.R.; Chapon, C.; Giorgio, S.; Goyhenex, C. Size Effects in Heterogeneous Catalysis. In Chemisorption and Reactivity on Supported Clusters and Thin Films: Towards an Understanding of Microscopic Processes in Catalysis; Lambert, R.M., Pacchioni, G., Eds.; Springer: Dordrecht, The Netherlands, 1997; pp. 117–152. [Google Scholar]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Zhang, B.; Zhao, H.; Kolb, U.; Zhu, Y.; Liu, L.; Han, Y.; Wang, G.; Wang, C.; et al. Sinter-resistant metal nanoparticle catalysts achieved by immobilization within zeolite crystals via seed-directed growth. Nat. Catal. 2018, 1, 540–546. [Google Scholar] [CrossRef]
- Kosinov, N.; Liu, C.; Hensen, E.J.M.; Pidko, E.A. Engineering of Transition Metal Catalysts Confined in Zeolites. Chem. Mater. 2018, 30, 3177–3198. [Google Scholar] [CrossRef]
- Kim, S.; Tsang, Y.F.; Kwon, E.E.; Lin, K.-Y.A.; Lee, J. Recently developed methods to enhance stability of heterogeneous catalysts for conversion of biomass-derived feedstocks. Korean J. Chem. Eng. 2019, 36, 1–11. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Confining isolated atoms and clusters in crystalline porous materials for catalysis. Nat. Rev. Mater. 2021, 6, 244–263. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Wang, L.; Wang, L.; Xiao, F.-S. Zeolite Fixed Metal Nanoparticles: New Perspective in Catalysis. Acc. Chem. Res. 2021, 54, 2579–2590. [Google Scholar] [CrossRef]
- Yang, X.; Toby, B.H.; Camblor, M.A.; Lee, Y.; Olson, D.H. Propene adsorption sites in zeolite ITQ-12: A combined synchrotron X-ray and neutron diffraction study. J Phys Chem B 2005, 109, 7894–7899. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Liu, C.; Halder, A.; Tyo, E.C.; Martinson, A.B.F.; Seifert, S.; Zapol, P.; Curtiss, L.A.; Vajda, S. Copper Cluster Size Effect in Methanol Synthesis from CO2. J. Phys. Chem. C 2017, 121, 10406–10412. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.-X.; Zijlstra, B.; Filot, I.A.W.; Zhou, Z.; Sun, S.; Hensen, E.J.M. Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 2018, 43, 200–209. [Google Scholar] [CrossRef]
- Borfecchia, E.; Beato, P.; Svelle, S.; Olsbye, U.; Lamberti, C.; Bordiga, S. Cu-CHA—A model system for applied selective redox catalysis. Chem. Soc. Rev. 2018, 47, 8097–8133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Du, X.; Qiao, B. Methane oxidation to methanol over copper-containing zeolite. Chem 2021, 7, 2270–2272. [Google Scholar] [CrossRef]
- Carter, J.H.; Dummer, N.F.; Chow, Y.K.; Williams, C.; Nasrallah, A.; Willock, D.J.; Hutchings, G.J.; Taylor, S.H. The Selective Oxidation of Methane to Oxygenates Using Heterogeneous Catalysts. In Heterogeneous Catalysis for Sustainable Energy; John Wiley & Sons: Hoboken, NJ, USA, 2022; pp. 183–201. [Google Scholar]
- Chai, Y.; Shang, W.; Li, W.; Wu, G.; Dai, W.; Guan, N.; Li, L. Noble Metal Particles Confined in Zeolites: Synthesis, Characterization, and Applications. Adv. Sci. 2019, 6, 1900299. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Shi, T.; Gu, J.; Cui, Y.; Zhang, Z.; Yang, C.; Chen, T.; Lin, M.; Wang, P.; Xue, N.; et al. CO2 Hydrogenation to Ethanol over Cu@Na-Beta. Chem 2020, 6, 2673–2689. [Google Scholar] [CrossRef]
- Cui, W.-G.; Li, Y.-T.; Yu, L.; Zhang, H.; Hu, T.-L. Zeolite-Encapsulated Ultrasmall Cu/ZnOx Nanoparticles for the Hydrogenation of CO2 to Methanol. ACS Appl. Mater. Interfaces 2021, 13, 18693–18703. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Wu, Z.; Iglesia, E. Mercaptosilane-Assisted Synthesis of Metal Clusters within Zeolites and Catalytic Consequences of Encapsulation. J. Am. Chem. Soc. 2010, 132, 9129–9137. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Wu, Z.; Zones, S.I.; Iglesia, E. Synthesis and Catalytic Properties of Metal Clusters Encapsulated within Small-Pore (SOD, GIS, ANA) Zeolites. J. Am. Chem. Soc. 2012, 134, 17688–17695. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Sun, Q.; Bai, R.; Li, X.; Guo, G.; Yu, J. In Situ Confinement of Ultrasmall Pd Clusters within Nanosized Silicalite-1 Zeolite for Highly Efficient Catalysis of Hydrogen Generation. J. Am. Chem. Soc. 2016, 138, 7484–7487. [Google Scholar] [CrossRef] [PubMed]
- Schumann, J.; Kröhnert, J.; Frei, E.; Schlögl, R.; Trunschke, A. IR-Spectroscopic Study on the Interface of Cu-Based Methanol Synthesis Catalysts: Evidence for the Formation of a ZnO Overlayer. Top. Catal. 2017, 60, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and CO Hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Mintova, S.; Olson, N.H.; Senker, J.; Bein, T. Mechanism of the Transformation of Silica Precursor Solutions into Si-MFI Zeolite. Angew. Chem. Int. Ed. 2002, 41, 2558–2561. [Google Scholar] [CrossRef]
- Medeiros-Costa, I.C.; Dib, E.; Nesterenko, N.; Dath, J.-P.; Gilson, J.-P.; Mintova, S. Silanol defect engineering and healing in zeolites: Opportunities to fine-tune their properties and performances. Chem. Soc. Rev. 2021, 50, 11156–11179. [Google Scholar] [CrossRef]
- Dib, E.; Costa, I.M.; Vayssilov, G.N.; Aleksandrov, H.A.; Mintova, S. Complex H-bonded silanol network in zeolites revealed by IR and NMR spectroscopy combined with DFT calculations. J. Mater. Chem. A 2021, 9, 27347–27352. [Google Scholar] [CrossRef]
- Bordiga, S.; Ugliengo, P.; Damin, A.; Lamberti, C.; Spoto, G.; Zecchina, A.; Spanò, G.; Buzzoni, R.; Dalloro, L.; Rivetti, F. Hydroxyls nests in defective silicalites and strained structures derived upon dehydroxylation: Vibrational properties and theoretical modelling. Top. Catal. 2001, 15, 43–52. [Google Scholar] [CrossRef]
- Clarke, D.B.; Bell, A.T. An Infrared Study of Methanol Synthesis from CO2 on Clean and Potassium-Promoted Cu/SiO2. J. Catal. 1995, 154, 314–328. [Google Scholar] [CrossRef]
- Robbins, J.L.; Marucchi-Soos, E. Evidence for multiple carbon monoxide hydrogenation pathways on platinum alumina. J. Phys. Chem. 1989, 93, 2885–2888. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Jensen, J.H.; Grassian, V.H. FTIR Spectroscopy Combined with Isotope Labeling and Quantum Chemical Calculations to Investigate Adsorbed Bicarbonate Formation Following Reaction of Carbon Dioxide with Surface Hydroxyl Groups on Fe2O3 and Al2O3. J. Phys. Chem. B 2006, 110, 12005–12016. [Google Scholar] [CrossRef]
- Meunier, F.C.; Kdhir, R.; Potrzebowska, N.; Perret, N.; Besson, M. Unravelling Platinum–Zirconia Interfacial Sites Using CO Adsorption. Inorg. Chem. 2019, 58, 8021–8029. [Google Scholar] [CrossRef]
- Meunier, F.C.; Dansette, I.; Eng, K.; Schuurman, Y. Differentiating the Reactivity of ZrO2-Bound Formates Formed on Cu/ZrO2 during CO2 Hydrogenation. Catalysts 2022, 12, 793. [Google Scholar] [CrossRef]
- Bando, K.K.; Sayama, K.; Kusama, H.; Okabe, K.; Arakawa, H. In-situ FT-IR study on CO2 hydrogenation over Cu catalysts supported on SiO2, Al2O3, and TiO2. Appl. Catal. A Gen. 1997, 165, 391–409. [Google Scholar] [CrossRef]
- Pritchard, J.; Catterick, T.; Gupta, R.K. Infrared spectroscopy of chemisorbed carbon monoxide on copper. Surf. Sci. 1975, 53, 1–20. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Hutchings, G.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial Gel Composition (Molar Ratio) | Hydrothermal Condition | Product Cu Content | |||||
|---|---|---|---|---|---|---|---|---|
| SiO2 | TPAOH | Cu(NO3)2 | en | H2O | Temp (°C) | t (h) | (wt.%) | |
| NS, nano-silicalite-1 | 1 | 0.3 | 0 | 0 | 19 | 100 | 30 | 0 |
| Cu1.1@NS | 1 | 0.3 | 0.014 | 0.13 | 19 | 100 | 48 | 1.10 |
| Cu2.1@NS | 1 | 0.3 | 0.028 | 0.25 | 19 | 100 | 54 | 2.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, R.; Fu, G.; Wang, S.; Yang, Y.; Lang, Q.; Zhao, H.; Yang, X.; Valtchev, V. The Activity of Ultrafine Cu Clusters Encapsulated in Nano-Zeolite for Selective Hydrogenation of CO2 to Methanol. Catalysts 2022, 12, 1296. https://doi.org/10.3390/catal12111296
Ding R, Fu G, Wang S, Yang Y, Lang Q, Zhao H, Yang X, Valtchev V. The Activity of Ultrafine Cu Clusters Encapsulated in Nano-Zeolite for Selective Hydrogenation of CO2 to Methanol. Catalysts. 2022; 12(11):1296. https://doi.org/10.3390/catal12111296
Chicago/Turabian StyleDing, Ruiqin, Guangying Fu, Songxia Wang, Yang Yang, Qiaolin Lang, Haonuan Zhao, Xiaobo Yang, and Valentin Valtchev. 2022. "The Activity of Ultrafine Cu Clusters Encapsulated in Nano-Zeolite for Selective Hydrogenation of CO2 to Methanol" Catalysts 12, no. 11: 1296. https://doi.org/10.3390/catal12111296
APA StyleDing, R., Fu, G., Wang, S., Yang, Y., Lang, Q., Zhao, H., Yang, X., & Valtchev, V. (2022). The Activity of Ultrafine Cu Clusters Encapsulated in Nano-Zeolite for Selective Hydrogenation of CO2 to Methanol. Catalysts, 12(11), 1296. https://doi.org/10.3390/catal12111296