Abstract
In this study, four monolithic, vanadium-based catalysts in granular (Vox/TiO2), honeycomb-type (Vox-Wox/TiO2 and Vox-MoOx/TiO2), and corrugated forms (Vox-Wox/TiO2) were investigated by multiple characterization methods (BET, XRF, XPS, XRD, H2-TPR, and NH3-TPD). Their catalytic performances were evaluated by the oxidation-reduction performance of ortho-dichlorobenzene (o-DCB) and NO/NH3. The modification of Wox and MoOx could promote catalytic activity by accelerating the transformation of V5+/V4+ and enriching the strong acid sites. The introduction of NO/NH3 significantly impaired the o-DCB oxidation, ascribed to the competitive adsorption of reactants on acid sites. The performance of Vox/TiO2 and Vox-MoOx/TiO2 catalysts indicated that strong acidity could enhance catalytic abilities over o-DCB and Nox. Nevertheless, the CE (conversion efficiency) of o-DCB was more related to a large BET surface area and a high amount of V5+ species, while the CE of Nox was more associated with redox ability and Vox surface density. The V4+/V5+ and OS-A/OS-L ratio increased prominently after the oxidation of o-DCB, indicating that it was the reoxidation of V4+ species, rather than the supplement of oxygen, that limited the reaction rate. This work revealed catalytic activity was positively affiliated with the surface area, amount of V5+ species, transformation rate of V4+/V5+, redox ability, and abundance of strong acid sites. Additionally, the results could guide the selectivity and improvement of industrial low-temperature catalysts for synergistic elimination of chloroaromatic organics and Nox.
1. Introduction
With the increasing amount of municipal and hazardous waste incineration plants in the past several years, the emissions of chloroaromatic organic pollutants have surged. Chloroaromatic organic molecules such as PCDD/Fs are carcinogenic and persistent; they can accumulate in food chains and result in cancer, immune system damage, hormone disruption, and issues with reproduction and development [1]. Nox, the conventional pollutant produced during the combustion process, is under strict regulation due to its damage on human health, causing lung tissue, breathing difficulties, and respiratory issues [2]. More stringent environmental regulations were introduced to limit the emission of NOx and PCDD/Fs. The selective noncatalytic reduction (SNCR) and selective catalytic reduction (SCR) technologies were often adopted simultaneously for denitrification; as for controlling PCDD/Fs emission, multiple control methods such as the injection of inhibitors [3], fabric filter [4], and catalytic oxidation [5] were applied to meet the 0.1 ng/Nm3 standard. Catalytic oxidation was preferred because it could destroy the PCDD/Fs and other chloroaromatic organic pollutants without secondary solid pollution. In addition, 1,2-dichlorobenzene (o-DCB), a representative chloroaromatic organic and reliable precursor of PCDD/Fs [6], was usually chosen as the model compound to evaluate the catalyst’s oxidation abilities on PCDD/Fs.
Catalytic oxidation on removing PCDD/Fs to achieve the 0.1 ng/Nm3 standard via SCR catalyst was first reported in 1989 [7] and further testified by Hagenmaler H. et al. [5,8]. Since then, the removal of PCDD/Fs using SCR catalysts has become the optimal choice for industrial applications because the simultaneous removal of Nox and chloroaromatic organic molecules can reduce the cost of flue gas treatment. Various SCR catalysts were developed or modified to apply in municipal incineration plants. These catalysts could be separated into two kinds based on the active species: noble metal and transient metal oxide catalysts. The noble metal-based catalysts using platinum and gold showed 99.7% I-TEQ conversion of dioxin at 150 °C [9]. However, the working life of noble metal catalysts was short due to the major poisoning problem [10]. Furthermore, the raw materials were overpriced, hindering further industrial application. The transient metal oxide catalysts were composed of one or two (sometimes three) active species such as Vox, MoOx, Wox, MnOx, etc. [11,12,13,14], and rich-pore carriers such as TiO2. They exhibited excellent oxidation abilities [15], high selectivity [16], good sulphate resistance [17], and a wide reaction temperature window [18]. Among these active species, Vox, Wox, and MoOx were the most popular active species due to their strong oxidation ability and good poisoning resistance [17,18,19]. Chin et al. [20] examined the o-DCB conversion using Vox/TiO2 nanoparticles. The catalytic oxidation rated at 150 °C and 200 °C were 46% and 95%. Huang et al. [21] investigated the catalytic oxidation of MoOx-doped Vox/TiO2 catalyst, and the chlorobenzene conversion reached over 95% at 250 °C; Wu et al. [22] determined that regarding the Vox catalysts supported by anatase, the former one could process more V5+-O-V5+ and hence had higher mobility of oxygen than the one supported by rutile TiO2. Gallastegi-Villa [23] investigated the simultaneous removal of Nox and o-DCB using different loading amounts of vanadium and revealed that the o-DCB was adsorbed mainly on Lewis acid sites at low temperatures. On the contrary, Albonetti et al. [24] showed that the Brønsted acid sites increased the conversion of o-DCB significantly by comparing Vox-Wox/TiO2 and Vox/TiO2. In contrast, the influence of Lewis acid sites was less prominent.
The catalysts used In previous studies were In power due to the preparation method. However, monolithic catalysts are reliable in industrial applications because of outstanding mechanical properties, long working life and low-pressure drops, and better adaptability [25,26,27]. Honeycomb-like, granular, and corrugated forms were the most common and mature monolithic types adopted by the SCR sector [27,28]. The investigations of honeycomb-like catalysts were widely reported [12,15,26,29]. Zhan et al. [26] reported that the powder-like catalysts had better degradation efficiency of PCDD/Fs in the stimulated flue gas; however, it was much lower in the practical flue gas than the honeycomb-like catalysts. The excellent removal efficiency (76.4% at 180 °C, 2000 h−1) of PCDD/Fs using Vox-Wox/TiO2 was investigated in the laboratory by Yu et al. [15]; another honeycomb Vox-Wox/TiO2 catalyst showed 95% removal efficiency at 150 °C in the SCR operation [29]. Moreover, the simultaneous removal of Nox and chloroaromatic organic molecules using monolithic catalysts were more concerning. The competitive adsorption of NH3, Nox, and PCDD/Fs (or o-DCB), especially at low temperatures, could jeopardize the further oxidation of organic compounds and reduction of the Nox process. Gan et al. [14] carried out a series of experiments on synergistic abatement of NO and chlorobenzene using a granular Vox/TiO2 catalyst; our team previously reported a field study that a granular Vox/TiO2 catalyst performed 94.59%, 90.57%, and 87.48% removal of PCDD/Fs, at 200 °C, 180 °C, and 160 °C, respectively, with the simultaneous NH3-SCR efficiency of 67.51%, 45.03%, and 42.74% [30]. Lu et al. [12] found that adding NH3 can slightly enhance the oxidation ability of honeycomb Vox-Wox/TiO2 catalysts.
However, there are few studies about the comparisons of synergistic removal of Nox and o-DCB over different monolithic catalysts, and the influence of the monolithic ways was not considered. In this study, we compared simultaneous removal on o-DCB and Nox using granular, honeycomb, and corrugated catalysts at low temperatures. The selectivity of N2O, and Cox, were also investigated to explore the relevance between acidity and catalytic oxidation and reduction.
2. Results and Discussion
2.1. CE of o-DCB and Nox, and the Selectivity of Cox/HCl/N2O
The conversion efficiency (CE) of o-DCB and Nox are displayed in Figure 1. The selectivity of CO2/CO (Cox), HCl, and N2O are exhibited in Figure 2. Before the NO/NH3 was introduced, the Vox/TiO2 (VT) catalyst showed excellent catalytic performance on the o-DCB oxidation (73%) at 150 °C and increased slightly as the temperature went up. The selectivity of Cox ranged from 59% to 68%, which indicated that the o-DCB could be eradicated. The honeycomb Vox-MoOx/TiO2 (VMT) catalyst showed less activity on o-DCB oxidation, with low CE (42%) and Cox selectivity (28%) at 200 °C. The honeycomb Vox-Wox/TiO2 (VWT1) catalyst performed a secondary oxidation ability compared to the VT catalyst, but a better mineralization ability with the highest selectivity of Cox (72%) at 200 °C. The corrugated Vox-Wox/TiO2 (VWT2) catalyst was more suitable to be applied at high temperatures, showing only 22% (CE) at 150 °C but 69% (CE) at 200 °C.
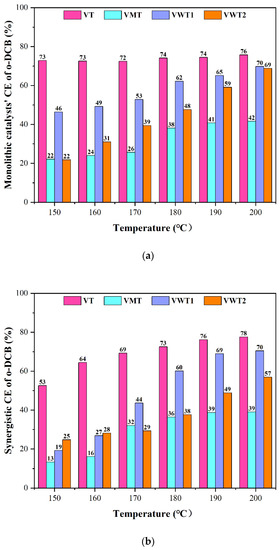
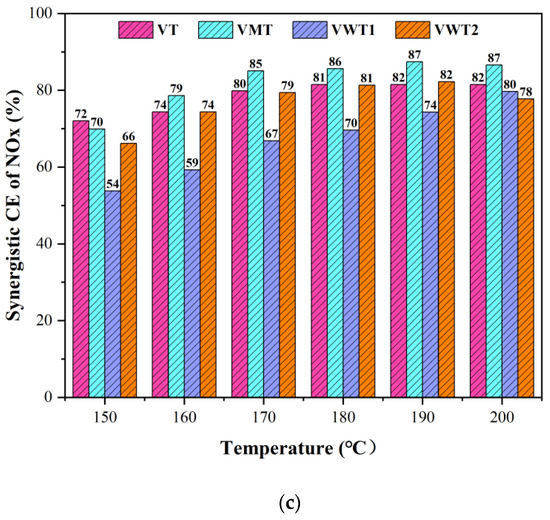
Figure 1.
(a) The CE of o-DCE of four catalysts at 6000 h−1, with an initial concentration of 100 ppm; (b) the synergistic CE of o-DCB of four catalysts with the introduction of 350 ppm NO and 350 ppm NH3; (c) the synergistic CE of NO.
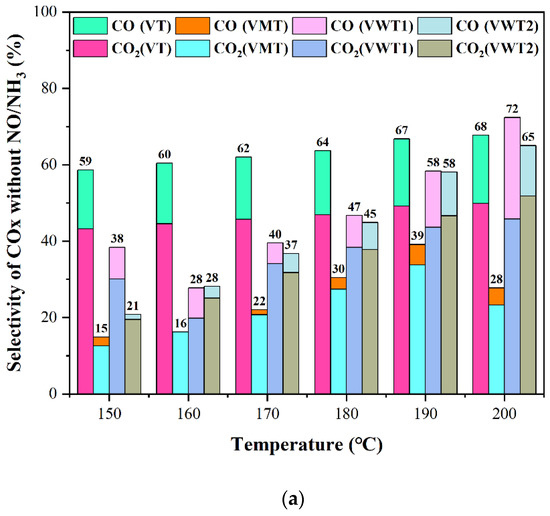
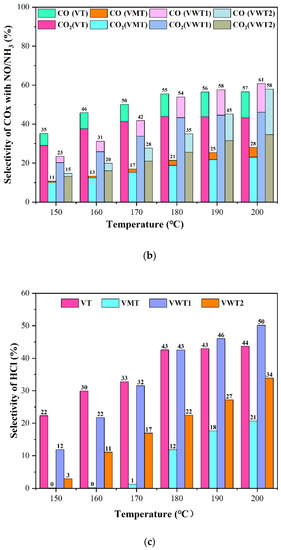
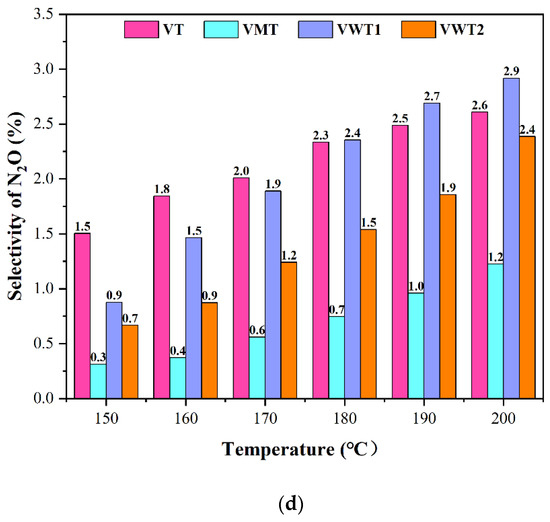
Figure 2.
(a) The selectivity of COx without NO/NH3; (b) The selectivity of COx without NO/NH3; (c) The selectivity of HCl with NO/NH3; (d) The selectivity of N2O.
Under the synergistic conditions (with the introduction of NH3:NO = 350 ppm: 350 ppm), the CE of o-DCB in Figure 1b decreased variously at low temperatures due to the competition between NH3 and o-DCB molecules [31]. It was reported that the catalyst acidity could significantly influence the conversion of o-DCB [24], and the NH3 is the strong adsorbate that occupied the acid sites and hindered the contact of o-DCB. VT and VWT1 exhibited better oxidation on o-DCB at relatively higher temperatures, 190 °C (76% and 69%, respectively), compared with no NO/NH3 (74% and 65%, respectively). This could be explained by the fact that as the temperature rose, NH3 adsorption diminished, and the H protons’ dissociation of the remaining NH3 became more active [31], supplying more Brønsted acid sites and speeding up the incomplete oxidation of o-DCB [24]. Additionally, the presence of NO2 during reoxidation made it simpler to rebuild the oxygen vacancy structure in the catalysts [32]. Nevertheless, the NH3 adsorbed by Brønsted acid sites was stimulated to escape as the temperature rose, whereas the one adsorbed by Lewis acid sites remained stable. The Cox selectivity in Figure 2a,b dropped by ca. 10% for the enhanced incomplete oxidation and compromised complete oxidation of o-DCB under synergistic conditions. The HCl selectivity (Figure 2c) was much lower than Cox selectivity due to the incomplete oxidation of o-DCB or intermediates’ chlorination.
VT, VMT, and VWT2 presented over 66% synergistic conversion efficiency of NOx at 150 °C and reached 70% at 200 °C. The VMT catalyst behaved worst in oxidizing o-DCB but performed well in removing NOx (70–87%). It benefited from the great acid sites provided by Mo = O [33]. On the contrary, the VWT1 catalyst, showing excellent performance on degrading o-DCB, became less incapable of removing NOx (54–67%) at low temperatures (150 °C–170 °C). The comparison between VWT1 and VMT illustrated that the mechanisms of NH3-SCR and oxidation of organic compounds are different at low temperatures. From the perspective of acidity, Gao et al. [33] regarded that the process mainly depended on coordinated NH3 absorbed on Lewis acid sites, while the conversion of o-DCB was decided primarily by Brønsted acid sites [24]. However, two typical NH3-SCR reactions, the “Eley-Rideal” (E-R) mechanism and the “Langmuir-Hinshelwood” (L-H) mechanism, can occur on both Lewis and Brønsted acid sites [34,35,36], which means competitive adsorption is unignorable. Compared with the reduction of NO, the oxidation of the hydroxyl group could be explained by Mars–van Krevelen (MVK) mechanism, in which the circulation of V5+ and V4+, as well as the mobility of oxygen, determined the catalytic process [37]. The N2O, the unwanted by-product of SCR reaction, is one of the regulated gases that causes the greenhouse effect [38]. The breaking of the N-H bond is the rate-determining step of the formation of N2O. Furthermore, Lewis acid sites are more active. NH3 was adsorbed on the Lewis acid sites and formed NH3 (ads), providing the weaker N-H bond [38]. N2O selectivity was displayed in Figure 2d. Among all the four catalysts, VWT1 displayed the highest N2O selectivity (2.9% at 200 °C), while VMT showed the lowest (1.2% at 200 °C).
The catalysts were meshed into powder to eliminate the influence of monolithic methods. It surprised us that VMT (CE was 6–25% in powder but 22–42% in honeycomb) and VWT2 (CE was 7–30% in powder and 22–69% in corrugated form) became less active in degrading o-DCB compared with their original forms listed in Figure S1a. Furthermore, COx selectivity reduced prominently, among which COx of VWT2 decreased from 15–65% to 10–20% at 150–200°C in Figure S1b. For the corrugated catalyst, VWT2, the SiO2 substrate was exposed after it was meshed into powder, reducing active sites. Moreover, compared with the powder structure, the corrugated catalyst could form microscopical channels for heat and mass transfer, enhancing the catalytic oxidation process. Another honeycomb, VWT1, had a close CE of o-DCB but lower COx selectivity in powder form, indicating the catalyst’s complete oxidation over o-DCB was compromised by the reduced fluid conditions. The granular VT catalyst showed excellent CE of o-DCB in both monolithic and powder forms, but better catalytic ability can be seen in the powder form (with CE of 72–87% and COx selectivity of 60–85%), indicating that the granular form could not be the preferential monolithic for VT catalyst. These outcomes showed that the various monolithic methods could affect the efficiency of catalysts because the structure of monolithic catalysts could benefit fluid dynamics and mass and heat transfer, which boosts the catalytic performance [27]. The corrugated method could significantly benefit the catalyst’s performance among granular, corrugated, and honeycomb monolithic methods, while granular monolithic methods may lower the catalytic performance.
As in Table 1, the outcomes could serve as a reference for statistical analysis and reflect their oxidation capacity from the perspective of surface kinetics. The detailed calculation steps were stated in the Supplementary Material. The Ea1 (apparent activation energy) of o-DCB over VT, VMT, VWT1, and VWT2 were calculated as 4.2 kJ/mol, 26.9 kJ/mol, 21.6 kJ/mol, and 35.6 kJ/mol, respectively, with a high correlation coefficient (R12) (0.92, 0.95, 0.99, and 0.99, respectively). As expected, Ea2, the apparent activation energy of o-DCB with the introduction of NH3/NO over four catalysts (24.3 kJ/mol, 44.4 kJ/mol, 62.9 kJ/mol, and 51.9 kJ/mol, respectively, with high R22 of 0.97, 0.91, 0.95, and 0.97, respectively) were much higher than Ea1. The calculated results were consistent with different CE of o-DCB under sole and synergistic conditions, indicating the oxidation of o-DCB was slowed down by the competition for active sites.

Table 1.
The activation energy of catalysts’ synergistic degradation and o-DCB degradation conditions. The subscripts of “1” and “2” denote o-DCB and synergistic conditions, respectively.
2.2. BET and XRF Analysis
The structural properties and chemical composition of catalysts are listed in Table 2. Attention must be paid to the other species in the VWT2 catalyst, which could reach 30.36%. On the contrary, the content of other species in VT was only 1.30%. The granular and honeycomb catalysts were the bulk catalyst extruded from the mixed raw materials using the metal module. They became self-supported after drying and calcining without a support frame. On the other hand, corrugated catalysts were formed by coating, which required an inserted monolithic backbone as the structure [27]. For the latter one, the dosages of active species were much lower. However, the working life would be much shorter due to the loss of active species. The surface area in Table 2 was a crucial indicator for the catalytic process. High specific surface area could provide more physisorption sites and expose more active and acid sites. With a high specific surface area of 91.98 m2/g and a low other species percentage, the VT catalyst could offer more active sites and oxygen vacancies to degrade o-DCB and NH3/NO [39]. VMT performed well in NO removal (87% at 200 °C) but much lower CE of DCB (39% at 200 °C) due to the ineffectiveness of low SBET (59.65 m2/g). The NO and NH3 were considerably smaller than o-DCB molecules [12], and the strong basicity of NH3 could make up for the incapable structure properties of VMT catalyst.
Table 2.
Basic information of catalysts.
To estimate vanadium dispersion, surface density (VOx) is introduced according to Equation (1):
where xv is the vanadium mass fraction, 6.022 × 1023 is the Avocado constant, MWv is the molecular weight of vanadium (g mol−1), and SBET is BET surface area (m2/g). The optimum vanadium loading falls between polyvanadate monolayer and crystalline V2O5 because the polymeric species possess the highest catalytic activity in the NH3-SCR reaction [40]. The vanadium density of VT (8.45 VOx/nm2) and VMT (7.19 VOx/nm2) were pretty close to the theoretical polyvanadate monolayer (7.50 VOx/nm2), which brought the superiority of CE (due to the dual-site E-R mechanism [23]). For the VWT1 (4.74 VOx/nm2) and VWT2 (4.30 VOx/nm2), the values were slightly higher than the theoretical monovanadate (2.3 VOx/nm2) and much lower than polyvanadate monolayer [23]. However, their catalytic abilities remained good due to the addition of WO3, which could provide new Lewis acid sites, according to Lietti et al. [41].
VOx/nm2 = −(xv × 6.022 × 1023)/(MWv × SBET)
2.3. XRD and XPS Analysis
Figure 3 showed the XRD results of four catalysts. Prominent peaks could be found at angles of 25.3°, 36.9°, 37.8°, 38.6°, 48.1°, 53.9°, 55.1°, 62.7°, 68.8°, 70.3°, 75.0°, and 82.7° (PDF#21-1272), respectively, in all catalysts which were ascribed to anatase TiO2 (PDF#21-1272), and no peak of V2O5 in VT and VWT1, which meant the vanadium disperses equably and no amorphous or microcrystalline form existed. The results were consistent with the calculated VOx surface density. The well-dispersed V2O5 benefited the formation of monovanadate and polyvanadate monolayer, which could enhance more exposed V = O and oxygen vacancies than crystalline V2O5 [23]. As a result, better oxidation of o-DCB (78% at 200 °C for VT, and 70% at 200 °C for VWT1) was found at synergistic conditions. As expected, the peaks of crystalline tungsten could be found from 23.1° to 24.4°, and 33.2° to 34.2° due to the high content of tungsten oxide (12.14%). Excessive tungsten species on the surface of titania could compress nearby vanadium species, resulting in the production of clustered VOx or crystalline V2O5 [42]. Just like the competition between WOx and VOx, the abundant MoOx (6.76%) could compete with the surface sites and increase the formation of polymeric, crystalline V2O5 [43]. Only one V2O5 peak was found at 30.96°, indicating the relatively low crystallinity. Though polyvanadate monolayers were more active in NH3-SCR, the crystalline V2O5 was also transformed to polymeric vanadyl species by the adsorption of NH3 [44], ensuring the excellent performance of VMT on NH3-SCR (70%–87%) under synergistic conditions. The large crystallite size of V2O5 (over 100 nm) was regarded as the less active site, but only a small amount of crystalline V2O5 can be observed in VMT. The crystallite sizes of TiO2 were relatively small in VT (25.1 nm), VMT (23.5 nm), VWT1 (23.7 nm), and VWT2 (27.0 nm), and no rutile TiO2 was observed, which meant no sintering effect happened during the catalyst calculation. The VWT2 catalyst showed large WO3 (35.3 nm) crystallite sizes, indicating the tungsten could not disperse well.
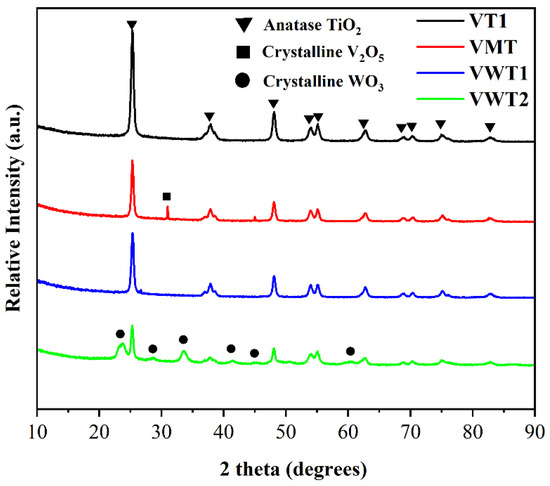
Figure 3.
X-ray power diffraction patterns of 4 catalysts, where the symbols ▼, ■, and ● denote anatase TiO2, crystalline V2O5, and WO3, respectively.
The XPS spectra of fresh catalysts and used catalysts in the o-DCB oxidation experiment were listed in Figure 4 and Table 3. Only V 2p 3/2 peaks were analyzed because the relative intensities of the V 2p 3/2 and V 2p 1/2 are constant. The vanadium species were mainly separated into V3+, V4+, and V5+, identified by the binding energy of 515.29 eV, 515.84 eV, and 517.26 eV [45]. As shown in Figure 4a, the intensities of peaks decreased in order of VT, VWT1, VMT, and VWT2, which were consistent with the vanadium contents in the catalysts. The distribution of V5+, V4+, and V3+ varied across different catalysts. As shown in Table 3, most vanadium existed as V5+ in VT (77.99%), VWT1 (62.77%), and VWT2 (65.86%). The crystalline V2O5 was reported to contribute to V4+; as a result, a significant proportion of V4+ (46.72%) could be seen in VMT [42]. The V=O provided by V5+ species was regarded as the leading catalytic site during the o-DCB degradation process [32,37] and the abatement of NOx [46]. The low V4+/V5+ ratio of VT (0.16) indicated that the large amount of V5+ species could ensure its excellent oxidation ability on o-DCB (53–78% under synergistic conditions). Yet further study also suggested that the V4+ species can provide more Brønsted acid sites, which can accelerate the “Fast-SCR” reaction at low temperatures [47,48]. Additionally, Brønsted acid sites were also crucial to the “L-H” and “E-R” reaction mechanisms [34,36,49]. The difference of V4+/V5+ contributed to the VMT’s different catalytic performance on o-DCB (13–39%) and NOx (70–87%). The higher binding energies of V5+, V4+, and V3+ (0.2–0.3 eV) were observed in VMT, VWT1, and VWT2. The interaction between MoO3 (WO3) and V2O5 could explain this condition. Most Mo existed as Mo6+ (over 94.6%) in both fresh and used catalysts, and only W6+ was observed in Figure S2a,d. The WO3 and MoO3 were reported to have poor catalytic activity without vanadium in TiO2-based catalysts [50], but the existence of Mo6+ and W6+ could ameliorate catalysts by replacing Ti4+ [16] and facilitating the formation of W-O-V or Mo-O-V [39], which had lower electronic cloud density with higher binding energy [51]. Figure 4b displays the valence changes of vanadium after the degradation of o-DCB with NH3/NO. The o-DCB molecules were absorbed by the surface lattice oxygen species (noted as OS-L) such as V=O and formed phenolate species [24]. Consequently, V5+ species were reduced to V4+. Later, the surface-adsorbed oxygen species (noted as OS-A) such as O− and O2− were consumed for further oxidation of phenolate species [37]. The oxygen in the flue gas could reoxidize V4+ into V5+ [31]. The rate of reoxidization was slower than oxidization. As a result, the V4+/V5+ ratio increased significantly (from 0.16 to 4.01, 1.73 to 2.39, 0.44 to 2.82, and 0.18 to 1.51 in VT, VMT, VWT1, and VWT2, respectively) after used.
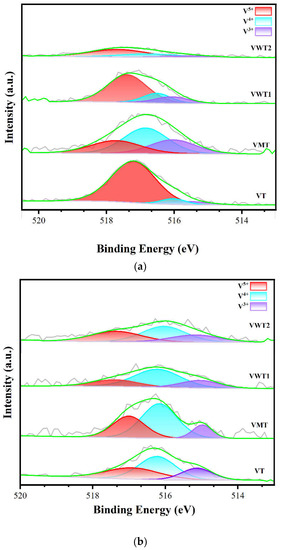
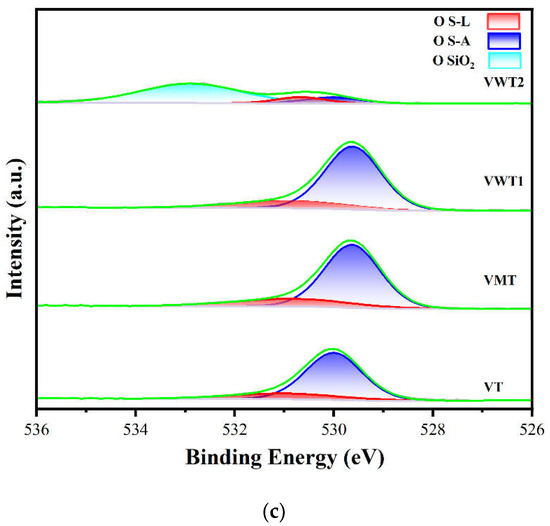
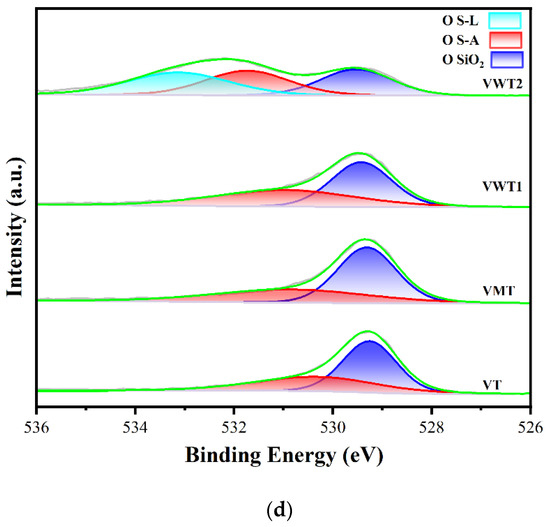
Figure 4.
(a) The valence distribution of vanadium in fresh catalysts, (b) in used catalysts; (c) the distribution of oxygen species in fresh catalysts, (d) in used catalysts.
Table 3.
The chemical states of vanadium and oxygen of fresh/used catalyst.
The distribution of OS-A and OS-L is displayed in Figure 4c,d. In VWT2, a prominent peak at 533.54 eV was observed and could be ascribed to SiO2. OS-A was reported to increase oxygen vacancies, boost redox capability, and promote the formation of Brønsted acid sites [52]. Moreover, OS-A that exists as O2− or O− has higher mobility and better oxygen-loaded capability [53]. OS-L was regarded as the lesser active oxygen than OS-A, but it also provided adsorption sites for o-DCB, which play a significant role in the MVK process [37,54]. The OS-A increased dramatically in the used catalysts in Figure 4d. The OS-A/OS-L in Table 3 increased from 0.24 to 0.64, 0.25 to 0.62, 0.34 to 0.87, and 0.91 to 1.10 in VT, VMT, VWT1, and VWT2, respectively. The augment of OS-A could be ascribed to the increasing V4+, which was reported to be the active site for forming O2−. O2− could replenish the consumed surface lattice oxygen and oxidize V4+ to V5+ [54,55], making the circulation of the redox reaction sustainable. The increasing proposition of V4+ and OS-A revealed that the reoxidation of V4+ species, rather than the supplement of oxygen, limited the oxidation rate of o-DCB.
2.4. H2-TPR and NH3-TPD Analysis
The results of H2-TPR are shown in Figure 5a. Three peaks were identified at 420.2 °C, 466.5 °C, and 713.5 °C in VT, corresponding to the formation of V6O13, VO2, and later V2O3, respectively [56]. VWT1 catalyst had similar peaks at the relatively higher temperature of 440.3 °C and 478.2 °C, which could be associated with the coreduction of V5+ to V4+ and W6+ to W4+ [57]. The decline between the two peaks was more stable, indicating that the addition of WO3 could accelerate the transformation from V6O13 to VO2, enhancing the circulation between V5+ and V4+. No peaks were observed at higher temperatures, demonstrating that WO3 improves catalytic performance by reducing V2O3 production and increasing the catalyst’s optimum reaction temperature range. The same redox peaks did not appear in VMT, though a considerable loading amount of vanadium (3.63%) was observed in VMT. The content of V4+ was relatively high (42.77%), while V5+ is at a low level (33.28%), making the transformation of V5+ to V4+ less prominent. Furthermore, the conversion of Mo6+ to Mo4+ occurred by redox at temperatures between 450 °C and 650 °C [58]. The redox peak of Mo6+ was so intense that the V5+ signal peak was covered. This meant the interaction between MoO3 and V2O5 could enhance the catalyst reducibility, leading to the extraordinary catalytic abilities of NH3/NO (70–87%). The H2-TPR profile of VWT2 showed no peaks at ca. 440 °C but had a sharper rise at 549.6 °C. The well-dispersed tungsten oxide had a lower reduction temperature than crystalline tungsten oxide [57]. The XRD results (Figure 3) showed a high proportion of crystalline WO3 in VWT2, which could cause the higher reduction temperature of W6+ to W4+. The prominent peak at 699.2 °C, 704.6 °C, and 713.5 °C represented the further reduction of V4+ and the formation of V2O3 [56]. The V4+/V5+ of VMT, VWT1, and VWT2 increased less prominently (1.73, 0.44, and 0.18 in the fresh catalysts, 2.39, 2.82, and 1.51 in the used catalysts) compared with VT (0.16 in the fresh catalyst and 4.01 in the used catalyst), indicating the addition of MoO3 and WO3 could accelerate the transformation from V4+ to V5+. Furthermore, the curves would surge as the temperature increased to 800 °C due to the consumption of H2 increasing during the redox process of W4+ (Mo4+) to W° (Mo°) (not shown in the figure completely) [57].
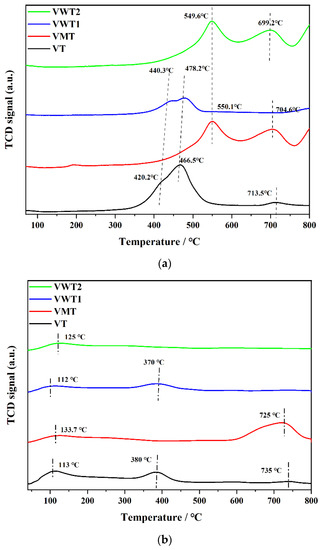
Figure 5.
(a) The H2-TPR profiles of four catalysts. (b) The NH3-TPD profiles of four catalysts.
The thermal stability of NH3 species adsorbed on the Brønsted acid sites was lower than that of species adsorbed on the Lewis acid site. Desorption peaks above 300 °C correlated with Lewis acid sites connected with NH3 molecules, whereas those below 200 °C were attributed to Brønsted acid sites associated with NH4+ ions [59]. Because it caused the nucleophilic substitution of the C-Cl and -OH group to generate HCl and promoted the subsequent ring cleavage, Brønsted acid was advantageous for the adsorption of o-DCB [24]. Moreover, the Lewis acid site was reported to be more significant in NH3 adsorption and H protons’ dissociation [49]. In Figure 5b, all catalysts showed a peak at ca. 120 °C, which could be attributed to Brønsted acid sites. No more peaks were observed at higher temperatures at VWT2, indicating that VWT2 had a limited number of Lewis acid sites. Table 4 lists the associated NH3 adsorption of different acid sites. The total acidity of VMT (0.80 µmol NH3/g) and VWT1 (0.85 µmol NH3/g) were relatively high, though the contents of V2O5 and BET surface area were lower than VT. This meant that the addition of MoOx and WOx could significantly increase the acid site [47,60]. Except for the VWT2, the total acidity did not increase with the introduction of WOx due to the existence form of crystalline WO3 in VWT2, causing low acidity compared with the monolayer WO3 [57].
Table 4.
The adsorption amount of NH3 at different temperatures.
The Brønsted acid sites in the VT were abundant (0.29 µmol NH3/g), while the Lewis acid sites were close (0.23 µmol NH3/g). Although VMT had a close Brønsted acidity (0.27 µmol NH3/g) and a significant quantity of Lewis acidity (0.53 µmol NH3/g), the CE of o-DCB was substantially lower. It could not even compete with VWT2, which had much lower Brønsted acidity (0.12 µmol NH3/g). The low BET surface area and a small amount of V5+ species, not acidity, could be the main hindrance for VMT to enhance the oxidation over o-DCB. Lewis acid sites benefited the oxidation of o-DCB [24] and the formation of N2O [38]. The VWT1 present rich Lewis acidity (0.74 µmol NH3/g), corresponding to the highest COx/HCl/N2O selectivity (61%, 50%, and 2.9% at 200 °C, respectively), in Figure 3.
3. Material and Methods
3.1. Catalysts Preparation and Characterization
Four monolithic catalysts were purchased from catalyst merchants. Their monolithic forms are listed in Table 2. Some characterization analyses were conducted in the study, and catalysts were crushed into 60–100 mesh powder before being tested.
X-ray fluorescence (XRF) spectrometer (Thermo Fisher Scientific, ADVANT’X 4200, Waltham, MA, USA) was used to determine the main catalyst components shown in Table 2. Specific surface area and pore size distribution of the catalysts were gained by N2-adsorption/desorption and Brunner–Emmet–Teller (BET) measurements (TRISTAR 3020, Micromeritics Instrument Corporation, Norcross, GA, USA). The H2-temperature-programmed reduction (H2-TPR, Atochem II 2920, Micromeritics Instrument Corporation, Norcross, GA, USA) experiment was carried out to analyze the reducibility of catalysts. Catalysts’ surface acidity was measured by temperature-programmed desorption of NH3 (NH3-TPD) performed on an Automatic Chemisorption Analyzer (BSD-Chem C200, Beijing, China). X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo Fisher Scientific Inc., Waltham, MA, USA) was used to determine the chemical states of the elements before/after experiments. Additionally, X-ray diffraction (XRD, X’Pert PRO, PANalytical B.V., Almelo, Overijssel, The Netherlands) was applied to analyze crystal morphology with a scan scope from 10° to 90°.
3.2. Catalytic Tests and Reaction Set-Up
The catalytic degradation efficiency (CE) and the selectivity of CO2 and CO (noted as COx) as well as HCl were used to measure the oxidation abilities over o-DCB and Nox, and the selectivity of N2O was measured to assess the reduction abilities over NOx. The catalysts were processed in the same outer volume of 2 cm3. The o-DCB was generated by the bubbling method, and the initial concentration was controlled at 100 ppm. The initial concentrations of NH3 and NO were both set at 350 ppm. The content of O2 was 11%, and the remaining mixing gas was N2. The total volume flow was set to 150 mL/min to ensure the gas hourly space velocity (GHSV) maintained 6000 h−1. The catalysts were placed in a tubular quartz reactor with an electric tube furnace heating, and the reaction temperatures were raised from 150 °C to 200 °C stepwise with an interval of 10 °C, among which 150 °C was also the balance temperature to determine the initial concentration of o-DCB. The schematic of the system is shown in Figure S3.
o-DCB in exhaust gas was measured by a gas chromatography-mass spectrometer (Trace 1300, Thermo Fisher Scientific, Waltham, MA, USA) equipped with a flame ionization detector (FID). The carbon dioxide/monoxide was analyzed by Nondispersive Infrared Carbon Dioxide/Monoxide Sensor (GYU-M500, L&L Technologies Inc., Romeo, HQ, USA). The NOx, HCl, and N2O in the flue gas were detected by Gasmet Portable FT-IR Gas Analyzer (Gasmet Dx4000, Gasmet Technologies Inc., Helsinki, Finland), and NH3 was only measured before placing catalysts. What is worth noting is that the mixed gas used contains carbon dioxide at the ppm level unavoidably due to the gas preparation process. A background calibration of CO2 was needed before the test to ensure measurement accuracy.
The calculation equations of the mentioned values are listed in Equations (2)–(6):
where the “in” and “out” represent the inlet and outlet concentration of COx, NOx, o-DCB, HCl, and N2O.
CE-o-DCB (%) = [ (o-DCB)in − (o-DCB)out]/(o-DCB)in × 100%
COx Selectivity (%) = [(CO + CO2) − (CO2)bg]/[6 × (o-DCB)in] ×100%
CE-NOx (%) = [ (NOx)in − (NOx)out]/(NOx)in × 100%
N2O Selectivity (%) = N2O/[0.5 × (NOx)in] × 100%
HCl Selectivity (%) = HCl/[2 × (o-DCB)in] ×100%
4. Conclusions
The synergistic removal of o-DCB and NH3/NO using four monolithic catalysts were investigated, and the characterizations of catalysts were analyzed. The granular VOx/TiO2 catalyst showed good catalytic performance on both o-DCB and NH3/NO removal due to its excellent SBET surface area (91.98 m2/g), high amount of V5+ species (77.2%), and good acidity (total acidity with 0.52 µmol NH3/g adsorption). The honeycomb MoOx-VOx/TiO2 catalyst displayed the lowest CE of o-DCB but the highest NE of NOx, indicating that NH3-SCR is more related to the high proportion of V4+ species, VOX surface density close to polyvanadium monolayer (7.50 VOx/nm2), and abundant acidity, while catalytic oxidation is more associated with pore structure and V5+ species. A V4+/V5+ and OS-A/OS-L ratio of four used catalysts revealed that the reoxidation of V4+ species limited the reaction rate. The comparisons between powder and monolithic catalysts revealed that monolithic ways took a significant effect on catalytic performance, among which the corrugated methods could benefit catalysts most. Further studies should focus on the working life and antipoisoning abilities of the catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12111342/s1, Figure S1: (a) The CE of o-DCE of four powder catalysts at 6000 h−1, with initial concentration of 100 ppm; (b) The COx selectivity of four powder catalysts.; Figure S2: (a) The valence distribution of molybdenum in fresh catalysts, (b) in used catalysts; (c) The distribution of tungsten species in fresh catalysts, (d) in used catalysts. Figure S3. Schematic of the synergistic removal of o-DCB and NH3/NO.
Author Contributions
J.L.: writing—original draft, writing—review and editing; Y.M.: data curation, software; J.W.: conceptualization, formal analysis; H.Y.: data curation; X.L. (Xiaodong Li): supervision, funding acquisition. X.L. (Xiaoqing Lin): methodology, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key Research and Development Program of China: 2020YFC1910100.
Data Availability Statement
Experimental data will be available for the request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bhavsar, S.P.; Reiner, E.J.; Hayton, A.; Fletcher, R.; Macpherson, K. Converting toxic equivalents (teq) of dioxins and dioxin-like compounds in fish from one toxic equivalency factor (tef) scheme to another. Environ. Int. 2008, 34, 915–921. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Kumar, A.; Ying, Q.; Vandenberghe, F.; Kleeman, M.J. Separately resolving nox and voc contributions to ozone formation. Atmos. Environ. 2022, 285, 119224. [Google Scholar] [CrossRef]
- Lu, S.; Xiang, Y.; Chen, Z.; Chen, T.; Lin, X.; Zhang, W.; Li, X.; Yan, J. Development of phosphorus-based inhibitors for pcdd/fs suppression. Waste Manag. 2021, 119, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Karademir, A.; Bakoglu, M.; Taspinar, F.; Ayberk, S. Removal of pcdd/fs from flue gas by a fixed-bed activated carbon filter in a hazardous waste incinerator. Environ. Sci. Technol. 2004, 38, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Fahlenkamp, H.; Mittelbach, G.; Hagenmaier, H.P.; Brunner, H.; Tichaczek, K.H. Catalytic oxidation. A technique for reducing pcdd/pcdf emissions from waste incinerators to less than 0.1 ng te/m3. VGB Kraftw. 1991, 71, 671–674. [Google Scholar]
- Mckay, G. Dioxin characterisation, formation and minimisation during municipal solid waste (msw) incineration: Review. Chem. Eng. J. 2002, 86, 343–368. [Google Scholar] [CrossRef]
- Hagenmaier, H. Katalystische oxidation halogenierter kohlenwasserstoffe unter besonderer berücksichtigung des dioxinproblems. VDI-Berichte 1989, 730, 239–254. [Google Scholar]
- Hagenmaier, H.; Horch, K.; Fahlenkamp, H.; Schetter, G. Destruction of PCDD and PCDF in refuse incineration plants by primary and secondary measures. Chemosphere 1991, 23, 1429–1437. [Google Scholar] [CrossRef]
- Okumura, M.; Akita, T.; Haruta, M.; Wang, X.; Kajikawa, O.; Okada, O. Multi-component noble metal catalysts prepared by sequential deposition precipitation for low temperature decomposition of dioxin. Appl. Catal. B Environ. 2003, 41, 43–52. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Uemichi, Y.; Ayame, A. Low-temperature hydrodechlorination mechanism of chlorobenzenes over platinum-supported and palladium-supported alumina catalysts. Appl. Catal. A Gen. 2005, 287, 89–97. [Google Scholar] [CrossRef]
- Cai, T.; Huang, H.; Deng, W.; Dai, Q.; Liu, W.; Wang, X. Catalytic combustion of 1,2-dichlorobenzene at low temperature over mn-modified co3o4 catalysts. Appl. Catal. B Environ. 2015, 166–167, 393–405. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Q.; Stevens, W.R.; Lee, C.W.; Gullett, B.K.; Zhao, Y. Study on the decomposition of trace benzene over v2o5–WO3/TiO2-based catalysts in simulated flue gas. Appl. Catal. B Environ. 2014, 147, 322–329. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Rivas, J.A.; Amiridis, M.D. Catalytic oxidation of 1,2-dichlorobenzene over supported transition metal oxides. J. Catal. 2000, 193, 264–272. [Google Scholar] [CrossRef]
- Gan, L.; Li, K.; Niu, H.; Peng, Y.; Chen, J.; Huang, Y.; Li, J. Simultaneous removal of nox and chlorobenzene on v2o5/TiO2 granular catalyst: Kinetic study and performance prediction. Front. Environ. Sci. Eng. 2020, 15, 70. [Google Scholar] [CrossRef]
- Yu, M.; Li, X.; Ren, Y.; Chen, T.; Lu, S.; Yan, J. Low temperature oxidation of pcdd/fs by TiO2-based v2o5/WO3 catalyst. Environ. Prog. Sustain. 2016, 35, 1265–1273. [Google Scholar] [CrossRef]
- Kuma, R.; Kitano, T.; Tsujiguchi, T.; Tanaka, T. Effect of molybdenum on the structure and performance of v2o5/TiO2–sio2–moo3 catalysts for the oxidative degradation of o-chlorotoluene. Appl. Catal. A Gen. 2020, 595, 117496. [Google Scholar] [CrossRef]
- Huang, L.; Zeng, Y.; Gao, Y.; Wang, H.; Zong, Y.; Chang, Z.; Zhang, S.; Han, P.; Yu, Y. Promotional effect of phosphorus addition on improving the so2 resistance of v2o5-moo3/TiO2 catalyst for nh3-scr of no. J. Phys. Chem. Solids 2022, 163, 110566. [Google Scholar] [CrossRef]
- He, C.; Cheng, J.; Zhang, X.; Douthwaite, M.; Pattisson, S.; Hao, Z. Recent advances in the catalytic oxidation of volatile organic compounds: A review based on pollutant sorts and sources. Chem. Rev. 2019, 119, 4471–4568. [Google Scholar] [CrossRef]
- Chen, C.; Cao, Y.; Liu, S.; Chen, J.; Jia, W. Review on the latest developments in modified vanadium-titanium-based scr catalysts. Chin. J. Catal. 2018, 39, 1347–1365. [Google Scholar] [CrossRef]
- Chin, S.; Jurng, J.; Lee, J.; Moon, S. Catalytic conversion of 1,2-dichlorobenzene using v2o5/TiO2 catalysts by a thermal decomposition process. Chemosphere 2009, 75, 1206–1209. [Google Scholar] [CrossRef]
- Huang, X.; Peng, Y.; Liu, X.; Li, K.; Deng, Y.; Li, J. The promotional effect of moo3 doped v2o5/TiO2 for chlorobenzene oxidation. Catal. Commun. 2015, 69, 161–164. [Google Scholar] [CrossRef]
- Wu, M.; Ung, K.C.; Dai, Q.; Wang, X. Catalytic combustion of chlorinated vocs over vox/TiO2 catalysts. Catal. Commun. 2012, 18, 72–75. [Google Scholar] [CrossRef]
- Gallastegi-Villa, M.; Aranzabal, A.; González-Marcos, M.P.; Markaide-Aiastui, B.A.; González-Marcos, J.A.; González-Velasco, J.R. Effect of vanadia loading on acidic and redox properties of vox/TiO2 for the simultaneous abatement of pcdd/fs and nox. J. Ind. Eng. Chem. 2020, 81, 440–450. [Google Scholar] [CrossRef]
- Albonetti, S.; Blasioli, S.; Bonelli, R.; Mengou, J.E.; Scirè, S.; Trifirò, F. The role of acidity in the decomposition of 1,2-dichlorobenzene over TiO2-based v2o5/WO3 catalysts. Appl. Catal. A Gen. 2008, 341, 18–25. [Google Scholar] [CrossRef]
- Cai, M.; Bian, X.; Xie, F.; Wu, W.; Cen, P. Formation and performance of monolithic catalysts for selective catalytic reduction of nitrogen oxides: A critical review. Chemistryselect 2021, 6, 12331–12341. [Google Scholar] [CrossRef]
- Zhan, M.X.; Fu, J.Y.; Ji, L.J.; Deviatkin, I.; Lu, S.Y. Comparative analyses of catalytic degradation of pcdd/fs in the laboratory vs. Industrial conditions. Chemosphere 2018, 191, 895–902. [Google Scholar] [CrossRef]
- Govender, S.; Friedrich, H.B. Monoliths: A review of the basics, preparation methods and their relevance to oxidation. Catalysts 2017, 7, 62. [Google Scholar] [CrossRef]
- Ma, Y.; Lai, J.; Lin, X.; Zhang, H.; Du, H.; Long, J.; Yan, J.; Li, X. Implication of operation time on low-temperature catalytic oxidation of chloroaromatic organics over vox/TiO2 catalysts: Deactivation mechanism analysis. J. Clean. Prod. 2022, 372, 133477. [Google Scholar] [CrossRef]
- Weber, R.; Sakurai, T.; Hagenmaier, H. Low temperature decomposition of pcdd/pcdf, chlorobenzenes and pahs by TiO2-based v2o5–WO3 catalysts. Appl. Catal. B Environ. 1999, 20, 249–256. [Google Scholar] [CrossRef]
- Ma, Y.; Lai, J.; Li, X.; Lin, X.; Li, L.; Jing, H.; Liu, T.; Yan, J. Field study on pcdd/f decomposition over vox/TiO2 catalyst under low-temperature: Mechanism and kinetics analysis. Chem. Eng. J. 2022, 429, 132222. [Google Scholar] [CrossRef]
- Xiao, G.; Guo, Z.; Lin, B.; Fu, M.; Ye, D.; Hu, Y. Cu-vwt catalysts for synergistic elimination of nox and volatile organic compounds from coal-fired flue gas. Environ. Sci. Technol. 2022, 56, 10095–10104. [Google Scholar] [CrossRef] [PubMed]
- Bertinchamps, F.; Treinen, M.; Blangenois, N.; Mariage, E.; Gaigneaux, E.M. Positive effect of nox on the performances of vox/TiO2-based catalysts in the total oxidation abatement of chlorobenzene. J. Catal. 2005, 230, 493–498. [Google Scholar] [CrossRef]
- Gao, F.; Tang, X.; Yi, H.; Zhao, S.; Li, C.; Li, J.; Shi, Y.; Meng, X. A review on selective catalytic reduction of nox by nh3 over mn–based catalysts at low temperatures: Catalysts, mechanisms, kinetics and dft calculations. Catalysts 2017, 7, 199. [Google Scholar] [CrossRef]
- Topsøe, N. Mechanism of the selective catalytic reduction of nitric oxide by ammonia elucidated by in situ on-line fourier transform infrared spectroscopy. Science 1994, 265, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Ramis, G.; Yi, L.; Busca, G.; Turco, M.; Kotur, E.; Willey, R.J. Adsorption, activation, and oxidation of ammonia over scr catalysts. J. Catal. 1995, 157, 523–535. [Google Scholar] [CrossRef]
- Su, W.; Chang, H.; Peng, Y.; Zhang, C.; Li, J. Reaction pathway investigation on the selective catalytic reduction of no with nh3 over cu/ssz-13 at low temperatures. Environ. Sci. Technol. 2015, 49, 467–473. [Google Scholar] [CrossRef]
- Jia, W.; Zhang, J.; Yu, X.; Feng, Y.; Wang, Q.; Sun, Y.; Tang, X.; Zeng, X.; Lin, L. Insight into the mars-van krevelen mechanism for production 2,5-diformylfuran over fenx@c catalyst. Biomass Bioenergy 2022, 156, 106320. [Google Scholar] [CrossRef]
- Zhu, M.; Lai, J.; Wachs, I.E. Formation of n2o greenhouse gas during scr of no with nh3 by supported vanadium oxide catalysts. Appl. Catal. B Environ. 2018, 224, 836–840. [Google Scholar] [CrossRef]
- Zhao, X.; Yan, Y.; Mao, L.; Fu, M.; Zhao, H.; Sun, L.; Xiao, Y.; Dong, G. A relationship between the v4+/v5+ ratio and the surface dispersion, surface acidity, and redox performance of v2o5–WO3/TiO2 scr catalysts. Rsc. Adv. 2018, 8, 31081–31093. [Google Scholar] [CrossRef]
- Went, G.T.; Leu, L.; Rosin, R.R.; Bell, A.T. The effects of structure on the catalytic activity and selectivity of v2o5/TiO2 for the reduction of no by nh3. J. Catal. 1992, 134, 492–505. [Google Scholar] [CrossRef]
- Lietti, L. Reactivity of v2o5&z.sbnd;WO3TiO2 de-nox catalysts by transient methods. Appl. Catal. B Environ. 1996, 10, 281–297. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, S.; Lee, M.; Ye, B.; Kim, T.; Kim, H.; Lee, J.W.; Lee, D.H. Effect of catalyst crystallinity on v-based selective catalytic reduction with ammonia. Nanomaterials 2021, 11, 1452. [Google Scholar] [CrossRef]
- Kompio, P.G.W.A.; Brückner, A.; Hipler, F.; Auer, G.; Löffler, E.; Grünert, W. A new view on the relations between tungsten and vanadium in v2o5WO3/TiO2 catalysts for the selective reduction of no with nh3. J. Catal. 2012, 286, 237–247. [Google Scholar] [CrossRef]
- Lian, Z.; Wei, J.; Shan, W.; Yu, Y.; Radjenovic, P.M.; Zhang, H.; He, G.; Liu, F.; Li, J.; Tian, Z.; et al. Adsorption-induced active vanadium species facilitate excellent performance in low-temperature catalytic nox abatement. J. Am. Chem. Soc. 2021, 143, 10454–10461. [Google Scholar] [CrossRef] [PubMed]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in xps analysis of first row transition metals, oxides and hydroxides: Sc, ti, v, cu and zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective catalytic reduction of nox with nh3 by using novel catalysts: State of the art and future prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, M.J.; Boyano, A.; Herrera, C.; Larrubia, M.A.; Alemany, L.J.; Moliner, R. Vanadium loaded carbon-based monoliths for the on-board no reduction: Influence of vanadia and tungsten loadings. Chem. Eng. J. 2009, 155, 68–75. [Google Scholar] [CrossRef]
- Wang, X.; Du, X.; Yang, G.; Xue, J.; Chen, Y.; Zhang, L. Chemisorption of no2 on v-based scr catalysts: A fundamental study toward the mechanism of “fast scr” reaction. J. Phys. Chem. C 2019, 123, 20451–20458. [Google Scholar] [CrossRef]
- Ramis, G.; Yi, L.; Busca, G. Ammonia activation over catalysts for the selective catalytic reduction of nox and the selective catalytic oxidation of nh3. An ft-ir study. Catal. Today 1996, 28, 373–380. [Google Scholar] [CrossRef]
- Debecker, D.P.; Bertinchamps, F.; Blangenois, N.; Eloy, P.; Gaigneaux, E.M. On the impact of the choice of model voc in the evaluation of v-based catalysts for the total oxidation of dioxins: Furan vs. Chlorobenzene. Appl. Catal. B Environ. 2007, 74, 223–232. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Z.; Wang, B.; Sun, Q.; Xu, W.; Zhu, T. Effects of mox (m=mn, cu, sb, la) on v–mo–ce/ti selective catalytic reduction catalysts. J. Rare Earths 2020, 38, 157–166. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Q.; Sun, J.; Liu, Q.; Zhao, D.; Gao, W.; Liu, L. Effects of mercury oxidation on v2o5–WO3/TiO2 catalyst properties in nh3-scr process. Catal. Commun. 2015, 59, 78–82. [Google Scholar] [CrossRef]
- Wang, P.; Gao, S.; Wang, H.; Chen, S.; Chen, X.; Wu, Z. Enhanced dual resistance to alkali metal and phosphate poisoning: Mo modifying vanadium-titanate nanotubes scr catalyst. Appl. Catal. A Gen. 2018, 561, 68–77. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, Q. Surface characterization studies on the interaction of v2o5–WO3/TiO2 catalyst for low temperature scr of no with nh3. J. Solid State Chem. 2015, 221, 49–56. [Google Scholar] [CrossRef]
- Zhang, S.; Li, H.; Zhong, Q. Promotional effect of f-doped v2o5–WO3/TiO2 catalyst for nh3-scr of no at low-temperature. Appl. Catal. A Gen. 2012, 435–436, 156–162. [Google Scholar] [CrossRef]
- Besselmann, S.; Freitag, C.; Hinrichsen, O.; Muhler, M. Temperature-programmed reduction and oxidation experiments with v 2 o 5/tio 2 catalysts. Phys. Chem. Chem. Phys. 2001, 3, 4633–4638. [Google Scholar] [CrossRef]
- Wang, C.; Yang, S.; Chang, H.; Peng, Y.; Li, J. Dispersion of tungsten oxide on scr performance of v2o5WO3/TiO2: Acidity, surface species and catalytic activity. Chem. Eng. J. 2013, 225, 520–527. [Google Scholar] [CrossRef]
- Dang, J.; Zhang, G.H.; Chou, K.C.; Reddy, R.G.; He, Y.; Sun, Y. Kinetics and mechanism of hydrogen reduction of moo3 to moo2. Int. J. Refract. Met. H 2013, 41, 216–223. [Google Scholar] [CrossRef]
- Hu, H.; Cai, S.; Li, H.; Huang, L.; Shi, L.; Zhang, D. In situ drifts investigation of the low-temperature reaction mechanism over mn-doped co3o4 for the selective catalytic reduction of nox with nh3. J. Phys. Chem. C 2015, 119, 22924–22933. [Google Scholar] [CrossRef]
- Qiu, Y.; Liu, B.; Du, J.; Tang, Q.; Liu, Z.; Liu, R.; Tao, C. The monolithic cordierite supported v2o5–moo3/TiO2 catalyst for nh3-scr. Chem. Eng. J. 2016, 294, 264–272. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).