Abstract
Until the year 2000, gold compounds were considered catalytically inert. Subsequently, it was found that they are able to promote the nucleophilic attack on unsaturated substrates by forming an Au–π-system. The main limitation in the use of these catalytic systems is the ease with which they decompose, which is avoided by stabilization with an ancillary ligand. N-heterocyclic carbenes (NHCs), having interesting σ-donor capacities, are able to stabilize the gold complexes (Au (I/III) NHC), favoring the exploration of their catalytic activity. This review reports the state of the art (years 2007–2022) in the nucleophilic addition of amines (hydroamination) and water (hydration) to the terminal and internal alkynes catalyzed by N-heterocyclic carbene gold (I/III) complexes. These reactions are particularly interesting both because they are environmentally sustainable and because they lead to the production of important intermediates in the chemical and pharmaceutical industry. In fact, they have an atom economy of 100%, and lead to the formation of imines and enamines, as well as the formation of ketones and enols, all important scaffolds in the synthesis of bioactive molecules, drugs, heterocycles, polymers, and bulk and fine chemicals.
1. Introduction
Copper, silver, and gold belong to Group 11 (Ib) of the periodic table. The metals of this group have the highest thermal and electrical conductivities. They are the most ductile and malleable and are also commonly called coinage metals. Their use goes back to prehistoric times. The oldest artifacts made with copper date to the Neolithic; silver was used to purify drinking water, and to create jewels and ornaments. In addition, gold was used mainly to make jewelry and to mint coins. For a long time, silver and gold were considered not useful for catalytic purposes; in fact, their use in catalysis is recent.
Nowadays, many complexes of these metals, stabilized by many kinds of ligands, are used as catalysts [,,,,,]. A class of ligands particularly studied and widely used in catalysis for its excellent chemical properties is constituted by N-heterocyclic carbenes. Their use dates back thirty years, when Arduengo, in 1991, was able to obtain and unequivocally characterize the first stable N-heterocyclic carbene (NHC): the 1,3-diadamantylimidazol-2-ylidene [].
Since then, other structurally modified NHC ligands have been synthesized, both cyclic and acyclic, by changing the nature of substituents on nitrogen atoms, or by inserting a heteroatom on the ring core or different substituents on the backbone (Figure 1).
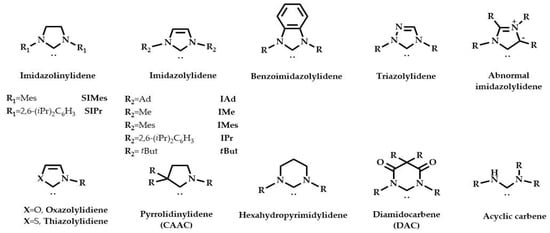
Figure 1.
Structure of the most used NHC ligands.
The most commonly used are imidazolinylidenes, five-membered ring imidazolylidenes, and triazolylidenes. NHC ligands have lone-pair electrons on the central carbon atom (sp2) that can coordinate to a metal. They are strong σ-donating ligands, and the π* molecular orbital (MO) of the NHCs can accept electron density from the filled d orbitals of the transition metal via a classical d → π* back-donation [,]. Frequently, NHCs have replaced phosphine ligands in the design and development of efficient catalysts.
It is worth noting that the chemical–physical properties of metal complexes (M-NHCs), e.g., thermal stability and tolerance against moisture and air, can be modulated by modifying the steric and electronic properties of substituents on the backbone and/or on the nitrogen atoms at the NHC ligand. NHCs are easily obtained upon deprotonation of an imidazolium salt. Their synthesis can be carried out following two methodologies: (i) a multicomponent reaction building up the heterocycle with the appropriate substituents in a one-pot reaction or (ii) nucleophilic substitution at the imidazole heterocycle.
The application of NHCs as ligands able to stabilize metal complexes active in catalysis includes all elements of the d-block from group 3 to group 12 []. The two main applications of NHCs–metal catalysts are the ruthenium-catalyzed olefin metathesis and the palladium-catalyzed cross-coupling reactions.
In this review, we describe the development of catalysis carried out using NHC–gold complexes in the hydroamination and hydration of alkynes, pointing out the advantages of using these complexes as catalysts.
2. Hydroamination of Alkynes
The reaction of hydroamination of alkynes is the addition of ammonia, primary or secondary amines to alkynes (Scheme 1).
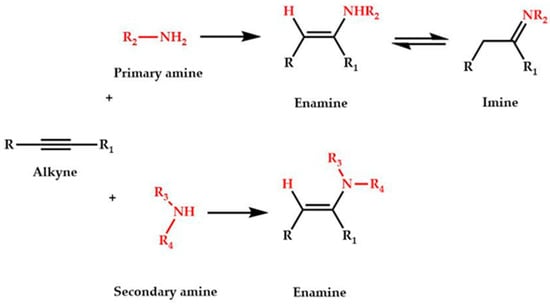
Scheme 1.
Hydroamination of alkynes.
The enamines and imines produced are useful key molecules for numerous nitrogen-containing compounds of biological, industrial, and agrochemical interest []. In general, in the hydroamination of alkynes, using as catalysts metal complexes stabilized by NHC ligands and having halides as counterions, a silver salt is added as co-catalyst, in order to remove the halides coordinated to the gold centers and generate the catalytic active species for substrate coordination.
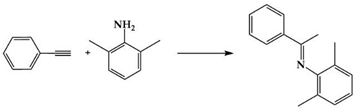
In 2007, Che et al. showed that (IPr)AuCl (Scheme 2) was active in the hydroamination of alkynes with primary arylamines [].
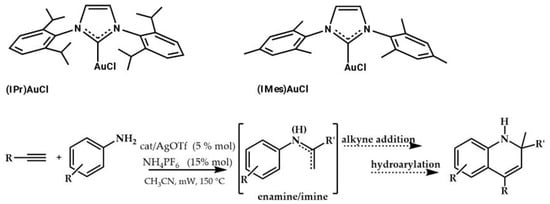
Scheme 2.
NHC–gold(I) catalyzed reactions between primary arylamines and alkynes.
This reaction, in the presence of 5 mol% of NHC–gold catalyst at 80–100 °C, gave 1,2-dihydroquinoline derivatives in up to 80% yields after 12–24 h. Experimental evidence demonstrated that enamine is an intermediate of the reaction. (Ipr)AuCl was found to be more efficient than (IMes)AuCl and as active as {(o-biphenyl)di-tert-butylphosphine}AuCl.
Bertrand et al., in 2008, synthesized and tested in catalysis of hydroamination of alkynes the complex 1 reported in Scheme 3 []. The authors obtained pyrroles from diynes for the addition of ammonia. In particular, double hydroamination occurred in the case of diynes.
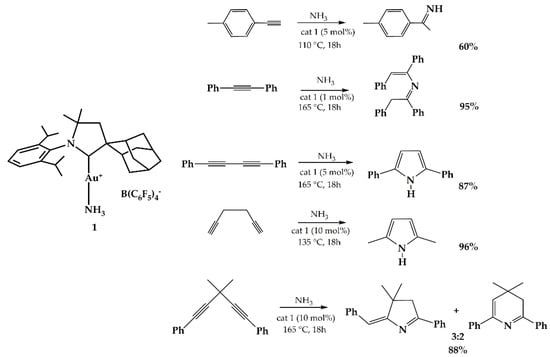
Scheme 3.
Cationic gold-catalyzed hydroamination of alkynes with NH3.
The study of the reaction mechanism revealed that it proceeds through an intermediate in which both the NH3 and the alkyne are bound to the acid Lewis gold complex.
In 2009, Bertrand and coworkers reported the catalytic activity of cationic η2-toluene gold(I) complex 2 (Figure 2) in the intermolecular hydroamination of alkynes with primary or secondary alkyl amines [,].
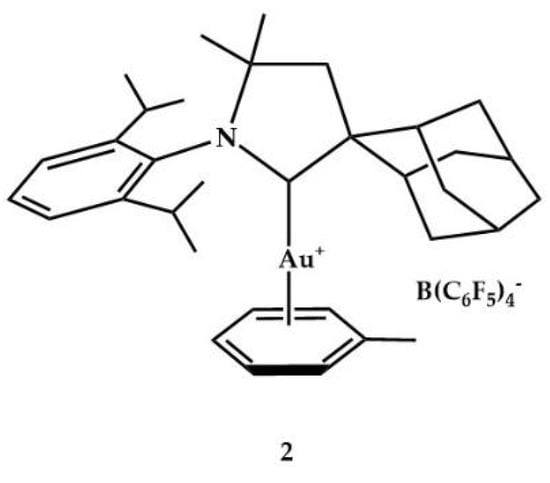
Figure 2.
Cationic gold(I) complex 2.
With the primary amines (2,4,6-trimethylaniline and tert-butylamine) and alkynes, using 5 mol% of 2 as catalyst, clean reactions occurred (81–94% yield) at temperatures between 40 °C and 140 °C and a reaction time between 10 and 24 h. The complex 2 was active (46–98% yield) also in the hydroaminations with secondary amines (e.g., diphenylamine, N-methylaniline, 1,2,3,4-tetrahydroisoquinoline, diethylamine); however, with phenylacetylene and diethylamine the yield was lower (23%). With methylphenylacetylene, the expected mixture of Markovnikov and anti-Markovnikov products was obtained. Instead, it was surprisingly observed that with diethylacetylene, the expected hydroamination adduct was only the minor product (21–43%) of the reaction, being the major product (79–57%) an isomer in which the unsaturation has been shifted.
In 2009, Bertrand et al. synthesized and characterized the complex 3 (Figure 3). The precatalyst was tested in hydroamination of internal alkynes with Et2NH []. The cyclic(alkyl)(amino)carbene (CAAC) ligand provided steric protection to the metal center; in addition, the replacement of one of the nitrogens by a carbon center makes CAACs slightly more nucleophilic, but considerably more electrophilic than NHCs.
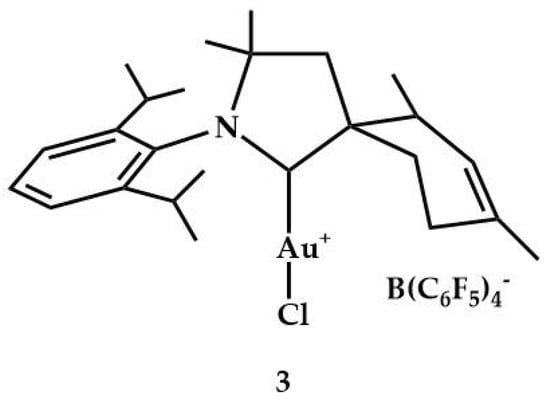
Figure 3.
Cationic gold(I) complex 3.
The precatalyst 3, in the presence of 1 equiv. of KB(C6F5)4, was active for the addition of diethylamine to three internal alkynes: 1,2-diphenylethyne (92% yield), 3-hexyne (93% yield), and 1-phenyl-1-propyne (92% yield). The authors found that 3 was as efficient as 2 (89–98% yield).
Ghosh et al., in 2010, synthesized a series of gold(I) and silver(I) precatalysts stabilized by N-heterocyclic carbenes based on 1,2,4-triazole derivatives (see Figure 4) [].
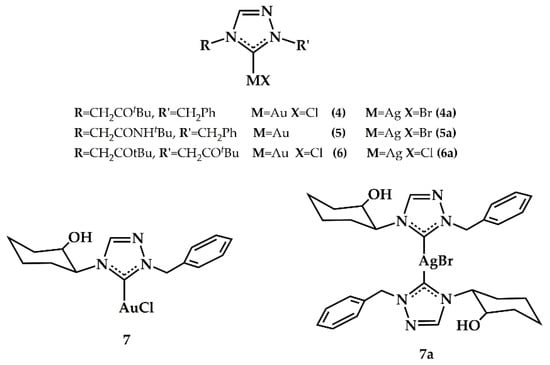
Figure 4.
Gold and silver precatalysts of 1,2,4-triazole based NHCs synthesized from Ghosh et al. [].
The silver(I) 4a–7a complexes were synthesized from the respective 1,2,4-triazolium halide salts by treatment with silver oxide in 43–64% yield, while the gold(I) complexes 4–7 were obtained by transmetalation reaction of the silver analogues 4a–7a with (Sme2)AuCl in 60–76% yield. The gold(I) 4–7 complexes displayed from good (42%) to high (93%) activity for the intermolecular hydroamination reaction (90 °C for 12 h, in air) of o/p-substituted aryl amines (2-methylaniline, 2,6-dimethylaniline, mesitylaniline, 2,6-diethylaniline, and 2,6-di-i-propylaniline) with terminal alkynes in CH3CN (Scheme 4).
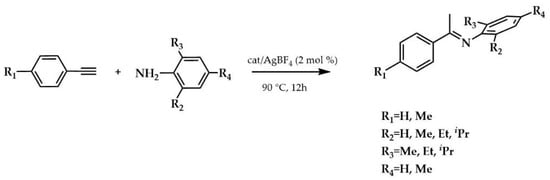
Scheme 4.
Hydroamination reaction of o/p-substituted aryl amines with terminal alkynes.
The yields (%) were determined by GC using diethylene glycol di-n-butyl ether as an internal standard. Complex 4 was more efficient in the reaction between 2,6-dimethylaniline or mesitylaniline and 4-ethynyltoluene with a yield of 93%, while complex 6 was more active (92% yield) in the reaction between 2,6-diethylaniline and phenylacetylene. The control experiments performed with (SMe2)AuCl/AgBF4 and also with the corresponding blank experiment carried out with only AgBF4 exhibited a significant increase of up to 85% under analogous conditions in the case of the gold 4–7 precatalysts.
The silver(I) 4a–7a complexes showed lower activity (from 0% to 29% yield) than the analogous gold(I) 4–7 complexes did. The reaction of hydroamination showed, for all the alkyne substrates, a Markovnikov-type N–H addition. The results obtained in this work underline the importance of gold complexes with respect to silver analogs in catalysis.
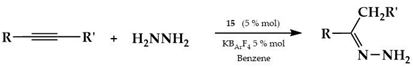
In 2011, Bertrand and coworkers reported that the two complexes in Figure 5 catalyzed the addition of hydrazine (H2NNH2) to a variety of non-activated alkynes and allenes to afford a diverse array of acyclic and cyclic nitrogen-containing compounds [].
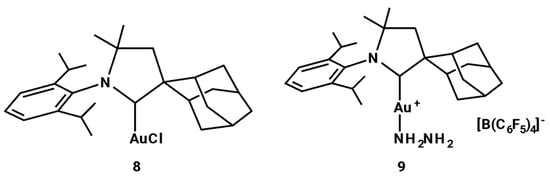
Figure 5.
Complexes studied from Bertrand.
The solid-state structure of 9 was determined by X-ray diffraction, which confirmed that only one amino group is coordinated to the cationic gold(I) to give a η1 Werner-like complex. The catalytic hydroamination of p-methoxyphenylacetylene with hydrazine, both complexes 8 and 9 to 5.0 mol% in the presence of KB(C6F5)4, led to the formation of hydrazone, which is the tautomer of the enamine resulting from a Markovnikov-type addition (yield 95%, at 100 °C for 30 min). By lowering the amount of complex 8 to 0.1 mol%, 95% conversion was observed when the reaction mixture was heated at 150 °C for 6 h. The reaction of hydrazine with terminal alkynes, i.e., n-butyne, benzyl acetylene, and 6midazole6ylene, using complex 8 (5 mol%) as catalyst, produced after 3 h at 100 °C the hydrazones resulting from a Markovnikov-type addition in high yields: 89%, 95%, and 92%, respectively. Both complexes 8 and 9 (5 mol%) gave high yields even in the reaction of 1,2-diphenylethyne with H2NNH2, although a longer reaction time (14 h) was required. Both complexes were also active in the catalysis of diynes with H2NNH2 for the synthesis of heterocycles.
In 2012, Lavoie et al. achieved a highly stereoselective Markovnikov-type addition of phenylacetylene to 2,6-dimethylaniline using gold(I) complexes of 1-[1-(2,6-dimethylphenylimino)alkyl]-3-(mesityl)imidazole-2-ylidene (C^ImineR), 1,3-dimesitylimidazol-2-ylidene (Imes) and the corresponding thione derivatives (S^ImineR and ImesS) complexes (Figure 6) [].
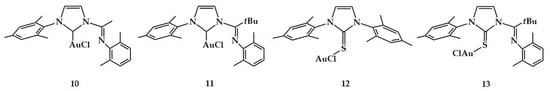
Figure 6.
Complexes of Lavoie et al. [].
The activity in catalysis of the gold complexes in the hydroamination of phenylacetylene with 2,6-dimethylaniline was evaluated and compared to control catalyst systems such as Au(Sme2)Cl (14), benchmark (Imes)AuCl, and AgBF4 (Table 1).

Table 1.
Catalyst screening for the hydroamination of phenylacetylene with 2,6-dimethylaniline.
The highest conversions were obtained with 11 (entry 4) and (Imes)AuCl complexes. All catalysts tested exclusively produced the product Markovnikov obtained for addition of the aniline with subsequent tautomerization of the enamine.
The different activity of complexes 11 and 10 found in catalysis suggested that the steric effects of the ligand play a relevant role. The 10, 11, and 13 gold complexes were used to study the steric and electronic effects of the substituents of the anilines (2,4,6-trimethylaniline, 2-tert-butylaniline, 2,6-diisopropylaniline, aniline, para-methylaniline, para-anisidine, para-(trifluoromethyl)aniline, para-nitroaniline) on the catalysis. The order of reactivity found was to be 11 > 13 > 10 using either sterically or electronically demanding anilines.
Ghosh et al., in 2012, studied the coordination pathways between two representative substrates, MeC≡CH and PhNH2, in the hydroamination reaction of gold(I) complex, namely, [1,3-dimethylimidazol-2-ylidene]gold chloride, using the density functional theory (DFT) []. The study found that the lowest activation barrier is for amine coordination compared to alkyne coordination. Furthermore, it was calculated that the hydroaminated enamine PhNHMeC=CH2 is more stable in its tautomeric imine form, PhN=Cme2.
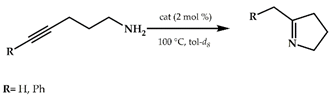
Bertrand et al., in 2013, synthesized the (carbene)gold chloride 15 (Scheme 5), which was isolated in 57% yield, and fully characterized also by a single-crystal X-ray diffraction study []. This catalyst promoted the hydroamination of 1-hexyne with hydrazine at room temperature.
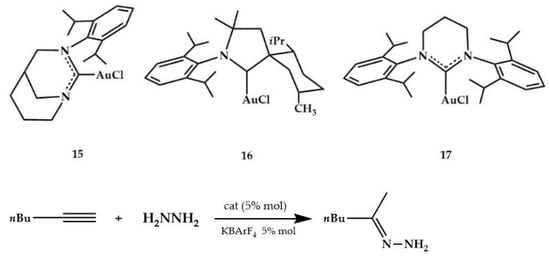
Scheme 5.
Hydroamination of 1-hexyne with hydrazine.
The reactions were carried out in different solvents, i.e., chloroform, dichloromethane, tetrahydrofuran, and benzene (entry 4). The latter gave the best results: 91% conversion after 3 h (Table 2).
Table 2.
Catalytic hydroamination of 1-hexyne with hydrazine.
Its catalytic activity was compared with the three benchmark metal complexes (16, 17, and 8) shown in the Scheme 5 and Figure 5, respectively. Under the same experimental conditions, only a 22% conversion was observed after 4 h with the complex 8 (entry 5), whereas complex 16 gave 10% conversion after 16 h (entry 6) and complex 17 proved to be even less effective (entry 7). In Table 3, the efficiency of the catalyst 15 using alkyl substituted terminal alkynes is reported. The catalytic activity decreased when the steric hindrance of the substrates increased.
Table 3.
Catalytic hydroamination of alkynes with hydrazine.
Peris et al. prepared a rigid, planar tris (NHC) based on a triphenylene core coordinated to palladium and gold [].
The obtained trimetallic gold 18 and 19 complexes (Figure 7) were tested in the hydroamination of phenylacetylene with several substituted anilines (see reaction of Table 1 and results in Table 4). Their catalytic activity was compared with 20 complex (Figure 7).
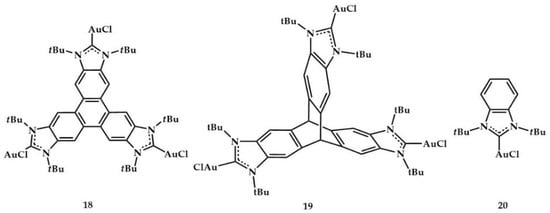
Figure 7.
Trimetallic gold 18 and 19 complexes and monometallic 20 complex.
Table 4.
Hydroamination of phenylacetylene with substituted anilines.
All the reactions catalyzed by the trimetallic complex 18 provided excellent yields (>90%), while the catalytic activity of the monometallic complex 20 was higher than that shown by the trimetallic complex 19, except for in the reactions carried out with o-methylaniline, for which the activities were similar.
Bertrand and coworkers, in 2016, reported the catalytic activity of gold complexes bearing anti-Bredt di(amino)carbenes ligands 21a–d (Figure 8) in the hydroamination of phenylacetylene with phenyl hydrazine or aniline (for reaction, see Table 1) [].
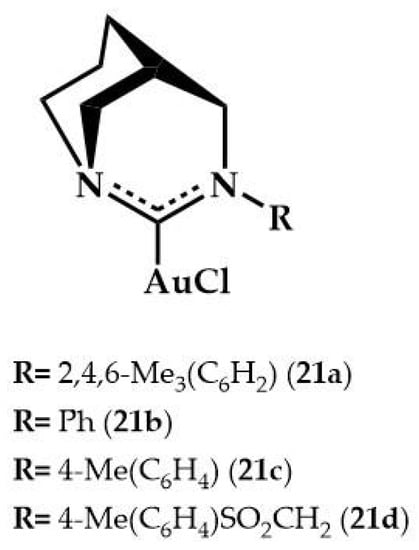
Figure 8.
Gold complexes with anti-Bredt ligands.
These ligands are significantly more π-accepting than classical N-heterocyclic carbenes are, while retaining their strong σ-donation to the metal []. The 21a complex (5 mol%) and potassium tetrakis(pentafluorophenyl)borate (KB(C6F5)4) were active in the reaction of phenylhydrazine with some arylacetylenes and 1-ethynylcyclohexene to afford quantitatively (84–99%) hydrazones at room temperature, after 24 h. The authors studied the hydroamination of phenylacetylene with aniline of the 21a–d complexes. Data in Table 5 show that all complexes were found to be very active at room temperature using KB(C6F5)4 as co-catalytic salt. The reaction rate proved to be insensitive to the N-substituent of the ligand.
Table 5.
Hydroamination of phenylacetylene with aniline.
The catalytic system 21a/KB(C6F5)4 was also tested in the reaction of aryl alkynes and 1-ethynylcyclohexene with various anilines bearing electron-withdrawing or electron-donating substituents. In all cases, almost complete conversion was observed after 24 h at room temperature.
In 2017, Peris et al. coordinated to gold a D3h-symmetry tris-N-heterocyclic carbene ligand that contains an electron-poor hexaazatriphenylene (HAT) core []. The tris–Au(I) 22 complex (Figure 9) was tested in the hydroamination of phenylacetylene with substituted anilines (for reaction, see Table 1) and in the three-component Strecker reaction.
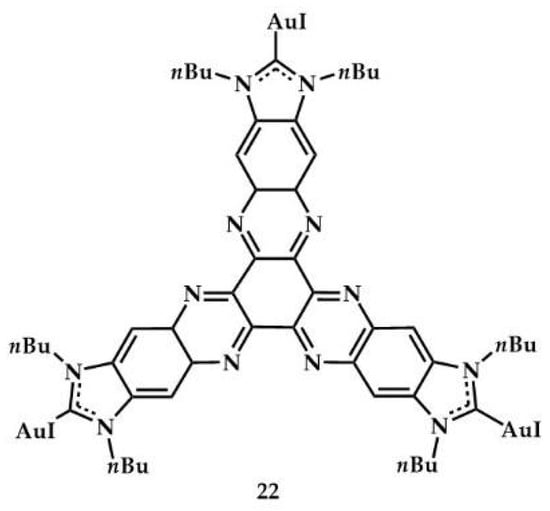
Figure 9.
Gold complex with hexaazatriphenylene core.
The catalytic activity in the hydroamination reaction was compared with that of the benchmark complex 20 (Table 6). The latter can be considered as one of the gold-containing branches of 22, but without the electron-poor pyrazine ring. With both gold complexes, AgBF4 was added as a chloride scavenger in order to activate the catalysts.
Table 6.
Hydroamination of phenylacetylene with substituted anilines.
In 2017, Peris and coworkers also synthesized three gold(I) complexes bearing NHC ligands with fused polycyclic aromatic hydrocarbons (Figure 10) [].
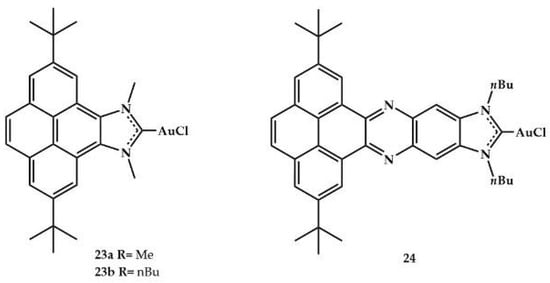
Figure 10.
Gold(I) complexes with fused polycyclic aromatic hydrocarbons as ligands.
The 23a–b and 24 complexes were tested as catalysts for the hydroamination of phenylacetylene (Table 7) with substituted anilines (for reaction see Table 1).
Table 7.
Hydroamination of phenylacetylene with substituted anilines.
They showed moderate-to-good conversion to products (21–90%). It was observed that the activities of the catalysts were enhanced by the addition of a little amount of pyrene, this was attributed to the formation of π−π stacking aggregates between the molecules of pyrene and the catalyst, which in turn partially prevents self-assembly of the catalyst.
In 2017, Messerle et al. reported the synthesis of an Au(III) complex containing a hemilabile bis-pyrazole carbene ligand, namely, (1,3-bis((1H-pyrazol-3-yl)methyl)-2,3-dihydro-1H-imidazole) [].
The 25 Au(III) complex was tested in dihydroalkoxylation, spirocyclization of alkynyl diols, and intra/intermolecular hydroamination reactions in the presence of sodium tetrakis(pentafluorophenyl)borate (NaB(C6F5)4). Its catalytic activity was compared with the analogous 26 gold(I) complexes (Figure 11) in the intramolecular hydroamination reactions (Table 8) and for hydroamination of phenylacetylene with aniline (Table 9) (for reaction see Table 1).
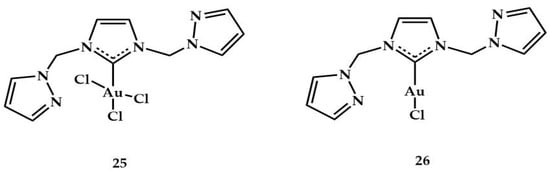
Figure 11.
Au(III) and Au(I) complexes synthesized by Messerle et al. [].
Table 8.
Intramolecular hydroamination reaction.
Table 9.
Gold-catalyzed intermolecular hydroamination reactions of phenylacetylene with Ar-NH2.
The catalytic system showed a good performance. The gold-catalyzed intermolecular hydroamination reactions between phenylacetylene and some substituted anilines were studied.
The intermolecular hydroamination between phenylacetylene and aniline gave a low conversion for both catalytic systems, 26% and 30% for 25 and 26, respectively. The complex 25 also promoted the reaction between phenylacetylene and mesitylamine or (trifluoromethyl)aniline, giving 61 and 49% conversion to product, respectively. Thus, the catalysts 25 and 26 proved more active for intra- than for intermolecular hydroaminations.
Peris, in 2018, reported the synthesis and the X-ray analysis of digold(I) complex with a pyrenebis (imidazolylidene) ligand (Figure 12) and its catalytic activity in the presence of AgBF4 in hydroamination of phenylacetylene (for reaction see Table 1) []. The authors studied the effects on the catalytic activity of 27 in the presence of a coronene.
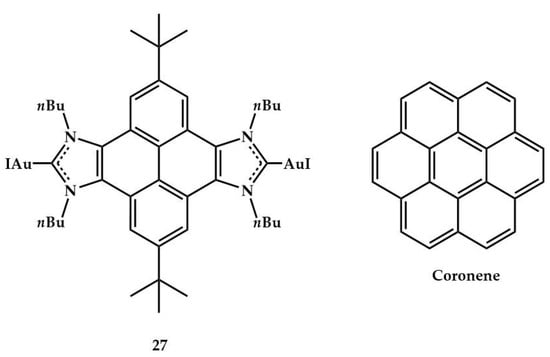
Figure 12.
Digold(I) complex 27 and coronene.
The results are given in Table 10.
Table 10.
Hydroamination of phenylacetylene with substituted anilines.
In 2018, Baron et al. investigated, in alkyne hydroamination and Suzuki cross-coupling reactions, the catalytic performance of dinuclear gold(I) complexes with bridging di(N-heterocyclic carbene) (diNHC) ligands of the general formula Au2Br2L1−9 (Figure 13) [].
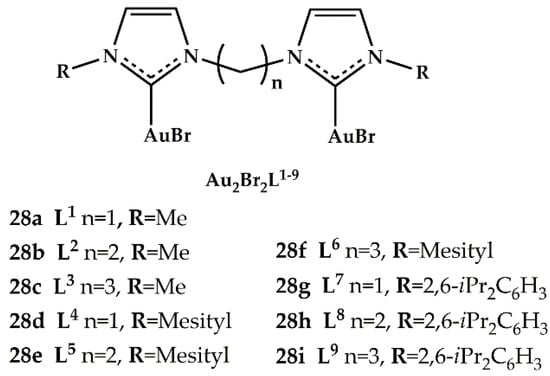
Figure 13.
Gold(I) diNHC complexes Au2Br2L1−9 of Baron et al. [].
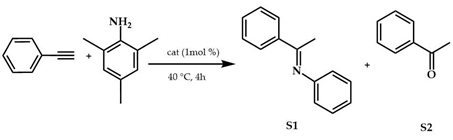
Single-crystal X-ray diffraction analyses have been made for Au2Br2L4, Au2Br2L5, and Au2Br2L6. The authors used 28i complex to optimize the reaction conditions of hydroamination of phenylacetylene with 2,4,6-trimethylaniline. The best reaction conditions found were neat conditions, 4 h and 2 mol% AgSbF6 as co-catalyst. The results are reported in Table 11. The catalytic activity of synthesized Au2Br2L1−9 complexes was compared with that of the benchmark (IPr)AuCl complex. Better performances were recorded with the N-methyl-substituted complexes (28a–c), which, together with benchmark (IPr)AuCl, reached complete alkyne conversions and very high hydroamination yields within the 4 h reaction time.
Table 11.
Hydroamination reaction of phenylacetylene with 2,4,6-trimethylaniline.
The reaction produced also a small amount of acetophenone. Its formation probably proceeded through two steps: (i) Au-catalyzed hydroamination of phenylacetylene and (ii) hydrolysis of the hydroamination product by water brought into the system with the silver salt co-catalyst, which is quite hygroscopic. It is worth noting that no hydroamination product was obtained with internal alkynes, i.e., phenylpropyne and diphenylacetylene and N-methylaniline (secondary arylamines), or with cyclohexylamine and morpholine (primary/secondary alkylamines), except with complexes bearing the N-2,6-diisopropylphenyl substituent.
In 2019, the group of Nolan reported the hydroamination reactions with the catalysts in Figure 14 [].
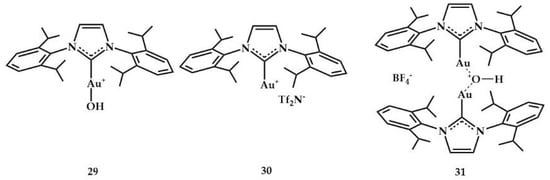
Figure 14.
Complexes tested by Nolan et al. in the hydroamination of internal alkynes.
The 29–31 complexes (0.5 mol%) were used to establish the best catalytic conditions in hydroamination of phenylacetylene with 1H-benzo[d][1,2,3]triazole; two methods were used: (i) a sand bath at 100 °C for 72 h and (ii) using microwave heating at 150 °C for 0.75 h. Complex 31 provided the best yields with both methods (66% and 96%, respectively, in neat conditions and in the presence of 5 mol% of Bu4NOTf as co-catalyst).
Subsequently, the authors used complex 31 with some alkynes (i.e., 1,2-diphenylethyne, dimethyl but-2-ynedioate, ethyl 3-phenylpropiolate, 3-phenylpropiolonitrile, prop-1-yn-1-ylbenzene, oct-4-yne) and azole nucleophiles (i.e., 1H-benzo[d][1,2,3]triazole, 4,5-dihydro-1H-1,2,3-triazole, 1-methylimidazolidin-2-one, 1H-pyrazole, 2H-indazole, 1H-1,2,4-triazole, 1H-imidazole) using both methods previously tested (Scheme 6).
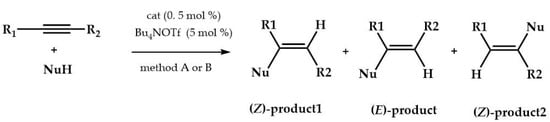
Scheme 6.
Hydroamination reactions.
The products obtained in almost all catalysis reactions were (Z)-enamines, as established on observing the NOE profiles after the full assignment of 1H/13C/15N chemical shifts.
Concerning the reaction mechanism using complex 31 as catalyst, the authors suggested that the Lewis acid [Au(IPr)][BF4] could coordinate to the alkyne and the Brønsted base [Au(IPr)(OH)] could deprotonate the azole nucleophile to generate the corresponding gold–azole complex and release a water molecule (Scheme 7).
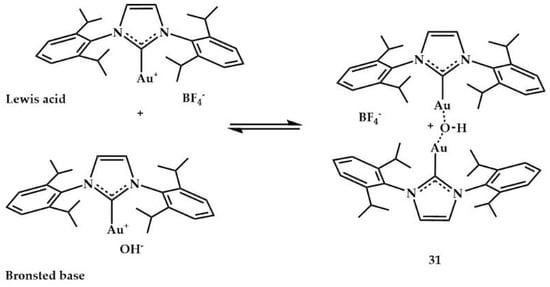
Scheme 7.
Species hypothesized for reaction mechanism.
Recently, Huynh et al. synthesized and characterized the first gold(III) complexes bearing expanded-ring NHCs (erNHCs) by oxidative addition of bromine to the respective gold(I) erNHC precursors (Figure 15) [].
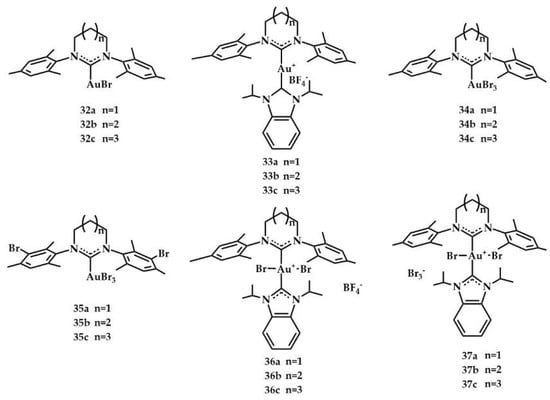
Figure 15.
Complexes synthesized by Huynh et al. [].
All Au erNHC complexes 32a–c–37a–c have been tested as precatalysts for the hydroamination of phenylacetylene with 2,4,6-trimethylaniline (for reaction, see Table 1). The simplest precatalyst, 32a, was chosen for the exploration of the best reaction conditions: phenylacetylene (0.3 mmol, 1.5 equiv.), 2,4,6-trimethylaniline (0.2 mmol, 1 equiv.), AgSbF6 (0.004 mmol, 2 mol%), and Au NHC (0.004 mmol, 2 mol%) in CH3CN (∼1 mL) heated at 90 °C in a screw cap tube under air for 14 h. Complex 32c was found to be the most active with 98% conversion (Table 12).
Table 12.
Hydroamination reaction.
Notably, all cationic heterobis (carbene) complexes 36a–c–37a–c were inactive regardless of the gold oxidation state, the nature of the counteranion, and the presence of the halide scavenger. The authors hypothesized that inactivity was due to two strong Au−NHCs bonds and the increased steric hindrance. Further, the complex 32c was tested in the intermolecular hydroamination of different alkynes (phenylacetylene, 4-methoxyphenylacetylene, 4-methylphenylacetylene, 4-fluorophenylacetylene, 4-(dimethylamino)-phenylacetylene, 1-hexyne) and amines (2,4,6-trimethylaniline, aniline, 2,6-diisopropylaniline, 4-methoxyphenylamine, 1-naphthylamine, butylamine). In all catalytic reactions, the complex 32c proved to perform well, except for in the hydroamination of phenylacetylene with butylamine and 1-hexyne with 2,4,6-trimethylaniline.
In 2022, Olmos and coworkers reported the synthesis of α-chloromethylketimines by hydroamination of aromatic and aliphatic 1-chloroalkynes with aromatic amines with (IPr)AuCl (Scheme 8) [].
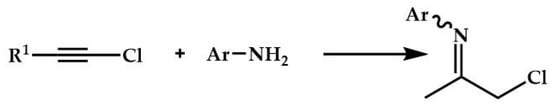
Scheme 8.
Hydroamination of aromatic and aliphatic 1-chloroalkynes with aromatic amine.
The authors optimized the reaction conditions using 1 mol% 1,3-bis(2,6-diisopropylphenyl-imidazol-2-ylidene)gold(I) chloride (IPr)AuCl activated by 1.5 mol% sodium tetrakis [3,5-bis(trifluoromethyl)phenyl]-borate (NaBArF) as catalyst in toluene (0.42 M) at 120 °C for 0.5 h. These selected conditions were used for the reaction of 1-chloroethynylbenzene with a series of primary amines. Efficiency was demonstrated by the excellent result obtained with the strongly hindered 2,6-diisopropylaniline (yield 98%) and with 4-aminophenol (yield 97%). The authors studied the reactivity of different 1-chloroalkynes (i.e., 2-(chloroethynyl)-1,3,5-trimethylbenzene, 1-(chloroethynyl)-4-methoxybenzene, 1-(chloroethynyl)-4-(trifluoromethyl)benzene, 1-chlorodec-1-yne and 2-(chloroethynyl)pyridine) with aniline. Excellent to very good yields were obtained in all cases, except for one, in which the presence of a pyridine inhibited the reaction.
Peris et al., in 2022, synthesized and characterized a gold complex with a naphthalene-di-imide-functionalized NHC ligand (Figure 16) [].
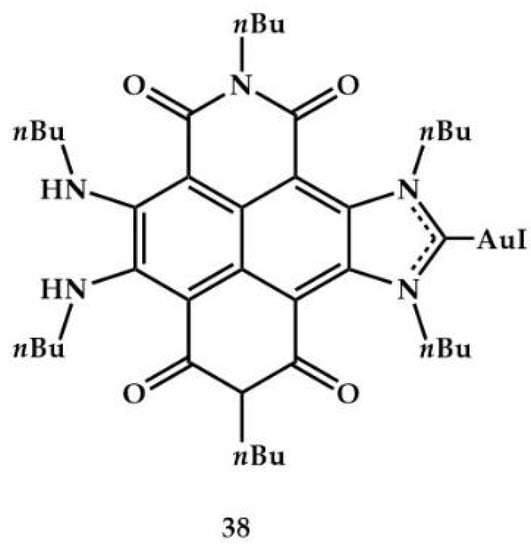
Figure 16.
Complex 38 of Peris et al. [].
The catalytic activity of complex 38 was tested in the hydroamination of phenylacetylene with four different amines (Table 13).
Table 13.
The activity of complex in the hydroamination of phenylacetylene with arylamine.
After 8 h of reaction, using a catalyst loading of 1 mol% produced yields of the imine products in the range of 75−82%. For the case of the reaction between phenylacetylene and aniline, a reduction of the catalyst loading to 0.5 mol% produced a significant decrease in the yield down to 49% (entry 5), while doubling the amount to 2 mol% increased the product yield up to 90% in just 5 h (entry 6).
3. Hydration of Alkynes
A synthetic method for generating carbonyl compounds is the hydration reaction of alkynes. This reaction is considered an example of both atom economy and an environmentally friendly synthetic method [,]. It is based on the addition of water to terminal alkynes giving either methyl ketone (Markovnikov addition) or an aldehyde (anti-Markovnikov addition; Scheme 9a), in addition, nonsymmetrical internal alkynes may give two regioisomeric ketones (Scheme 9b).
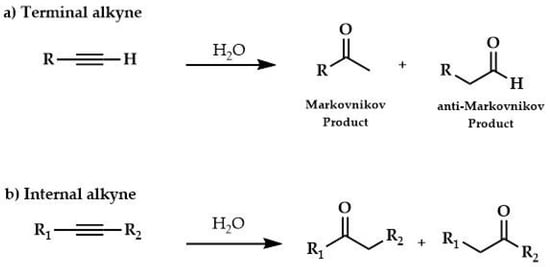
Scheme 9.
Hydration of (a) terminal and (b) internal alkynes.
The use of a catalyst is required, because it promotes the attack of water to the alkyne unit. Among the catalytic systems used, gold-based compounds occupy a prominent place due to their high versatility. In fact, they can act as σ- and π-Lewis acids, which has allowed the activation of a wide diversity of functional groups, including unsaturated carbon systems (i.e., alkenes, allenes, and alkynes).
The hydration of alkynes can be promoted by L–Au–X compounds (L = an ancillary ligand, and X− = a counterion). In this review, we focused on N-heterocyclic carbenes (NHCs) [,] as neutral ligands, because they have already proven to be particularly effective in stabilizing gold complexes used in homogeneous catalysis [,], and they have resulted in the development of highly efficient systems at very low catalyst loadings [,,] under mild conditions.
In 2008, Nolan and coworkers reported an in-depth investigation of a catalytic system for the hydration of alkynes []. They concentrated on the study of the hydration of diphenylacetylene with (NHC)AuCl complexes (NHC = IPr or IMes or ItBu), examining different parameters, such as temperature, concentration, solvent, water/alkyne ratio, and the nature of silver(I) salt, used as co-catalyst, and finally, of the NHC ligand. The reaction conditions were optimized (Scheme 10).
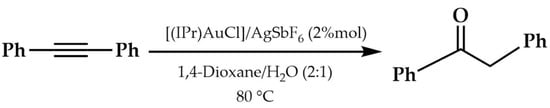
Scheme 10.
Optimization of the reaction of hydration.
Among the NHC ligands used, IPr was the most reactive, especially in equimolar quantities of (IPr)AuCl and AgSbF6, with a conversion of 97%. Other silver salts (AgBF4 and AgOAc) are less active compared to AgSbF6. 1,4-dioxane, in a 2:1 ratio with water, turned out to be the best solvent. Moreover, (IPr)AuCl at 0.1 mol%, at 120 °C, gave a ketone yield of 77%.
Using methanol as solvent, instead of 1,4-dioxane, Nolan obtained very interesting results with terminal alkynes. In fact, while with diphenylacetylene the conversion into ketone was much slower in methanol than it was with 1,4-dioxane, with terminal alkynes, such as phenylacetylene, this relation was the opposite. Probably, these results were due to different mechanisms, and methanol could play an active role in this reaction. To verify this hypothesis, the reactivity of several terminal alkynes was investigated, and a faster conversion was obtained when the reactions were performed in methanol instead of in 1,4-dioxane.
In 2016, Silbestri and coworkers developed new NHC–gold complexes 39–43 (Figure 17) soluble in water and able to catalyze the hydration of alkynes [,].
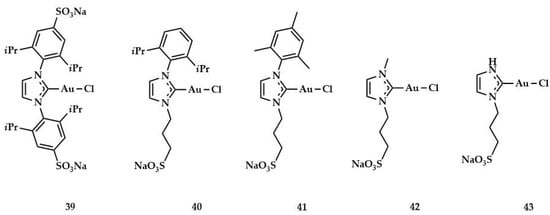
Figure 17.
Structure of gold(I) sulfonated NHC complexes of Silvestri et al. [,].
The synthesis of gold complexes was obtained by reaction between imidazolium compounds and silver oxide [,], then the Au complexes were obtained via transmetallation, using [AuCl(tht)] (tht = tetrahydrothiophene) as gold precursor [] (Scheme 11).
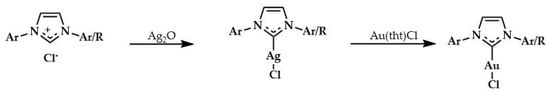
Scheme 11.
Synthesis of Gold(I) sulfonated NHC complexes.
The study started with the hydration of phenylacetylene using complex 39 as catalyst and pure water as solvent. The results are reported in Table 14. When carrying out the reaction at 80 °C with 1 mol% of 40, acetophenone in a quantitative yield was obtained after 5 h (entry 1). The effects of some parameters such as the use of silver salts [], the catalyst load, and the reaction temperature were studied. No improvement was obtained by adding 1 mol% of AgSCN (entry 2), but the time of reaction was reduced when using 2 mol of AgSCN or when increasing the amount of catalyst (2 mol%, entry 3 and 4) or increasing the temperature (entry 6 and 7). Instead, the addition of 1 mol% of AgSCN and AgSbF6, at 100 °C, caused a decrease in the catalytic activity (entry 8 and 9), while the time of reaction was reduced to 30 min when the reaction was carried out with 2 mol% of catalyst, without silver salt, at 100 °C in pure water.
Table 14.
Hydration of phenylacetylene promoted by complex 39.
An analogous study was performed by carrying out the reaction in a mixture of methanol and water (1:1) [] and analyzing the effects of temperature, different loadings of catalyst, and the addition of silver salts on reaction time. The decrease in temperature (from 100 to 80 °C) caused an increment in the reaction time (from 30 to 45 min, entry 11 and 12). Instead, when using 2 mol% of catalyst, the reaction time was reduced to 15 min (entry 13), while the introduction of 1 mol% of AgSbF6 and 1 mol% 39 decreased the reaction time to 8 min (entry 16). Considering that the reactions in water were slower than those in water:methanol 1:1, Silbestri and coworkers carried out the reaction in methanol with stoichiometric amounts of water; however, the time was increased, and the amount of ketone was reduced (entry 17).To evaluate the effects of the catalyst, a series of experiments were carried out both in water and in water:methanol, at 100 °C, reducing the amounts of catalyst (from 1 to 0.05 mol%), in the absence of silver salts. The results are shown in Table 15.
Table 15.
Optimization of catalyst loading.
In all cases, the catalyst was found to be active, and when halving the catalyst loading, it was necessary to double the reaction time to obtain the same results. As already noted from the previous results, the reaction carried out in water (from 1.17 to 13 h, entries 1–5) was slower than that in water:methanol (from 30 min to 7 h, entries 6–10). Another important aspect is that the catalyst was active at 0.5 mol% and it was recycled and reused five times without losing its activity.
Other studies have investigated the steric effect of NHC ligands on catalyst activities, focusing on the hydration of phenylacetylene with less bulky NHC–gold complexes (40–43) using 1 mol%. All reactions were carried out in pure water and water:methanol, in both the presence and absence of silver salts, with opportune temperature. The results are shown in Table 16. No complexes exhibited activity in water. Furthermore, complexes 40 and 42 did not react, despite the longer time reaction (24 h) and increasing the loads of Au (5 mol%) or adding different silver salts (entries 1–5, 18–22). Complexes 41 and 42 were unstable in water; in fact, they totally decomposed in 3 h and 5 h, respectively, as they did when using a lower temperature (30 °C) or when loading 10 mol% of silver salt (entries 10 and 11). On the other hand, all reactions in water:methanol gave positive results. In fact, when using only 1 mol% of 40, the reaction was quantitative in 13 h (entry 6), while adding 1 mol% of silver salts (AgOTs and AgSbF6) caused the reaction time to decrease (6 h and 7.5 h, respectively) (entries 7–8); this effect was probably due to counterion with gold(I) catalysts [,]. Complex 41 showed a good yield after 48 h, and the addition of silver salts [AgSCN, AgOTs, and AgSbF6] did not determine beneficial effects on conversion (35%, 80%, and 56%, respectively) in the same reaction time (entries 13–15). Regarding complexes 42 and 43, they gave a moderate conversion in 60 h. The addition of silver salts did not produce any benefit for the reaction with complex 42, while with complex 43 it produced a positive effect (entries 23–25, 29–31).
Table 16.
Hydration of alkynes catalyzed by 40–43.
Other information about the influence of steric hindrance around a metal center was given by kinetic experiments, by means of which Silbestri and coworkers confirmed the importance of this parameter both in the stability and in the catalytic activity of gold (I) complexes. The bulkier NHC complexes were the most effective catalysts in these reactions. Other authors reported similar results in different reactions carried out in conventional organic solvents [,,,]. The lower activity of complexes 40 and 43 was due to sulfonated moiety, which may coordinate the metal or interfere with the reaction center. These limitations could be eliminated at an acidic pH; for example, the complex 43 was more active at pH = 2 than at a pH between 6.4 and 7.2. This achievement supports the hypothesis that there was a coordination between the metal and the sulfonate group. The influence of methanol is no less important. In fact, the use of methanol as a co-solvent could lead to a more active precatalyst being obtained. In support of this hypothesis, two other complexes with methanol coordinated were synthesized (44 and 45, Scheme 12), according to the procedures described [,].
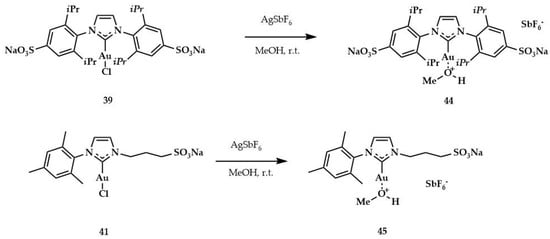
Scheme 12.
Synthesis of the catalysts 44 and 45 by corresponding gold sulphonated NHC complexes.
Comparing the catalytic activity of 39 and 44 and of 41 and 45, either in water or water:methanol, it was observed that 44 was more active than 39 was in water (85% vs. 42%), but they showed similar activity in water:methanol (90% vs. 88%). The same results were obtained with 45 vs. 41. Considering the excellent activity of complex 39, Silbestri and coworkers studied the reactivity of selected terminal and internal alkynes in water at 100 °C, without silver salts (see Scheme 9). The results are shown in Table 17. The hydration of terminal alkynes gave the corresponding Markovnikov ketone in quantitative yields and in an acceptable reaction time (entries 1–6). On the other hand, the reactions with internal enyne gave a mixture of ketone (entry 7). Finally, the reaction conducted with prop-2-yn-1-ol and but-3-yn-1-ol, in the same condition (100 °C), did not give the expected ketone, but the corresponding hydroxy ketone was obtained when carrying out the reaction with AgSbF6 at 60 °C.
Table 17.
Hydration of terminal and internal alkynes promoted by complex 39 in water.
Zuccaccia et al., in 2016, developed an efficient methodology for the hydration of alkynes by NHC gold complexes using solvent-, silver-, and acid-free conditions, focusing their attention on the pivotal role of the counterion []. They analyzed the activity of (IPr)Au-X (X− = BArF−, BF4−, SbF6−, ClO4−, OTf−, NTf2−, OTs−, TFA−, BArF = tetrakis(3,5-bis(trifluoromethyl)-phenyl) borate) complexes in neat condition and without using silver salts in the hydration of alkynes at room/mild temperature, with suitable ionic additives. The synthesis of complexes was carried out according to literature procedures [], and they studied the hydration of internal and terminal alkynes. Some experiments carried out on 3-hexyne (see Scheme 9b, R1 = R2 = Et, Table 18) showed that the complexes in which the counterions were BArF−, BF4−, SbF6−, ClO4−, OTs−, and TFA− did not promote the reaction after 24 h; probably it was due to the poor basic properties and low coordinating ability of BArF−, BF4−, SbF6−, and ClO4−- or to strong coordinating ability and basic properties of OTs− and TFA− (entries 1–3 and 6–8). Instead, OTf− and NTf2− gave a quantitative conversion (entries 4–5).
Table 18.
Hydration of 3-hexyne catalyzed by (IPr)Au-X complexes.
Moreover, the TOF value ((TOF (h−1) = (mol of product)/((mol of catalyst) (time elapsed)); Table 18)] obtained by NHC–Au–OTf and NHC–Au–NTf2 was about 64 h−1 higher than those reported in the literature under neat conditions and at room temperature []. The theoretical mechanisms proposed by Zuccaccia and coworkers for an alkyne hydration reaction catalyzed by [NHC-Au]+ catalyst in the presence of the OTf− emphasizes the importance of the counterion role (Scheme 13).
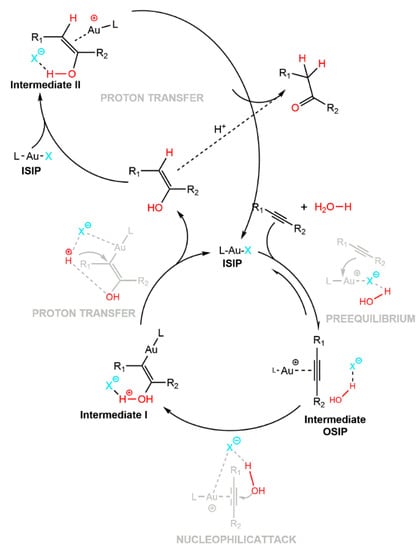
Scheme 13.
Mechanism of hydration of Au(I)NHC. Reprinted with permission from Ref. []. Copyright 2016 American Chemical Society.
They showed that, during nucleophilic attack, the anion plays two useful roles: (i) it locks the water molecule in the correct position for the outer-sphere addition; (ii) it increases the nucleophilicity of the attacking water through the HOH···OTf− hydrogen bond, which in turn polarizes the oxygen.
Zuccaccia and coworkers, afterwards, paid attention to the importance of the ligand and understanding its effect during different steps in the hydration of alkynes [,]. For this purpose, they synthetized several L–Au–X complexes where L = 1,3-bis(2,6-di-isopropylphenyl)-imidazol-2-ylidene {IPrNHC}, tris(3,5-bis(trifluoromethyl)phenyl)phosphine {PArF}, bis(imino)acenaphtene-supported-1,3-bis(2,6-diisopropylphenyl) dihydroimidazol-2-ylidene {BIAN}, 1,3-bis-(2,6-di-isopropyl-phenyl)dihydroimidazol-2-ylidene {NHCCH2}, bis(tert-butylamino)methylidene {NAC}, 2-di-tertbutyl(o-diphenyl)phosphine {JohnPhos}, tricyclohexylphosphine {PCy3}, triphenylphosphine {PPh3}, tris(2,4-di-tertbutylphenyl) phosphite {POR3} (Figure 18) and X− = Cl−, OTf−, OTs−. The structure of the ligand modulates the acidic character of the metal center and influences the stability of intermediates [,,,].
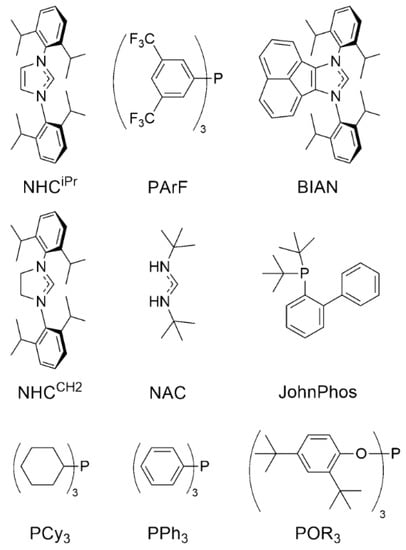
Figure 18.
Structure of ligands used by Zuccaccia et al. Reprinted with permission from Ref. [] Copyright 2016 American Chemical Society.
Furthermore, they examined the effect of ionic additive compounds (for example, NBu4Otf), silver salts, and the temperature on the hydration of alkynes. Their principal aim was the development of an efficient methodology for the hydration of unreactive diphenylacetylene in neat condition and without silver salt and acid using only 0.1 mol% of catalyst. The complexes were tested as catalysts in the hydration of 3-hexyne and diphenylacetylene (Table 19 and Table 20) in solvent- and acid-free conditions.
Table 19.
L–Au–X (0.1 mol%) catalyzed hydration of 3-hexyne at 30 °C in the presence of NBu4OTf.
Table 20.
NHCIPr–Au–OTf-catalyzed hydration of diphenylacetylene.
When utilizing L−Au−Cl/AgOTf systems, where L is PCy3, PArF, POR3, PPh3, and NAC (Table 19, entries 5 and 7−10), no formation of product was observed within 24 h, because the decomposition of catalyst in neat conditions occurred []. In addition, the complexes having NHC ligands showed a decomposition process promoted by silver salt in neat and acid-free conditions. Under silver-free conditions, the complexes with phosphine-based ligands were also decomposed (entries 16–19), except for JohnPhos (conversion of 74% in 5 h, entry 15). The complexes with NHC ligands and -OTf (IPrNHC–Au–OTF) were more active compared to IPrNHC–Au–OTs (entry 1 vs. 11), because the coordinating ability of OTs− was higher than that of OTf−.
The same results were obtained in the hydration of diphenylacetylene; in fact, the IPrNHC-Au-OTf was more effective compared to the analogous complex with OTs- as counterion (Table 20, entries 3–7). In this work, it was possible to reduce, for the first time, the catalyst loading to 0.01 mol% in solvent-, silver-, and acid-free conditions, with high TON (3400) and TOF (435 h−1) thanks to the synergy effect of ligand and counterion []. The neat conditions are not applicable when the reagents are solid or viscous.
For this reason, in a recent work, Zuccaccia et al. focused on the use of green solvents in the hydration of alkyne []. In the literature, many organic processes, promoted by transition metals, were carried out in bio-based solvents [] (i.e., water [], glycerol [], D-limonene and p-cymene [,], γ-valerolactone (GVL) [,], lactic acid and its derivates []) instead of in volatile organic solvents. They found that the reaction of hydration of alkynes promoted by (IPr)Au–OTf proceeded very well in some of the alternative solvents, such as ethyl lactate, glycerol, propylene carbonate, D-limonene, γ-valerolactone, and p-cymene. In addition, they noted that the activity of the catalyst, in terms of TOF, seemed to be inversely related to the polarity of the solvent. In fact, when increasing the dielectric constant, the equilibrium between the ion pair and free ions was shifted toward free ions, and this reduced the useful effect of OTf- during the nucleophilic attack of methanol (water). The presence of proton acceptor/donor functionalities in some solvents (e.g., cyclohexanone and glycerol) increased the rate of the reaction, even if their dielectric constant was medium–high. At the same time, solvents having coordinated functional groups (e.g., D-limonene C=C, propionitrile C≡N, and DMSO) decreased the rate of reaction due to their coordination to the metal center. More recently, the Ujaque group [] studied the reaction mechanism for the hydration to some alkynes (terminal and internal), alkenes, and allenes catalyzed by (IPr)Au+ complex (see Scheme 2) by means of DFT calculations. In Scheme 14, the reactions studied are summarized [].
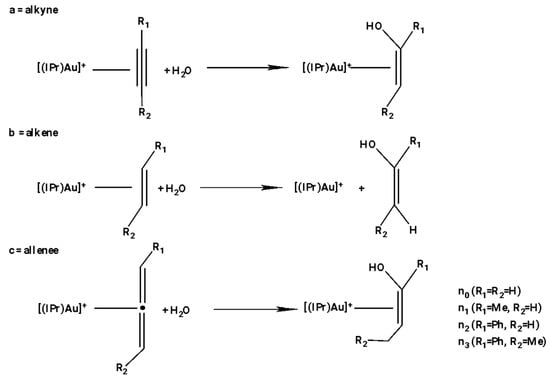
Scheme 14.
IPrAu+-catalyzed hydration of alkynes, alkenes, and allenes.
The catalytic cycle for the three substrates can be described by three steps: (i) π coordination of reactant to the Au(I) complex, (ii) nucleophilic addition of water, and (iii) protodeauration (this is the step with subtle differences among the reactants). DFT calculations showed that alkynes and allenes undergo the reaction of hydration without any problems, while the alkenes show a scarce reactivity. This different reactivity is not due to the nucleophilic attack of water, although it goes with different energies (lower energies with alkynes and allenes, higher energy for alkenes), but is attributed to the high-energy intermediate formed in the protodeauration step. However, it could be argued that the terminal functional groups are always more reactive compared to the internal functional groups as they are often difficult to access; unsubstituted alkyne (ethyne, a0) and alkene (ethene, b0) have higher barriers to water addition compared to the species with terminal groups, this effect being due to the favorable presence of the Me and Ph substituents on the Markovnikov addition []. Regarding regioselectivity, addition on the most substituted carbon (Markovnikov addition) was always favorable for all unsaturated functional groups evaluated. Regarding alkenes, the situation becomes more complicated, because nucleophilic addition was significantly higher in terms of the Gibbs energy barrier than it was in the case of alkynes and allenes. The high energy of this species is related to the strength of the C–O bond in the presence or absence of a π-bond in the organic fragment and not to the Au(I)–CC system interaction.
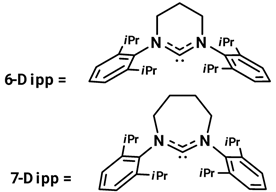
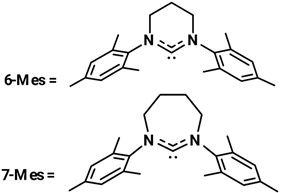
Asachenko and coworkers studied the effect of ring sizes and substituents in NHC ligands in some (NHC)Au(I) complexes in the reaction of hydration of alkynes, especially focusing their study on regioselectivity of asymmetrical diarylacetylene and alkylarylacetylenes []. The results are reported in Table 21.
Table 21.
Results of Au(I) NHC precatalysts with different ring size.
In both hydration reactions, the 7-DippAuCl demonstrated the best regioselectivity, even if its efficiency was not outstanding. The results show that Au(I) complexes having Dipp (2,6-diisopropylphenyl) as NHC ligands are more active than those with Mes (2,4,6-trimethylphenyl) substituted. However, in this kind of reaction, the regioselectivity was almost independent of the NHC ligand; in fact, all complexes demonstrated low selectivity towards the Markovnikov product. The complex 7-DippAuCl was used to investigate the effect of different substituents in the aromatic rings (i.e., 1-methyl-4-(phenylethynyl)benzene, 1-methyl-2-(phenylethynyl)benzene, 1,3,5-trimethyl-2-(phenylethynyl)benzene, 1-methoxy-4-(phenylethynyl)benzene, 1-(phenylethynyl)-3-(trifluoromethyl)benzene) of the alkynes on the regioselectivity of the hydration reaction. They obtained always Markovnikov products. However, in the study of the reaction of alkylarylacetylenes, under the same condition, the regioselectivity was anti-Markovnikov and highly controlled by the ligand. These results are more important because it is very difficult to obtain anti-Markovnikov products by other methods, so this reveals the synthetic importance of this research []. More probably this unusual selectivity is due to the difference in steric repulsion between aryls of arylalkylacetylenes and the NHC ligand in the transition state of the water addition step.
4. Conclusions
Ever since Arduengo isolated the first N-heterocyclic carbene, the use of these ligands in the organometallic field has received a lot of attention from the scientific community. NHC complexes of gold, unlike other transition metals, have received interest only more recently. Many works have shown that NHC gold (I/III) complexes are powerful catalysts for intermolecular and intramolecular hydroamination and hydration of terminal and internal alkynes, through π activation of the CC multiple bond.
This review highlighted the catalytic activity of the NHC complexes of gold (I/III) in the formation of C–N and C–O bonds, by adding amines and water to the alkynes. The high focus on this type of reaction is attributable to the atom economy and sustainable conditions. Despite all these studies, there is still work to be done, focusing, for example, on the following: (1) achieving low-loadings catalytic NHC systems; (2) enhancing the catalytic activity of gold catalysts turned on by ligands and counteranions. Furthermore, given the interesting results obtained by (NHC)Au(III) complexes in hydroamination reactions, it is also possible to imagine that new higher-performing gold (III) complexes will be designed and tested in these and other types of reactions.
The aim of this review was to summarize the most interesting results obtained by the Au(I/III) NHC complexes in the addition of amines and water to alkynes, and secondly, to encourage the synthesis of new N-heterocyclic carbene ligands for the development of increasingly active gold catalysts.
Author Contributions
Conceptualization and supervision: P.L. and A.M.; writing—original draft preparation: A.M. and R.T.; review, editing, and schemes: A.M., M.S., R.T., S.R. and P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research required no external funding.
Data Availability Statement
This study did not report any data.
Acknowledgments
The authors are grateful to the Department of Science of Basilicata University and to the Department of Chemistry and Biology of Salerno University, Italy, for facilities of this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Homogeneous Gold Catalysis Beyond Assumptions and Proposals-Characterized Intermediates. Angew. Chem. Int. Ed. 2010, 49, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Naodovic, M.; Yamamoto, H. Asymmetric Silver-Catalyzed Reactions. Chem. Rev. 2008, 108, 3132–3148. [Google Scholar] [CrossRef]
- Weibel, J.-M.; Blanc, A.; Pale, P. Ag-Mediated Reactions: Coupling and Heterocyclization Reactions. Chem. Rev. 2008, 108, 3149–3173. [Google Scholar] [CrossRef]
- Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Silver-Mediated Synthesis of Heterocycles. Chem. Rev. 2008, 108, 3174–3198. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Frenking, G.; Solà, M.; Vyboishchikov, S.F. Chemical Bonding in Transition Metal Carbene Complexes. J. Organomet. Chem. 2005, 690, 6178–6204. [Google Scholar] [CrossRef]
- Falivene, L.; Cavallo, L. Theoretical NMR Spectroscopy of N-Heterocyclic Carbenes and Their Metal Complexes. Coord. Chem. Rev. 2017, 344, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Ding, P.; Huang, J.-S.; Che, C.-M. Synthesis of Substituted 1,2-Dihydroquinolines and Quinolines from Aromatic Amines and Alkynes by Gold(I)-Catalyzed Tandem Hydroamination−Hydroarylation under Microwave-Assisted Conditions. Org. Lett. 2007, 9, 2645–2648. [Google Scholar] [CrossRef]
- Lavallo, V.; Frey, G.D.; Donnadieu, B.; Soleilhavoup, M.; Bertrand, G. Homogeneous Catalytic Hydroamination of Alkynes and Allenes with Ammonia. Angew. Chem. Int. Ed. 2008, 47, 5224–5228. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Frey, G.D.; Kousar, S.; Bertrand, G. A Cationic Gold(I) Complex as a General Catalyst for the Intermolecular Hydroamination of Alkynes: Application to the One-Pot Synthesis of Allenes from Two Alkynes and a Sacrificial Amine. Chem.-Eur. J. 2009, 15, 3056–3060. [Google Scholar] [CrossRef]
- Lavallo, V.; Frey, G.D.; Kousar, S.; Donnadieu, B.; Bertrand, G. Allene Formation by Gold Catalyzed Cross-Coupling of Masked Carbenes and Vinylidenes. Proc. Natl. Acad. Sci. USA 2007, 104, 13569–13573. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Frey, G.D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Synthesis of a Simplified Version of Stable Bulky and Rigid Cyclic (Alkyl)(Amino)Carbenes, and Catalytic Activity of the Ensuing Gold(I) Complex in the Three-Component Preparation of 1,2-Dihydroquinoline Derivatives. J. Am. Chem. Soc. 2009, 131, 8690–8696. [Google Scholar] [CrossRef] [Green Version]
- Dash, C.; Shaikh, M.M.; Butcher, R.J.; Ghosh, P. Highly Convenient Regioselective Intermolecular Hydroamination of Alkynes Yielding Ketimines Catalyzed by Gold(I) Complexes of 1,2,4-Triazole Based N-Heterocyclic Carbenes. Inorg. Chem. 2010, 49, 4972–4983. [Google Scholar] [CrossRef]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef]
- Alvarado, E.; Badaj, A.C.; Larocque, T.G.; Lavoie, G.G. N-Heterocyclic Carbenes and Imidazole-2-Thiones as Ligands for the Gold(I)-Catalysed Hydroamination of Phenylacetylene. Chem.-Eur. J. 2012, 18, 12112–12121. [Google Scholar] [CrossRef]
- Katari, M.; Rao, M.N.; Rajaraman, G.; Ghosh, P. Computational Insight into a Gold(I) N-Heterocyclic Carbene Mediated Alkyne Hydroamination Reaction. Inorg. Chem. 2012, 51, 5593–5604. [Google Scholar] [CrossRef]
- López-Gómez, M.J.; Martin, D.; Bertrand, G. Anti-Bredt N-Heterocyclic Carbene: An Efficient Ligand for the Gold(i)-Catalyzed Hydroamination of Terminal Alkynes with Parent Hydrazine. Chem. Commun. 2013, 49, 4483. [Google Scholar] [CrossRef] [PubMed]
- Gonell, S.; Poyatos, M.; Peris, E. Triphenylene-Based Tris(N-Heterocyclic Carbene) Ligand: Unexpected Catalytic Benefits. Angew. Chem. Int. Ed. 2013, 52, 7009–7013. [Google Scholar] [CrossRef]
- Hu, X.; Martin, D.; Bertrand, G. Room Temperature Hydroamination of Alkynes with Anilines Catalyzed by Anti-Bredt Di(Amino)Carbene Gold(i) Complexes. New J. Chem. 2016, 40, 5993–5996. [Google Scholar] [CrossRef]
- Martin, D.; Canac, Y.; Lavallo, V.; Bertrand, G. Comparative Reactivity of Different Types of Stable Cyclic and Acyclic Mono- and Diamino Carbenes with Simple Organic Substrates. J. Am. Chem. Soc. 2014, 136, 5023–5030. [Google Scholar] [CrossRef]
- Ibáñez, S.; Poyatos, M.; Peris, E. A D3h-Symmetry Hexaazatriphenylene-Tris-N-Heterocyclic Carbene Ligand and Its Coordination to Iridium and Gold: Preliminary Catalytic Studies. Chem. Commun. 2017, 53, 3733–3736. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, S.; Poyatos, M.; Peris, E. Gold Catalysts with Polyaromatic-NHC Ligands. Enhancement of Activity by Addition of Pyrene. Organometallics 2017, 36, 1447–1451. [Google Scholar] [CrossRef]
- Nair, A.G.; McBurney, R.T.; Gatus, M.R.D.; Binding, S.C.; Messerle, B.A. Gold(III) NHC Complexes for Catalyzing Dihydroalkoxylation and Hydroamination Reactions. Inorg. Chem. 2017, 56, 12067–12075. [Google Scholar] [CrossRef]
- Nuevo, D.; Poyatos, M.; Peris, E. A Dinuclear Au(I) Complex with a Pyrene-Di-N-Heterocyclic Carbene Linker: Supramolecular and Catalytic Studies. Organometallics 2018, 37, 3407–3411. [Google Scholar] [CrossRef]
- Baron, M.; Battistel, E.; Tubaro, C.; Biffis, A.; Armelao, L.; Rancan, M.; Graiff, C. Single-Step Synthesis of Dinuclear Neutral Gold(I) Complexes with Bridging Di(N-Heterocyclic Carbene) Ligands and Their Catalytic Performance in Cross Coupling Reactions and Alkyne Hydroamination. Organometallics 2018, 37, 4213–4223. [Google Scholar] [CrossRef]
- Michon, C.; Gilbert, J.; Trivelli, X.; Nahra, F.; Cazin, C.S.J.; Agbossou-Niedercorn, F.; Nolan, S.P. Gold(i) Catalysed Regio- and Stereoselective Intermolecular Hydroamination of Internal Alkynes: Towards Functionalised Azoles. Org. Biomol. Chem. 2019, 17, 3805–3811. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, C.; Tinnermann, H.; Huynh, H.V. Gold(I) and Gold(III) Complexes of Expanded-Ring N-Heterocyclic Carbenes: Structure, Reactivity, and Catalytic Applications. Organometallics 2020, 39, 172–181. [Google Scholar] [CrossRef]
- Sarmiento, J.T.; Cárcel, M.; Ramírez de Arellano, C.; Varea, T.; Asensio, G.; Olmos, A. Straightforward Synthesis of α-Chloromethylketimines Catalyzed by Gold(I). A Clean Way to Building Blocks. J. Org. Chem. 2022, 87, 3114–3122. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Zambrana, C.; Poyatos, M.; Peris, E. A Redox-Switchable Gold(I) Complex for the Hydroamination of Acetylenes: A Convenient Way for Studying Ligand-Derived Electronic Effects. ACS Catal. 2022, 12, 4465–4472. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom−Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Hintermann, L.; Labonne, A. Catalytic Hydration of Alkynes and Its Application in Synthesis. Synthesis 2007, 2007, 1121–1150. [Google Scholar] [CrossRef] [Green Version]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef]
- de Frémont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, Properties, and Organometallic Chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Arcadi, A. Alternative Synthetic Methods through New Developments in Catalysis by Gold. Chem. Rev. 2008, 108, 3266–3325. [Google Scholar] [CrossRef]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligand Effects in Homogeneous Au Catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Crabtree, R.H.; Mata, J.; Peris, E. Chelating Bis-Carbene Rhodium(Iii) Complexes in Transfer Hydrogenation of Ketones and IminesElectronic Supplementary Information (ESI) Available: Spectroscopic Data for the Rhodium(Iii) Complexes. Chem. Commun. 2002, 1, 32–33. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. [(NHC) 2 Cu]X Complexes as Efficient Catalysts for Azide-Alkyne Click Chemistry at Low Catalyst Loadings. Angew. Chem. Int. Ed. 2008, 47, 8881–8884. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Brouwer, C.; He, C. Gold-Catalyzed Organic Transformations. Chem. Rev. 2008, 108, 3239–3265. [Google Scholar] [CrossRef]
- Marion, N.; Ramón, R.S.; Nolan, S.P. [(NHC)Au I ]-Catalyzed Acid-Free Alkyne Hydration at Part-per-Million Catalyst Loadings. J. Am. Chem. Soc. 2009, 131, 448–449. [Google Scholar] [CrossRef]
- Fernández, G.A.; Chopa, A.B.; Silbestri, G.F. A Structure/Catalytic Activity Study of Gold(i)–NHC Complexes, as Well as Their Recyclability and Reusability, in the Hydration of Alkynes in Aqueous Medium. Catal. Sci. Technol. 2016, 6, 1921–1929. [Google Scholar] [CrossRef]
- Fernández, G.A.; Picco, A.S.; Ceolín, M.R.; Chopa, A.B.; Silbestri, G.F. Synthesis and Structural Characterization of Water-Soluble Gold(I) N-Heterocyclic Carbene Complexes. An X-Ray Absorption Fine Structure Spectroscopy (XAFS) Study. Organometallics 2013, 32, 6315–6323. [Google Scholar] [CrossRef]
- Wang, H.M.J.; Lin, I.J.B. Facile Synthesis of Silver(I)−Carbene Complexes. Useful Carbene Transfer Agents. Organometallics 1998, 17, 972–975. [Google Scholar] [CrossRef]
- de Frémont, P.; Scott, N.M.; Stevens, E.D.; Ramnial, T.; Lightbody, O.C.; Macdonald, C.L.B.; Clyburne, J.A.C.; Abernethy, C.D.; Nolan, S.P. Synthesis of Well-Defined N -Heterocyclic Carbene Silver(I) Complexes. Organometallics 2005, 24, 6301–6309. [Google Scholar] [CrossRef]
- Lin, I.J.B.; Vasam, C.S. Preparation and Application of N-Heterocyclic Carbene Complexes of Ag(I). Coord. Chem. Rev. 2007, 251, 642–670. [Google Scholar] [CrossRef]
- Leyva, A.; Corma, A. Isolable Gold(I) Complexes Having One Low-Coordinating Ligand as Catalysts for the Selective Hydration of Substituted Alkynes at Room Temperature without Acidic Promoters. J. Org. Chem. 2009, 74, 2067–2074. [Google Scholar] [CrossRef]
- Sanz, S.; Jones, L.A.; Mohr, F.; Laguna, M. Homogenous Catalysis with Gold: Efficient Hydration of Phenylacetylene in Aqueous Media. Organometallics 2007, 26, 952–957. [Google Scholar] [CrossRef]
- Zhdanko, A.; Maier, M.E. Explanation of Counterion Effects in Gold(I)-Catalyzed Hydroalkoxylation of Alkynes. ACS Catal. 2014, 4, 2770–2775. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. [Google Scholar] [CrossRef]
- Martin, A.R.; Makida, Y.; Meiries, S.; Slawin, A.M.Z.; Nolan, S.P. Enhanced Activity of [Ni(NHC)CpCl] Complexes in Arylamination Catalysis. Organometallics 2013, 32, 6265–6270. [Google Scholar] [CrossRef]
- Collado, A.; Balogh, J.; Meiries, S.; Slawin, A.M.Z.; Falivene, L.; Cavallo, L.; Nolan, S.P. Steric and Electronic Parameters of a Bulky yet Flexible N-Heterocyclic Carbene: 1,3-Bis(2,6-Bis(1-Ethylpropyl)Phenyl)Imidazol-2-Ylidene (IPent). Organometallics 2013, 32, 3249–3252. [Google Scholar] [CrossRef]
- Jacquemard, U.; Harpainter, P.; Roland, S. Introduction of Bulky Tert-Butyl Substituents on the Core of N,N′-Diaryl N-Heterocyclic Carbenes through the Corresponding Vicinal Diamines. Tetrahedron Lett. 2013, 54, 4793–4795. [Google Scholar] [CrossRef]
- Weber, S.G.; Zahner, D.; Rominger, F.; Straub, B.F. Mechanistic Investigations of a Stable, Highly Active, Extremely Sterically Shielded Molecular Gold Catalyst. ChemCatChem 2013, 5, 2330–2335. [Google Scholar] [CrossRef]
- de Frémont, P.; Stevens, E.D.; Fructos, M.R.; Mar Díaz-Requejo, M.; Pérez, P.J.; Nolan, S.P. Synthesis, Isolation and Characterization of Cationic Gold(i) N-Heterocyclic Carbene (NHC) Complexes. Chem. Commun. 2006, 19, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Ricard, L.; Gagosz, F. Synthesis and Reactivity of Air-Stable N-Heterocyclic Carbene Gold(I) Bis(Trifluoromethanesulfonyl)Imidate Complexes. Organometallics 2007, 26, 4704–4707. [Google Scholar] [CrossRef]
- Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Solvent-, Silver-, and Acid-Free NHC-Au-X Catalyzed Hydration of Alkynes. The Pivotal Role of the Counterion. ACS Catal. 2016, 6, 7363–7376. [Google Scholar] [CrossRef]
- Gatto, M.; Del Zotto, A.; Segato, J.; Zuccaccia, D. Hydration of Alkynes Catalyzed by L–Au–X under Solvent- and Acid-Free Conditions: New Insights into an Efficient, General, and Green Methodology. Organometallics 2018, 37, 4685–4691. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, X.; Shao, J.; Yang, G.; Wu, Y.; Zhang, Z. Hydration of Alkynes at Room Temperature Catalyzed by Gold(i) Isocyanide Compounds. Green Chem. 2015, 17, 532–537. [Google Scholar] [CrossRef]
- Wang, W.; Hammond, G.B.; Xu, B. Ligand Effects and Ligand Design in Homogeneous Gold(I) Catalysis. J. Am. Chem. Soc. 2012, 134, 5697–5705. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Gold η 2 -Coordination to Unsaturated and Aromatic Hydrocarbons: The Key Step in Gold-Catalyzed Organic Transformations. Organometallics 2010, 29, 2–23. [Google Scholar] [CrossRef]
- Brooner, R.E.M.; Widenhoefer, R.A. Cationic, Two-Coordinate Gold π Complexes. Angew. Chem. Int. Ed. 2013, 52, 11714–11724. [Google Scholar] [CrossRef]
- Liu, L.-P.; Hammond, G.B. Recent Advances in the Isolation and Reactivity of Organogold Complexes. Chem. Soc. Rev. 2012, 41, 3129–3139. [Google Scholar] [CrossRef]
- Cordón, J.; López-de-Luzuriaga, J.M.; Monge, M. Experimental and Theoretical Study of the Effectiveness and Stability of Gold(I) Catalysts Used in the Synthesis of Cyclic Acetals. Organometallics 2016, 35, 732–740. [Google Scholar] [CrossRef]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: On Alternative Counter Anions. Adv. Synth. Catal. 2018, 360, 3949–3959. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Hydration and Alkoxylation of Alkynes Catalyzed by NHC–Au–OTf. Green Chem. 2018, 20, 2125–2134. [Google Scholar] [CrossRef]
- Gu, Y.; Jérôme, F. Bio-Based Solvents: An Emerging Generation of Fluids for the Design of Eco-Efficient Processes in Catalysis and Organic Chemistry. Chem. Soc. Rev. 2013, 42, 9550–9570. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Mathers, R.T.; McMahon, K.C.; Damodaran, K.; Retarides, C.J.; Kelley, D.J. Ring-Opening Metathesis Polymerizations in d-Limonene: A Renewable Polymerization Solvent and Chain Transfer Agent for the Synthesis of Alkene Macromonomers. Macromolecules 2006, 39, 8982–8986. [Google Scholar] [CrossRef]
- Alzari, V.; Nuvoli, D.; Sanna, D.; Ruiu, A.; Mariani, A. Effect of Limonene on the Frontal Ring Opening Metathesis Polymerization of Dicyclopentadiene. J. Polym. Sci. Part Polym. Chem. 2016, 54, 63–68. [Google Scholar] [CrossRef]
- Tian, X.; Yang, F.; Rasina, D.; Bauer, M.; Warratz, S.; Ferlin, F.; Vaccaro, L.; Ackermann, L. C–H Arylations of 1,2,3-Triazoles by Reusable Heterogeneous Palladium Catalysts in Biomass-Derived γ-Valerolactone. Chem. Commun. 2016, 52, 9777–9780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strappaveccia, G.; Luciani, L.; Bartollini, E.; Marrocchi, A.; Pizzo, F.; Vaccaro, L. γ-Valerolactone as an Alternative Biomass-Derived Medium for the Sonogashira Reaction. Green Chem. 2015, 17, 1071–1076. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Wang, C.; Wan, J.-P.; Wen, C. Bio-Based Green Solvent Mediated Disulfide Synthesis via Thiol Couplings Free of Catalyst and Additive. RSC Adv. 2013, 3, 21369–21372. [Google Scholar] [CrossRef]
- Sciortino, G.; Muñoz-López, S.; Lledós, A.; Ujaque, G. Comparative Mechanistic Study on the [Au(NHC)]+-Catalyzed Hydration of Alkynes, Alkenes, and Allenes. Organometallics 2020, 39, 3469–3479. [Google Scholar] [CrossRef]
- Muñoz-López, S.; Couce-Rios, A.; Sciortino, G.; Lledós, A.; Ujaque, G. Mechanistic Insights on the Hydration of Terminal and Internal Allenes Catalyzed by [(NHC)Au]+. Organometallics 2018, 37, 3543–3551. [Google Scholar] [CrossRef]
- Couce-Rios, A.; Lledós, A.; Fernández, I.; Ujaque, G. Origin of the Anti-Markovnikov Hydroamination of Alkenes Catalyzed by L–Au(I) Complexes: Coordination Mode Determines Regioselectivity. ACS Catal. 2019, 9, 848–858. [Google Scholar] [CrossRef]
- Rzhevskiy, S.A.; Philippova, A.N.; Chesnokov, G.A.; Ageshina, A.A.; Minaeva, L.I.; Topchiy, M.A.; Nechaev, M.S.; Asachenko, A.F. Ring Size and Nothing Else Matters: Unusual Regioselectivity of Alkyne Hydration by NHC Gold(i) Complexes. Chem. Commun. 2021, 57, 5686–5689. [Google Scholar] [CrossRef]
- Yuan, L.-Z.; Zhao, G.; Hamze, A.; Alami, M.; Provot, O. Chlorotrimethylsilane and Sodium Iodide: A Useful Combination for the Regioselective Deoxygenation of Arylalkyl-α-Diketones. Adv. Synth. Catal. 2017, 359, 2682–2691. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).