Abstract
Bloom (BLM) helicase is an important member of the RecQ family of DNA helicases that plays a vital role in the maintenance of genomic stability. The defect of BLM helicase leads to a human genetic disorder called Bloom syndrome, characterized by genomic instability, specific phenotypic features, and a predisposition to many types of cancer. The predisposition to cancer caused by BLM helicase is due to defects in important DNA metabolic pathways such as replication, recombination, and repair. Therefore, the aim of this work was to investigate the effects of two prenylated chalcones, WZH-10 and WZH-43, on the expression of BLM helicase in prostate cancer cells, as well as the biological activity of the purified BLM helicase from cancer cells. This might lead to a better understanding of the role of BLM helicase in the aforementioned DNA metabolic pathways that directly influence chromosomal integrity leading to cancer. The results indicated that the two prenylated chalcones inhibited the growth of prostate cancer cells PC3 by inducing apoptosis and arresting the cell cycle. However, they only inhibited the protein expression of BLM helicase without regulating its transcriptional expression. In addition, they did not significantly regulate the expression of the homologous family members WRN and RECQL1, although the DNA unwinding and ATPase activity of BLM helicase were inhibited by the two prenylated chalcones. Finally, a negligible effect was found on the DNA-binding activity of this enzyme. These results demonstrated that prenylated chalcones can be an effective intervention on the expression and function of the BLM helicase protein in cancer cells to inhibit their growth. Therefore, they might provide a novel strategy for developing new anti-cancer drugs targeting the genomic stability and DNA helicase.
1. Introduction
The stability of the genetic material in the DNA guarantees the existence and continuity of the species. The genetic information is copied from the parental cells to the daughter cells by the DNA semi-conservative replication, which guides the transcription and translation. In this process, the double-stranded DNA (dsDNA) needs to be separated into two single-stranded DNA (ssDNA) by DNA helicase, providing the templates for DNA replication [1]. The DNA helicase works as a molecular motor, using the energy deriving from the catalytic hydrolysis of nucleotide triphosphate (NTP) to translocate along the nucleic acid strand and separate the complementary strands of the dsDNA [2]. DNA helicases are important for other functions of cell metabolism including genome replication, DNA repair, recombination, transcription, and telomere maintenance [2,3], as well as playing a key role in chromosome stability [3,4,5].
The RecQ helicase family, as an important member of DNA helicase, plays an important role in chromosome maintenance [3,4,5,6]. Defects in human RecQ helicases Bloom (BLM), Werner (WRN), and RECQL4 are associated with BLM syndrome, WRN syndrome, and Rothmund–Thomson syndrome, respectively [6,7]. The BLM helicase is associated with DNA damage repair factors such as RECQ1, WRN (RECQ3), RECQ4, and RECQ5. BLM syndrome, as a rare autosomal recessive genetic disease, is related to the functional defect of BLM helicase [8]. It is characterized by genomic instability and susceptibility to cancer [9]. BLM helicase is ATP-dependent and has a 3′, 5′orientation that unlocks Watson–Crick double-stranded DNA in vitro [10]. Based on the unique role of human RecQ helicase in genomic stability, strategies are being explored to combat multiple types of cancer by regulating the function of human RecQ helicase or its interacting factors [11,12,13,14]. Helicase-dependent DNA metabolic pathways in cancer cells are selectively inactivated, thus accelerating the occurrence and development of tumors [9,15]. Thus, the human RecQ helicases may be suitable targets for cancer therapy [12,13]. Indeed, many small molecules are designed to modulate the expression and function of the RecQ helicases for their use in cancer treatment [7].
Chalcone compounds are a class of natural organic compounds found in medicinal plants, which are precursors for the synthesis of flavonoids in plants. In addition, they are widely present in plants as secondary metabolites [16,17]. The basic skeleton structure of chalcone compounds is 1,3-diarylpropenone, containing two phenyl rings (the A- and B-rings) connected by a three-carbon α,β-unsaturated carbonyl bridge [18]. Its molecule has great flexibility, and can bind to different receptors, making it have an important pharmacological effect, including anti-bacterial, anti-inflammation, and anti-cancer [19]. Our previous studies indicated that the design and synthesized prenylated chalcone WZH-10 could cause Fli-1 expression inactivation through targeted promoter activity regulation for inhibiting growth and metastasis of erythroleukemia cells. In this paper, we investigated the effect of two prenylated chalcones WZH-10 and WZH-43 (the chemical structure of these minor groove binders is shown in Figure 1A) on the expression and biological activity of BLM helicase (DNA-binding, DNA unwinding, and ATPase activity) in vitro on prostate cancer cells (PC3) using methods including fluorescence polarization, free phosphorus detection, and gel blocking technology. Our results revealed that these two compounds inhibited the growth of prostate cancer cells PC3 by inducing apoptosis and arresting the cell cycle, only inhibited the protein expression of BLM helicase without regulating its transcriptional expression, and inhibited the helicase function, including the unwinding and ATPase activity, but did not affect the DNA-binding activity. Therefore, this study could provide an important approach for developing new anti-cancer drugs targeting DNA helicase.
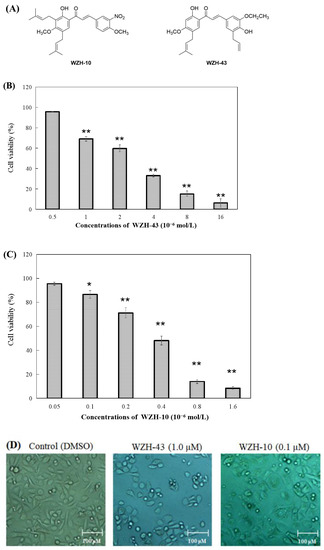
Figure 1.
Effect of two prenylated chalcones on the proliferation of PC3 cells. (A) Chemical structure of WZH-43 and WZH-10. Cell viability of PC3 cells in vitro after the treatment with WZH-43 (B) and WZH-10 (C). (D) morphology of PC3 cells after the exposure to the two compounds for 48 h by optical microscope. Cells (1 × 104 cells/mL) were treated with compounds at different concentrations for 48 h. The cell viability was calculated by MTT assay and the IC50 values were determined. Values are expressed as mean ± SD of three independent experiments. Scale bar = 100 μM in all images. * p < 0.05, ** p < 0.01 compared with the control.
2. Results
2.1. Prenylated Chalcones Inhibited the Proliferation of PC3 Cells
The percentage of cell survival was significantly decreased with the increase in the concentration of the compounds (Figure 1B,C), indicating a dose-dependent inhibitory activity. The evaluation of the IC50 values of the two compounds on the proliferation of PC3 cells revealed that the inhibitory activity of WZH-10 (with an IC50 of 0.375 ± 0.025 µmol/L) on the growth of the PC3 cell was more than (p < 0.01) that of WZH-43 (with an IC50 of 3.863 ± 0.937 µmol/L). In addition, the decrease in cell number was concentration-dependent, as shown in Figure 1B,C, as the number of the cells decreased with the increase in the concentration of the compounds. Moreover, some apoptotic bodies were observed in the cells treated with different concentrations of the compounds at 48 h, suggesting that both of them may induce apoptosis in PC3 cells (Figure 1D).
2.2. Prenylated Chalcones Induced Apoptosis and Arrested the Cell Cycle in PC3 Cells
The apoptosis of PC3 cells was significantly induced by the highest concentration of the two compounds, as demonstrated by flow cytometry (p < 0.01) (Figure 2A,B). However, only a slight but still significant apoptotic effect was observed in the cells treated with the lower concentration of the two compounds (p < 0.05). In addition, the effect of the two compounds on the nuclear chromatin of PC3 cells (Figure 2C) revealed a significant morphological change. The nucleus of the untreated cells was less bright, and the color was more homogeneous than that of the compounds-treated cells, suggesting that the two compounds caused chromatin condensation that is the hallmark of apoptosis. The quantification of the effect of both compounds at different concentrations (WZH-43: 0.5 and 1.0 µM, WZH-10: 0.05 and 0.1 µM) for 48 h on the cell cycle revealed that the number of cells in G1 and S phases of PC3 were significantly (p < 0.01) decreased, while the G2 phase was significantly (p < 0.01) increased compared to the control, suggesting that the two prenylated chalcones significantly blocked the cell cycle of prostate cancer cells PC3 (Figure 3A,B).
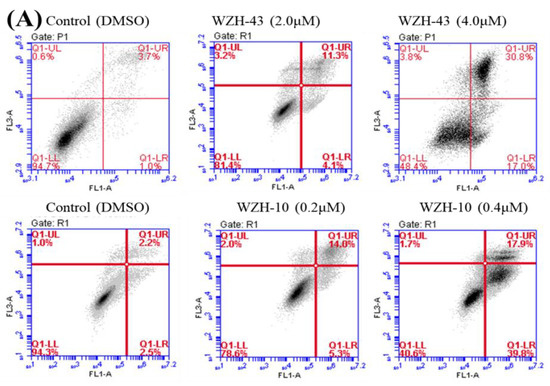
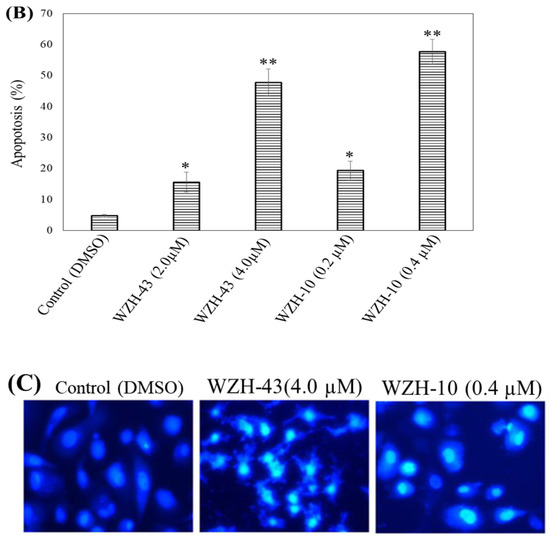
Figure 2.
Cell apoptosis induced by two prenylated chalcones for 48 h. (A) Cell apoptosis induced by two prenylated chalcones detected with flow cytometry. (B) The quantification of the results in (A). Apoptotic cells were counted as percentage of total gated events. (C) Two prenylated chalcones induced apoptosis as visualized by Hoechst 33,258 staining. Chromatin condensation and fragmentation were observed by a fluorescent inverted microscope after the treatment with 4.0 μM WZH-43 and 0.4 μM WZH-10 for 48 h. Scale bar = 100 μM in all images. All experiments were performed in triplicate. Results are presented as mean ± SEM. * p < 0.05, ** p < 0.01 (n = 3) compared with the control.
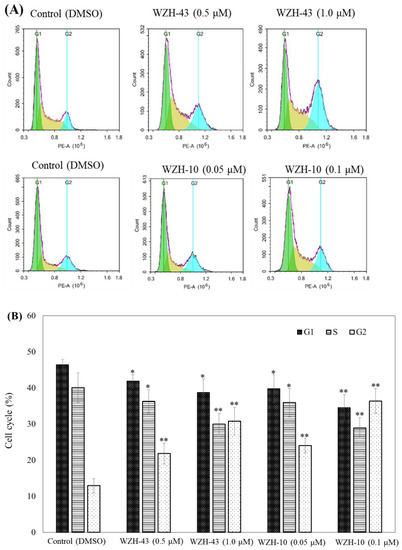
Figure 3.
Cell cycle analysis of PC3 cells treated with different concentrations of the two prenylated chalcones for 48 h. (A) Cell cycle changes induced by the two prenylated chalcones by flow cytometry. (B) The quantification of the results in (A). The G1, G2, and S percentages were related to total diploid cells. Results are presented as mean ± SEM. * p < 0.05, ** p < 0.01 (n = 3) compared with the control.
2.3. Prenylated Chalcones Down-Regulated the Protein Expression of BLM Helicase Gene in PC3 Cells
Cell cycle arrest and cell apoptosis of cancer cells after the compounds treatment are accompanied by the changes in the expression of many proteins involved in DNA metabolism; thus, the gene and protein expression of human RecQ family DNA helicase in PC3 cells was evaluated after drug treatment. The treatment of PC3 cells with 1.0 μmol/L of WZH-43 and 0.1 μmol/L of WZH-10 (Figure 4) showed that the two prenylated chalcones did not significantly modify the transcriptional expression of BLM helicase (Figure 4A) but significantly (p < 0.01) down-regulated its protein expression (Figure 4B), and did not significantly regulate the mRNA and protein expression of WRN and RECQL1, the other two members of the RecQ family. These results indicated that the two prenylated chalcones inhibited the growth of prostate cancer cells by regulating the protein expression of BLM helicase.
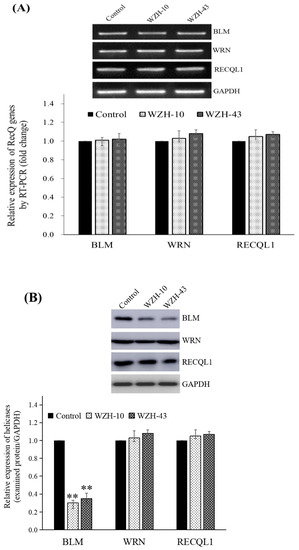
Figure 4.
Effect of the two prenylated chalcones on the expression of BLM helicase in PC3 cells. (A) Expression of the BLM gene in PC3 cells after the treatment with the two agents. (B) BLM helicase protein expression in PC3 cells by Western blotting. Cells were treated with 1.0 µM WZH-43 and 0.1 µM WZH-10 for 48 h. The results were normalized to the expression of GAPDH, and the fold change was relative to PC3 cells incubated in medium alone (time matched). Results are presented as mean ± SEM. ** p < 0.01 (n = 3) compared with the control.
2.4. Effects of Two Prenylated Chalcones on the DNA-Binding Activity of BLM Helicase
The effect of WZH-43 and WZH-10 on the DNA-binding activity of BLM helicase, obtained from prostate cancer cells by recombinant cloning, induced expression, isolation, and purification, was analyzed using dsDNA as substrates and fluorescence polarization (Figure 5). The DNA binding activity showed that WZH-43 reacted directly with the helicase and BLM-DNA complex (Figure 5A). This binding was slightly increased with increasing WZH-43 concentrations. A similar increase in binding activity was also found in the reactions of WZH-10 and the BLM helicase and BLM-DNA complex (Figure 5B). The effects of the two compounds on the anisotropy of dsDNA were further analyzed (Figure 5C) by fluorescence polarization technology. The anisotropy of dsDNA also changed with the increasing concentration of both prenylated chalcones, but it was not statistically significant. However, the anisotropy was not increased with the increase in the drug concentration when the DNA substrate was ssDNA (Figure 5D). These results demonstrated that the DNA-binding activity of BLM helicase was not affected by WZH-43 and WZH-10 treatment. The result was further confirmed by gel retardation technology (Figure 6A) and the change in Kd values (Figure 6B) after two prenylated chalcones treatment.
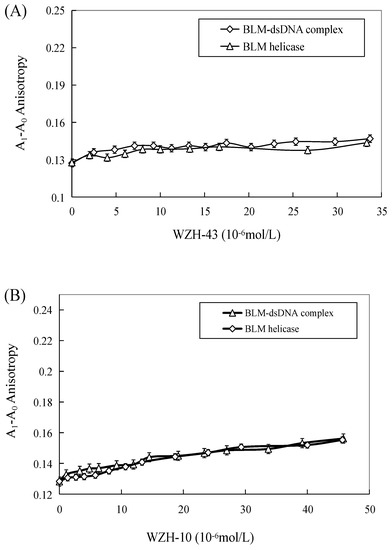
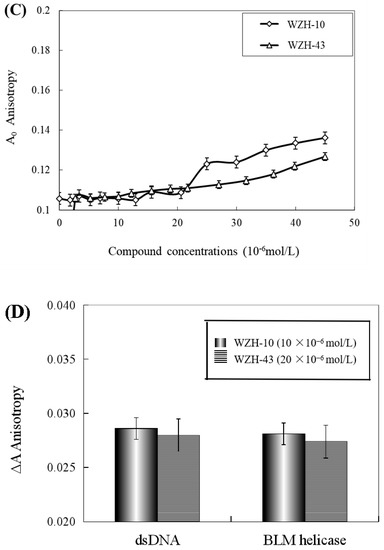
Figure 5.
Effect of WZH-43 (A) and WZH-10 (B) on the DNA-binding activity of BLM helicase bound to dsDNA and on the anisotropy of dsDNA (C) and ssDNA (D). A0: fluorescence anisotropy of the fluorescein-labeled DNA. A1: fluorescence anisotropy of the miscible liquid of the helicase at different concentrations of WZH-43 and WZH-10.

Figure 6.
Effect of the two prenylated chalcones on the DNA-binding activity of BLM helicase. (A) Effect of WZH-43 and WZH-10 on the DNA-binding activity of the helicase by EMSA. (B) Effect of WZH-43 and WZH-10 on the Kd values of the helicase and DNA substrate.
2.5. Effect of the Two Prenylated Chalcones on the DNA-Unwinding Activity of BLM Helicase
The results revealed that the DNA unwinding was increased at the lower concentration of the two compounds (0.4 μmol/L), indicating the promotion of DNA unwinding activity by the two prenylated chalcones at concentrations lower than 0.4 μmol/L. However, the anisotropy (Figure 7A) and Kobs values (Figure 7B) of dsDNA unwinding were decreased with the two compounds at concentrations higher than 0.4 μmol/L, indicating the inhibition of DNA unwinding activity by two compounds. The half inactivating concentration (Ci) of WZH-43 associated with the DNA unwinding activity was 0.83 ± 0.24 μmol/L, similar to WZH-10 with a Ci value of 0.85 ± 0.18 μmol/L.
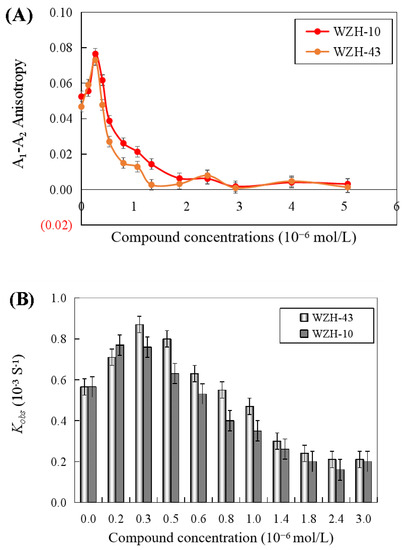
Figure 7.
Effect of the two prenylated chalcones on the DNA unwinding activity of BLM helicase by fluorescence polarization (A) and change in Kobs values of DNA unwinding activity of BLM helicase treated by different concentrations of compounds (B).
2.6. Effect of the Two Prenylated Chalcones on the ATPase Activity of BLM Helicase
Both two prenylated chalcones inhibited the ATPase activity in a dose-dependent manner (Figure 8A), and no significant difference was found in the inhibitory effect of the two compounds on the activity of the helicase. The Ci values of WZH-43 and WZH-10 for the ATPase activity were 2.65 ± 0.25 μmol/L and 1.35 ± 0.31 μmol/L mol/L, respectively, indicating that the inhibition of WZH-10 was stronger (p < 0.05) than that of WZH-43. The inhibitory effect of WZH-43 (Figure 8B) and WZH-10 (Figure 8C) was time-dependent. According to the results from the Michaelis–Menten equation (Table 1), the Km, Vmax, and Kcat constants decreased with the compounds concentration, indicating that the compounds were behaving as anticompetitive inhibitors for the ATPase activity of BLM helicase. We selected ML216, a BLM helicase inhibitor, as a reference agent.
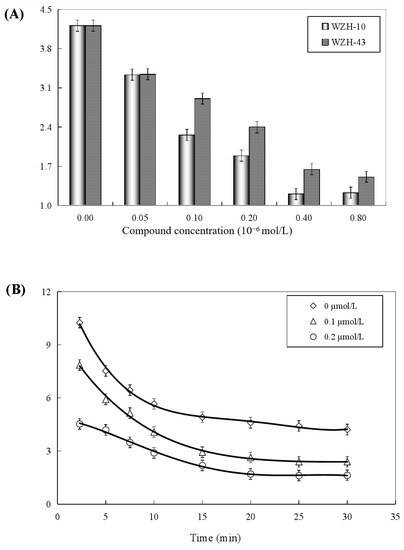

Figure 8.
Concentration-dependent (A) and time-dependent inhibitory effect of WZH-43 (B) and WZH-10 (C) on the ATPase activity of BLM helicase.

Table 1.
Effects of the two prenylated chalcones on the ATPase activity of BLM helicase.
3. Discussion
The mechanism of action of numerous anti-cancer drugs is associated with their regulation on the DNA metabolism, resulting in the inhibition of the rapid growth and metastasis of tumor cells [20]. The consequence of the perturbations due to these drugs can be represented by the inhibition of DNA synthesis during DNA replication/repair or transcription. The introduction into cells of certain chemicals that interfere with DNA metabolism results in the formation of ssDNA and/or dsDNA strand breaks [21]. Especially, small molecules working as anti-tumor agents can inactivate DNA metabolism-related enzymes [15,20]. Therefore, it is a new strategy to fight cancer for investigating small novel chemical molecules that can cause the inactivation of proteins that unwind dsDNA, consequently inhibiting cell proliferation [22,23,24]. A limited number of studies revealed the effects of small chemical compounds used as chemotherapeutic drugs on the unwinding activity of human DNA helicases [20,25]. Therefore, this study investigated the effects of two novel prenylated chalcones on the expression of BLM helicase in prostate cancer cells, as well as the biological activity of purified BLM helicase from the same cancer cells. Our results indicated that the two chalcones inhibited the growth of prostate cancer cell PC3 by inducing apoptosis and arresting the cell cycle. In addition, the two chalcones only inhibited the protein expression of BLM helicase, without inhibiting its transcriptional expression, and without significantly changing the expression of the homologous family members WRN and RECQL1. The DNA unwinding and ATPase activities of BLM helicase were inhibited by the two compounds, while a negligible effect was detected on the DNA-binding activity of the helicase. These results demonstrated that prenylated chalcones might be an effective intervention on the protein expression and function of BLM helicase in cancer cells to inhibit the growth of cancer cells. Thus, this approach might provide a new strategy for the development of new anti-cancer drugs targeting the genomic stability and DNA metabolic pathways.
The two prenylated chalcones used in this work only inhibited the protein expression of BLM helicase in prostate cancer cells, but no significant change in the gene expression of BLM helicase was observed, suggesting that the two compounds might effectively regulate the translation process of the DNA unwinding protein BLM helicase. However, no previous reports described which key functional genes are involved in regulating the translation of BLM helicase and the mechanism involved. The findings of this work provided a valuable guidance to further study the molecular mechanisms of BLM helicase regulating DNA replication, transcription, repair, and telomere stability. BLM helicase may be functionally regulated by multiple factors during translation. Some of the results of an ongoing study in our research group confirmed that BLM could specifically unwind the G4DNA structure with 3′ terminal ssDNA [26], and after the reduction in BLM mRNA expression using siRNA, proteomics detection revealed that a variety of genes involved in the translation process were differentially expressed in cells, such as promoting the transcriptional activity of the promoter factor ZEB1, promoter regulatory protein RFPL3, hTERT precursor mRNA selective splicing regulator BRM, and the interaction proteins PCDH10, MDM2, and FOXO3a. Other multiple inflammatory factors such as TNF-α, IL-1β, IL-6, and IL-8, as well as hTERT and their associated target proteins, were differentially expressed in cells after BLM deletion (unpublished results). These results might suggest that the translation process of BLM helicase is involved in the regulation of several key genes, and these regulatory effects directly or indirectly regulate a variety of DNA metabolic processes in the cells.
BLM helicase is one of the important motor proteins in the process of DNA unwinding, leading to the release of the chemical energy by catalytic hydrolysis of the nucleoside triphosphate to unwind the hydrogen bond between the two strands of the dsDNA, to transform it into ssDNA and provide a template for DNA replication [27]. All DNA helicases discovered so far have DNA-dependent ATPase activity. Therefore, BLM helicase is a multifunctional protein with DNA-binding activity, DNA unwinding activity, and ATPase activity [10]. This work mainly studied the effect of two novel prenylated chalcones on the three biological activities of BLM helicase, aiming to prove whether the two chalcones not only inhibit the protein expression of BLM helicase but also regulate its biological activities. The effects of two prenylated chalcones on DNA-binding activity were also studied, as well as the DNA-unwinding and ATPase activities of BLM helicase in vitro. The results showed that these two compounds could not significantly affect the DNA-binding activity but could significantly affect the DNA-unwinding and ATPase activities. The same significant differences were observed between the two drugs and two activities of the helicase: the inhibitory effect of WZH-43 was more than that of WZH-10 regarding the unwinding activity of the helicase of prostate cancer cells, while the inhibition level of the two drugs was not much different from the Ci value of the two compounds.
The effect of prenylated chalcones on the biological activity of other DNA helicases is also scarcely reported. General strategies in some specific and selective DNA helicase inhibitors are based on the solved structures and functional mechanism of the enzymes. Drugs targeting the biological activity of the helicase include one or more of the following mechanisms: (1) The combination of the inhibitors and minor groove of dsDNA substrates results in the arrest of the effect of helicase on dsDNA. (2) Some inhibitors can promote the formation of a hairpin structure of ssDNA to prevent the translocation of DNA helicase. (3) DNA helicase inhibitors are directly bound to ssDNA, inhibiting the binding of the helicase with dsDNA substrates. (4) DNA helicase inhibitors are directly bound to the helicase, affecting the structure and function of the helicase, further altering the bioactivity of the helicase [7,15,20,25]. Our results showed that BLM protein expression is upregulated in human prostate cancer PC3 cells, and WZH-43 and WZH-10 did not directly interact with the helicase, as revealed by the analysis of the change in the observed anisotropy. In addition, the hypothesis of the formation of the DNA hairpin structure was based on the complementarity of the paired base and the unmatched base in the center, and the DNA sequences in this study did not have the structure on the ssDNA in the 3′-terminal of the dsDNA. Therefore, our speculation was that the mechanism used by the two prenylated chalcones affecting the biological activity of the helicase was that the drug molecule bound with the minor groove of the dsDNA to prevent the translocation of the helicase on dsDNA. Nowadays, the basic research of DNA helicase as a potential anti-cancer effect has always been an international spotlight. A study on the molecular mechanism of RecQ helicase as an anti-cancer approach can be helpful to develop a new therapeutic target for different human diseases to provide a new strategy in the design of anti-cancer drugs.
4. Materials and Methods
4.1. Cells and Reagents
The human prostate cancer cell line PC3 was purchased from the Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from hicylone (Carlsbad, CA, USA). ATP was purchased from Sigma-Aldrich (St. Louis, MO, USA). DNA hybrid buffer was composed of 20 mM Tris-HCl, 100 mM NaCl, 5 mM KCl, and pH 7.9. The same buffer used for binding and unwinding was composed of 20 mM Tris-HCl (pH 7.9), 20 mM NaCl, 3 mM MgCl2, and 0.1 mM dithiothreitol (DTT). The pH values were measured using an ORION pH meter and a 0079 microelectrode (ORION, St. Louis, MO, USA) with a precision of ±0.01 pH unit at 25 °C. SYBR Premix Ex Taq™ IIKit (Perfect Real Time) was purchased from Takara Bio Inc. (Kyoto, Japan). Polyclonal rabbit anti-rat BLM, WRN, and RECQL1 were purchased from Abcam (Cambridge, UK; dilution of 1:1000) and GADPH from GenScript (Nanjing, China; dilution of 1:50,000).
4.2. DNA Substrates
The sequences and lengths of the DNA substrates are listed in Table 2, as described in previous study [28]. The PAGE-purified, fluorescein-unlabeled, and labeled synthetic oligonucleotides were purchased from Shanghai Biological Engineering Technology Service Cooperation (Shanghai, China). dsDNA was made in DNA hybrid buffer (20 mM Tris-HCl, 100 mM NaCl, and pH 7.4) by adding an equal amount of two types of ssDNA. The mixture was heated to 85 °C for 5 min and annealing was allowed by slow cooling to room temperature. The duplexes were used as the DNA substrates in fluorescence polarization assay.
Table 2.
Oligonucleotide sequences of the substrates.
The plasmid DNA (3 kilobase) used as the substrates of EMSA was purified using the Plasmid DNA Purification Kit. Linear PMD-18T dsDNA was produced by the incubation in water of the plasmid DNA with Hind III for 3 h at 37 °C. The concentration of dsDNA was determined using an ultraviolet (UV) spectrophotometer (GE Healthcare, St. Louis, MO, USA) equipped with a 1.0 cm cuvette.
4.3. Cell Culture
The prostate cancer cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 1% streptomycin (Sijiqing, Hangzhou, China), and incubated at 37 °C under a humidified atmosphere of 5% CO2.
4.4. Cell Proliferation Assay
Cell proliferation was assessed using the MTT assay. Cells were cultured in DMEM supplemented with 10% FBS and reached 1 × 104 cells per well of a 96-well plate. Then, the cells were treated with the compounds at different concentrations (up to 20 µmol/L) and incubated for 48 h. Next, the cells were treated with 5 mg/mL of MTT and incubated for 4 h. After removal of the MTT solution, 150 µL/well of dimethyl sulfoxide (DMSO) were added, and the formed crystals were dissolved gently 2 to 3 times by pipetting. The absorbance was measured at a wavelength of 570 nm. The growth inhibition rate was calculated according to a previous study [29].
4.5. Cell Apoptosis Assay
4.5.1. Hoechst 33,258 Staining
The Hoechst 33,258 staining method was used to identify cell apoptosis caused by the compounds [29]. Prostate cancer PC3 cells were treated with 1 µmol/L of each of the two active compounds for 48 h. Cells were harvested and washed with PBS, stained with 1 μL of Hoechst 33,258 (1 mg/mL in ddH2O), and incubated for 10 min. Nuclear morphological changes were observed by fluorescence microscopy.
4.5.2. Flow Cytometry Assay
Cell apoptosis was detected using annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining kits (BD Pharmingen, San Diego, CA, USA). Apoptotic cells were defined as FITC-positive. The cells were digested with trypsin, centrifuged at 1000 rpm for 5 min, washed with PBS twice, and suspended again. FITC and PI (Sigma, St. Louis, MO, USA) were added to the cell suspension, incubated in the dark for 15 min, and analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
4.6. Cell Cycle Analysis
The treated cells were treated with trypsin, washed with PBS, centrifuged at 200× g for 5 min, and fixed with 75% ethanol. Ethanol was removed by centrifugation at 4 °C for 1 h. The cell suspension was added with RNase, followed by a 37 °C water bath for 30 min and PI staining for 15 min. Flow cytometry analysis was performed using FACSArray (BD Biosciences, Haryana, India). The histogram of PI signal intensity was generated, and the percentage of G0, G1, S, and G2/M phase cells was determined.
4.7. Differential Expression of RecQ Helicases
4.7.1. mRNA Expression
The mRNA expression of the human RecQ helicases (BLM, WRN, and RECQL1) in the cells treated with agents was determined by reverse transcriptase-polymerase chain reaction (RT-PCR) and quantitative real-time PCR. Total RNA was extracted from the treated cancer cells using Trizol reagent and was transcribed at 37 °C for 1 h in a volume of 20 μL containing 5 × RT buffer, 10 mmol/L of dNTPs, 40 units of RNase inhibitor, 200 units of M-MLV reverse transcriptase, and 100 pmole of oligo-dT primer. Subsequently, 0.8 μL of the reaction mixture from each sample was amplified with 10 pmole of each oligonucleotide primer (Table 3) designed using Primer 5 (Premier, Vancouver, BC, Canada), 0.2 mmol/L of dNTPs, 1.5 mmol/L of MgCl2, and 1.25 units of Taq DNA polymerase, at a final volume of 25 μL. PCR was performed as follows: one cycle of 95 °C for 2 min, followed by 35 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 15 s, and extension at 72 °C for 15 s. The number of amplification cycles was optimized in preliminary experiments to ensure that the PCR did not reach a plateau. PCR products were subjected to a 2% (w/v) agarose gel electrophoresis, and analyzed by ChemiDoc XRS (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed using SYBR Premix Ex Taq (Takara) and the Real-Time PCR detection system (Bio-Rad). The PCR analysis for human RecQ helicases was performed as follows: initial denaturation at 95 °C for 10 min, amplification by denaturation at 95 °C for 10 s, annealing at 58 °C for 15 s, and extension at 72 °C for 15 s for 20 cycles. GAPDH was used as the endogenous control, and the relative gene expression was calculated by the 2−ΔΔCT method.
Table 3.
List of primer sequences.
4.7.2. BLM Protein Expression
BLM helicase protein expression in the cells was determined by Western blotting. PC3 cells were lysed in 50 mmol/L of Tris-HCl (pH 7.6) buffer containing 0.15 mol/L of NaCl, 1 mmol/L of EDTA, protease inhibitor cocktail, and 1% Triton X-100. Proteins in the cell lysates were separated by 10% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked using phosphate-buffered saline containing 0.05% (v/v) Tween 20 and 5% (w/w) skim milk. Then, the membrane was treated with the primary antibody and incubated overnight at 4 °C, followed by the incubation with HRP-conjugated secondary antibody at room temperature for 1 h. The detection was performed using Chemi DocTM XRS+ with the image system (Bio-Rad).
4.8. Biological Activity Assay
4.8.1. Recombinant BLM Helicase
Using pET15b expression plasmid, 6 cloned from PC3 cells were expressed in E. coli strain BL21 (DE3) × His labeled BLM helicase [28]. That is, the overexpressed helicase was purified by chelating Ni2+ Sepharose (GE Healthcare) in a rapid flow affinity chromatography column at 4 °C, and then purified by an FPLC size-exclusion chromatography Superdex 200 (Amersham, Buchs, Switzerland) [26]. The purity of the helicase was above 95% according to the blue-stained 10% SDS-PAGE analysis.
4.8.2. DNA-Binding Activity Assays
Two different approaches were used: (1) BLM and dsDNA were incubated together to form the BLM-dsDNA complex, and then the compounds were added. (2) BLM helicase was treated with the compounds, and then dsDNA was added. DNA binding activity was evaluated using a Beacon 2000 fluorescence polarizer (Pan Vera Corp., Madison, WI, USA) [26,28]. An amount of 2 nM fluorescein-labeled dsDNA (A1A2) or ssDNA (A2) was added to the reaction buffer (20 mmol/L of Tris-HCl, 20 mmol/L of NaCl, 3 mmol/L of MgCl2, 0.1 mM of dithiothreitol, pH 7.9). It was placed in a temperature-controlled tube at 25 °C. The measurement of anisotropy was rapid and continuous until it stabilized. BLM helicase was treated with WZH-43 or WZH-10 at different concentrations for 2 min, and then the miscible solution was added into a colorimetric tube with a total volume of 150 mL to quickly determine the change in the sample. According to Dou’s report, the dissociation constant (apparent Kd) of the binding in the presence of drugs is calculated by Equations (1) and (2) [30]:
DT is the total molarity of DNA, PT is the total molarity of helicase, A is the fluorescence anisotropy at a given concentration of helicase, Amax is the anisotropy at saturation, and Amin is the initial anisotropy.
In addition, the interaction between the compounds and the BLM-DNA complex was evaluated. Two nanomoles of fluorescein-labeled dsDNA or ssDNA were titrated by the helicase until saturation at 25 °C. Then, the complex was titrated by 1 mmol/L of WZH-43 or WZH-10, and the changes in the anisotropy were recorded every 8 s until it was stabilized.
4.8.3. DNA Unwinding Activity Assay
DNA unbinding activity was evaluated by referring to DNA binding assay [26]. The 2 nM fluorescein-labeled dsDNA was added to the reaction buffer in a 25 °C temperature-controlled test tube, and the anisotropy of the DNA substrate was recorded every 8 s until it stabilized. BLM uncyclase was treated with WZH-43 or WZH-10 at different concentrations and incubated for 2 min. Then, miscible liquid was added to the test tube to rapidly evaluate the anisotropy changes until stability. An amount of 1 mM ATP was rapidly added into a colorimetric dish with a total volume of 150 mL, and anisotropic changes were recorded every 8 s until it stabilized. According to Equation (3) [25], DNA untangling kinetic data can be obtained:
where At is the anisotropy at time t, kobs is the observed rate constant, and A1 is the anisotropy when the dsDNA substrate was completely bound to the helicase.
4.8.4. ATPase Activity Assay
According to the colorimetric method of inorganic phosphate produced by ATP enzymatic hydrolysis [31], the ATPase activity assay kit was used to measure the activity of ATPase. The helicase was diluted and reacted in different concentrations of WZH-43 or WZH-10 for 2 min. Atpase activity assay buffer (0.5 mol/L of Tris, pH 7.9, 10 mmol/L of ATP, 4 nmol of ssDNA (B1), 0.1 mol/L of MgCl2) was added to trigger the reaction. The reaction mixture, 200 μL in total, was collected at the following time points (2.5, 5, 7.5, 10, 15, 20, 25, 30 min) and mixed with 50 μL of Gold mixture to stop ATP hydrolysis. Each sample was added with 50 μL of stabilizer and reacted for 30 min. The OD650 of the sample was determined by a UV spectrophotometer. The enzyme activity (activity unit · mL−1 · min−1) was expressed as one unit, i.e., the amount of enzyme catalyzing 1 μM of substrate reaction per minute. According to the kit instructions, the ATPase activity of the undiluted enzyme was calculated using Equation (4):
A is the concentration of the free phosphorus ion (μmol/L) determined from the standard curve; B is the assay time (min); and C is the reciprocal of the enzyme dilution factor.
The steady-state kinetic parameters (Km and Kcat of ATP) of WZH-43 and WZH-10 were determined at concentrations of 0, 40, and 100 μmol/L using 10 nmol/L of helicase. The ATP concentration varied from 0.1 to 1 mmol/L. According to the Michaelis–Menten equation of the reaction, Km and Kcat values are obtained.
4.8.5. Agarose Gel Mobility Shift Assay
The binding and activity of helicase can be confirmed by agarose gel mobility determination [32]. When the total volume of DNA buffer is 25 µL at room temperature, the complex can be formed. Using WZH-43 or WZH-10 of different concentrations (0–600 µmol/L), 60 µmol/L of hindIII restricted 3-kilobase double-stranded DNA was mixed with 10 µmol/L of helicase and incubated for 30 min. An amount of 5 µL of loading buffer (40 mmol/L of triacetate, pH 7.5, 50% glycerol, and 0.25% (w/v) bromophenol blue) was added to each sample. The complexes were separated by electrophoresis with 0.8% agarose gel in 100 V Tae buffer (40 mm of Tris acetate, 1 mm of EDTA, pH 8.3) for 1 h. The bands of the complex were observed by gold field staining under ultraviolet light. The unwinding activity was determined in the same way, but the difference was that 10 mM ATP was added to the mixture of 60 µmol/L of double-stranded DNA and 10 µmol/L of helicase at the above concentration of WZH-43 or WZH-10.
4.9. Statistical Analysis
The results were evaluated by the F-test in SPSS 18.0 (SPSS 18.0, SPSS Inc., Chicago, IL, USA) statistical software, and the data were analyzed by the general linear model. The results are expressed as mean ± standard error (SEM) or standard deviation (SD). When p < 0.05, the results were considered statistically significant.
Author Contributions
Conceptualization, B.-F.S., X.-H.Z., B.-X.X. and H.L.; methodology, B.-F.S., J.H. and X.-H.Z.; validation, Y.-K.Q., J.Y. and S.C.; formal analysis, B.-F.S. and X.-H.Z.; data curation, Y.-K.Q., L.-L.L.; writing—original draft preparation, B.-F.S. and H.L.; writing—review and editing, B.-F.S., F.-J.S. and H.L.; article revision, H.L., L.-L.L.; supervision, H.L. and B.-X.X.; project administration, H.L.; funding acquisition, H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundations of China (81760573), the Science and Technology Support Plan of Guizhou Province (No. QKHZC [2022]194) and the main projects of the Natural Science Foundation of Guizhou province (No. QKHJC -ZK [2022]ZD031).
Data Availability Statement
Data sharing not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brosh, R.M., Jr.; Matson, S.W. History of DNA Helicases. Genes 2020, 11, 255. [Google Scholar] [CrossRef]
- Pham, X.H.; Tuteja, N. Potent inhibition of DNA unwinding and ATPase activities of pea DNA helicase 45 by DNA-binding agents. Biochem. Biophys. Res. Commun. 2002, 294, 334–339. [Google Scholar] [CrossRef]
- Bachrati, C.Z.; Hickson, I.D. RecQ helicases: Suppressors of tumorigenesis and premature aging. Biochem. J. 2003, 374 Pt 3, 577–606. [Google Scholar] [CrossRef]
- Amor-Guéret, M. Bloom syndrome, genomic instability and cancer: The SOS-like hypothesis. Cancer. Lett. 2006, 236, 1–12. [Google Scholar] [CrossRef]
- Wong, I.; Moore, K.J.; Bjornson, K.P.; Hsieh, J.; Lohman, T.M. ATPase activity of Escherichia coli Rep helicase is dramatically dependent on DNA ligation and protein oligomeric states. Biochemistry 1996, 35, 5726–5734. [Google Scholar] [CrossRef]
- Hickson, I.D.; Davies, S.L.; Li, J.L.; Levitt, N.C.; Mohaghegh, P.; North, P.S.; Wu, L. Role of the Bloom’s syndrome helicase in maintenance of genome stability. Biochem. Soc. Trans. 2001, 29 Pt 2, 201–204. [Google Scholar] [CrossRef]
- Sharma, S.; Doherty, K.M.; Brosh, R.M., Jr. DNA helicases as targets for anti-cancer drugs. Curr. Med. Chem. Anticancer Agents 2005, 5, 183–199. [Google Scholar] [CrossRef]
- German, J.; Roe, A.M.; Leppert, M.F.; Ellis, N.A. Bloom syndrome: An analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc. Natl. Acad. Sci. USA 1994, 91, 6669–6673. [Google Scholar] [CrossRef]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Janscak, P.; Garcia, P.L.; Hamburger, F.; Makuta, Y.; Shiraishi, K.; Imai, Y.; Ikeda, H.; Bickle, T.A. Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J. Mol. Biol. 2003, 330, 29–42. [Google Scholar] [CrossRef]
- Hickson, I.D. RecQ helicases: Caretakers of the genome. Nat. Rev. Cancer 2003, 3, 169–178. [Google Scholar] [CrossRef]
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452. [Google Scholar] [CrossRef]
- Van Maldergem, L.; Siitonen, H.A.; Jalkh, N.; Chouery, E.; De Roy, M.; Delague, V.; Muenke, M.; Jabs, E.W.; Cai, J.; Wang, L.L.; et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 2006, 43, 148–152. [Google Scholar] [CrossRef]
- Xi, X.G. Helicases as antiviral and anticancer drug targets. Curr. Med. Chem. 2007, 14, 883–915. [Google Scholar] [CrossRef]
- Ahmad, W.; Jantan, I.; Jasamai, M. Syed Nasir Abbas Bukhari. Review of Methods and Various Catalysts Used for Chalcone Synthesis. Mini-Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar] [CrossRef]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes. Nutr. 2011, 6, 125–147. [Google Scholar] [CrossRef]
- Wen, Z.; Zhang, Y.; Wang, X.; Zeng, X.; Hu, Z.; Liu, Y.; Xie, Y.; Liang, G.; Zhu, J.; Luo, H.; et al. Novel 3′,5′-diprenylated chalcones inhibited the proliferation of cancer cells in vitro by inducing cell apoptosis and arresting cell cycle phase. Eur. J. Med. Chem. 2017, 133, 227–239. [Google Scholar] [CrossRef]
- Peng, F.; Wang, G.; Li, X.; Cao, D.; Yang, Z.; Ma, L.; Ye, H.; Liang, X.; Ran, Y.; Chen, J.; et al. Rational design, synthesis, and pharmacological properties of pyranochalcone derivatives as potent anti-inflammatory agents. Eur. J. Med. Chem. 2012, 54, 272–280. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Karow, J.K.; White, E.J.; Shaw, N.D.; Hickson, I.D.; Bohr, V.A. Potent inhibition of werner and bloom helicases by DNA minor groove binding drugs. Nucleic Acids Res. 2000, 28, 2420–2430. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Zou, G.; Peng, S.; Liu, C.; Zhou, X. Detection and Application of 5-Formylcytosine and 5-Formyluracil in DNA. Acc. Chem. Res. 2019, 52, 1016–1024. [Google Scholar] [CrossRef]
- Ni, L.; Yamada, T.; Nakatani, K. Assembly of ruthenium complexes on double stranded DNA using mismatch binding ligands. Chem. Commun. 2020, 56, 5227–5230. [Google Scholar] [CrossRef]
- Moraca, F.; Amato, J.; Ortuso, F.; Artese, A.; Pagano, B.; Novellino, E.; Alcaro, S.; Parrinello, M.; Limongelli, V. Ligand binding to telomeric G-quadruplex DNA investigated by funnel-metadynamics simulations. Proc. Natl. Acad. Sci. USA 2017, 114, E2136–E2145. [Google Scholar] [CrossRef]
- Pfitzer, L.; Moser, C.; Gegenfurtner, F.; Arner, A.; Foerster, F.; Atzberger, C.; Zisis, T.; Kubisch-Dohmen, R.; Busse, J.; Smith, R.; et al. Targeting actin inhibits repair of doxorubicin-induced DNA damage: A therapeutic approach for combination therapy. Cell Death Dis. 2019, 10, 302. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, A.H.; Chen, L.; Xi, X.G. Inhibition of DNA helicase, ATPase and DNA-binding activities of E. coli RecQ helicase by chemotherapeutic agents. J. Biochem. 2008, 143, 773–779. [Google Scholar] [CrossRef]
- Zhu, X.; Sun, B.; Luo, M.; Yu, J.; Zhang, Y.; Xu, H.Q.; Luo, H. Bloom helicase explicitly unwinds 3’-tailed G4DNA structure in prostate cancer cells. Int. J. Biol. Macromol. 2021, 180, 578–589. [Google Scholar] [CrossRef]
- Xue, C.; Daley, J.M.; Xue, X.; Steinfeld, J.; Kwon, Y.; Sung, P.; Greene, E.C. Single-molecule visualization of human BLM helicase as it acts upon double- and single-stranded DNA substrates. Nucleic Acids Res. 2019, 47, 11225–11237. [Google Scholar] [CrossRef]
- Luo, H.; Xu, H.Q.; Chen, X.; Ding, M.; Yang, Q.X.; Li, K. Potent in vitro interference of fleroxacin in DNA-binding, unwinding and ATPase activities of Bloom helicase. Biomed. Environ. Sci. 2013, 26, 231–242. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, B.; Yu, J.; Huang, L.; Zeng, X.; Shen, X.; Ren, C.; Ben-David, Y.; Luo, H. Fli-1 Activation through Targeted Promoter Activity Regulation Using a 3′,5′-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2216. [Google Scholar] [CrossRef]
- Dou, S.X.; Wang, P.Y.; Xu, H.Q.; Xi, X.G. The DNA binding properties of the Escherichia coli RecQ helicase. J. Biol. Chem. 2004, 279, 6354–6363. [Google Scholar] [CrossRef]
- George, J.W.; Ghate, S.; Matson, S.W.; Besterman, J.M. Inhibition of DNA helicase II unwinding and ATPase activities by DNA-interacting ligands. Kinetics and specificity. J. Biol. Chem. 1992, 267, 10683–10689. [Google Scholar] [CrossRef]
- Xu, H.Q.; Zhang, A.H.; Auclair, C.; Xi, X.G. Simultaneously monitoring DNA binding and helicase-catalyzed DNA unwinding by fluorescence polarization. Nucleic Acids Res. 2003, 31, e70. [Google Scholar] [CrossRef] [PubMed][Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).