
Determinants for an Efficient Enzymatic Catalysis in Poly(Ethylene Terephthalate) Degradation

Abstract
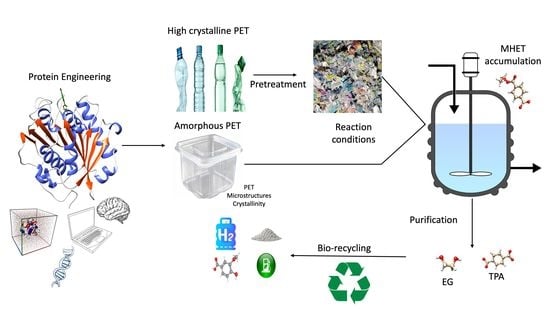
:
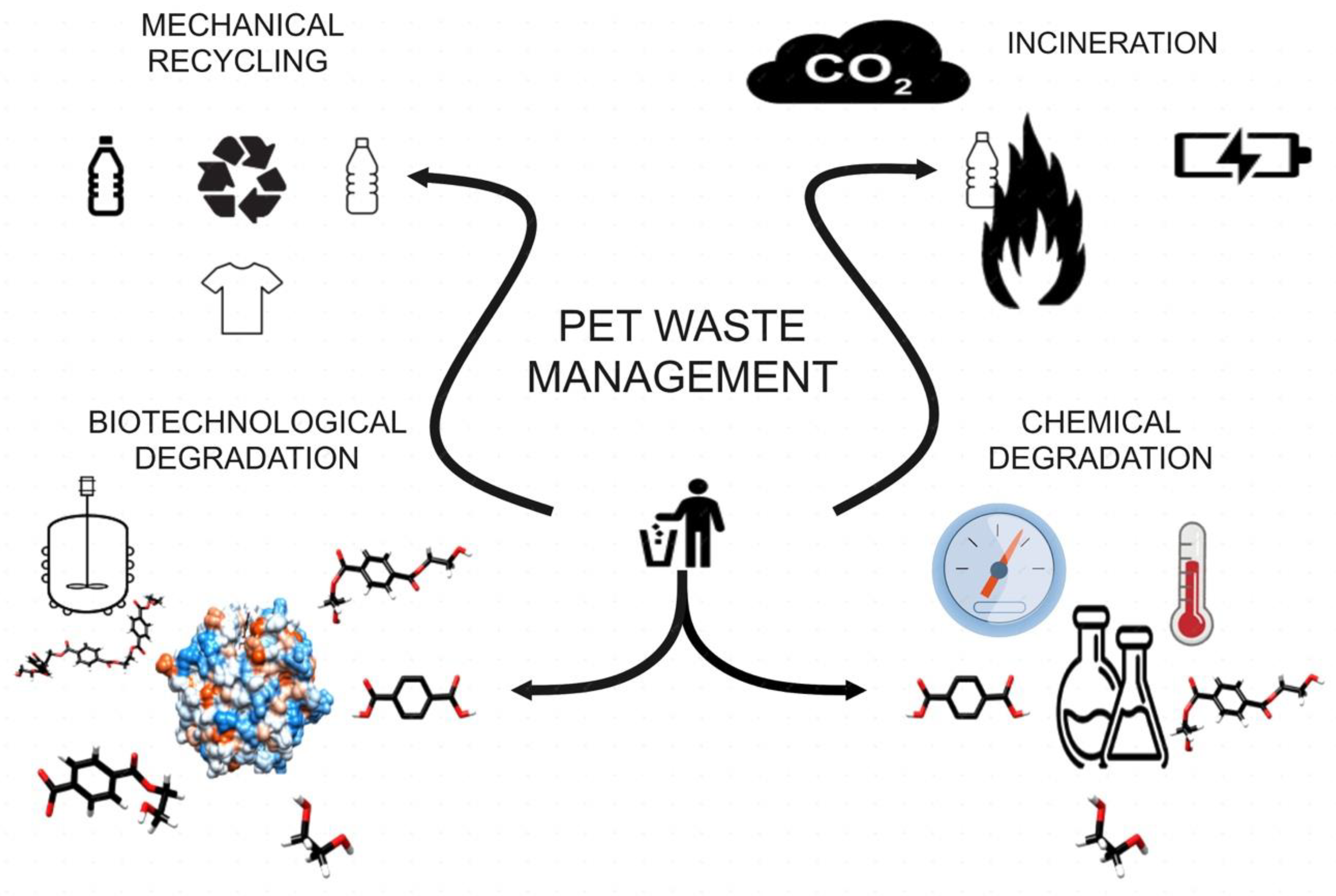
1. Introduction
- (i)
- Mechanical recycling, which has limitations due to the loss of PET’s properties.
- (ii)
- Chemical degradation, which uses and produces compounds harmful to the environment. The specifics of these technologies have been reviewed elsewhere [17,18]. Briefly, some available chemical treatments are: hydrolysis (acidic, neutral, or basic) at high temperature and pressure, to render TPA; glycolysis at 180–240 °C under an inert atmosphere, to produce BHET; aminolysis at 20–200 °C, to yield terephthalic acid diamides plus oligomeric diacids; ammonolysis, consisting in PET treatment with zinc acetate at 70–180 °C under high pressure, to generate terephthalamide; and methanolysis, using methanol at 180–280 °C, and under 20 to 40 atm, to release EG and dimethyl terephthalate (DMT). Nevertheless, the need for product purification, their poor selectivity, and the possible contamination of catalysts by the presence of other plastics, impact the cost of these forms of recycling.
- (iii)
- PET incineration, which generates energy, but has the drawback of CO2 emission, and may release some toxic fumes [19].
- (iv)
- Recycling by biotechnological degradation (Figure 1).
2. Enzymes Acting on PET
3. Important Aspects Related to the Use of These Enzymes in Degradation and Bio-Recycling of PET
- (i)
- Does catalytic efficiency limit PET hydrolysis by these enzymes?
- (ii)
- Are the interfacial diffusion rate and the affinity for the PET surface critical steps in the degradation process?
- (iii)
- Are the reaction conditions required for optimal TPA production yet to be found?
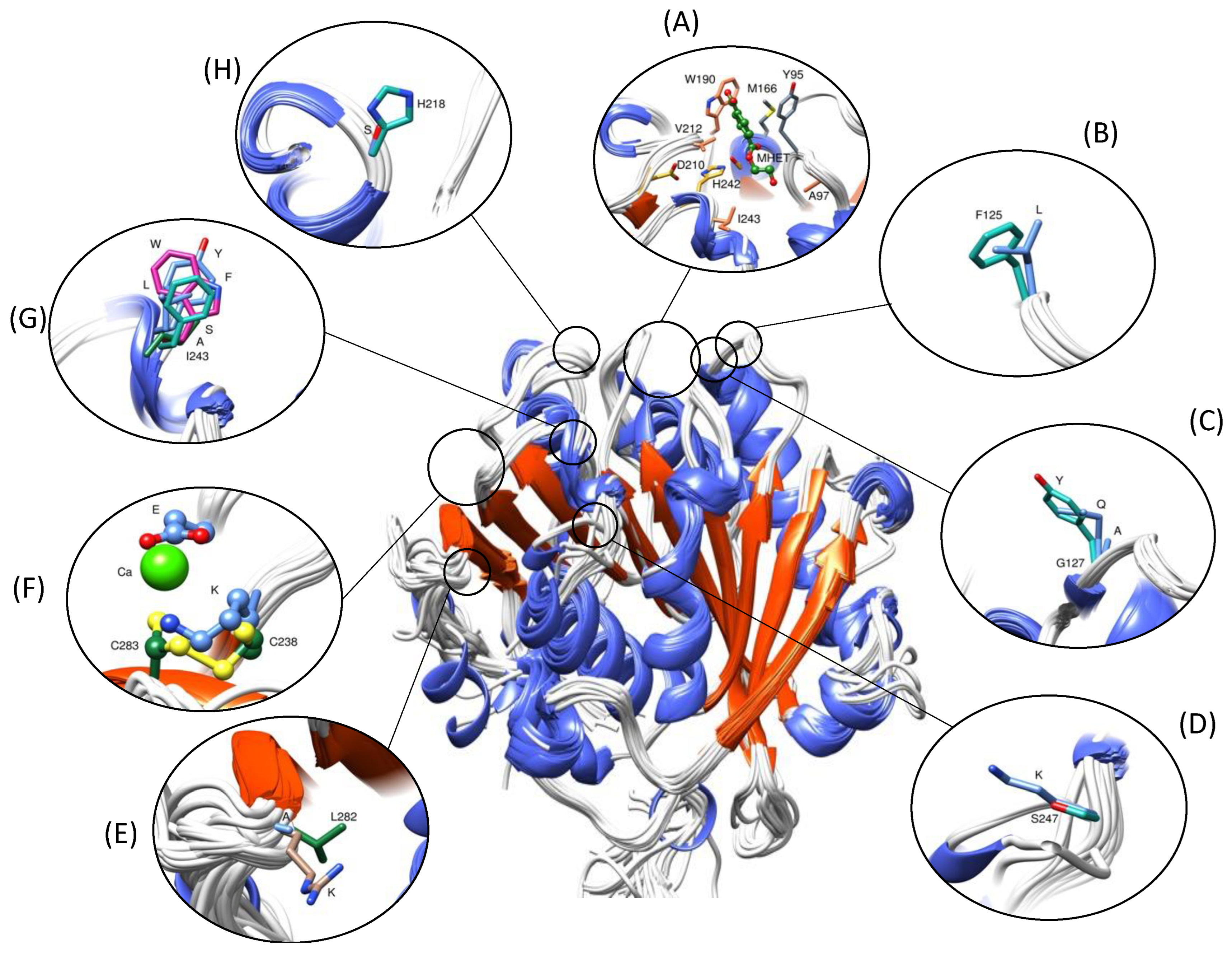
3.1. Protein Improvements through Selection or Engineering
3.1.1. Enzyme Kinetics
3.1.2. Thermostability
- Ions and co-solvents
- Design of disulfide bridges
- Other stabilizing mutations
3.1.3. Catalytic Efficiency
3.1.4. Search for Novel PET Hydrolases
3.1.5. Enzyme Combinations and Chimeras to Solve MHET Hydrolysis
4. Strategies to Improve Biocatalyst-Polymer Interaction and Monomers Yield
4.1. PET Pretreatment
4.2. Effect of Reaction Medium
4.3. Mass Transfer Problems
5. Challenges and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Plastics—The Facts 2022. 2022. Available online: https://plasticseurope.org/wp-content/uploads/2022/10/PE-PLASTICS-THE-FACTS_V7-Tue_19-10-1.pdf (accessed on 6 November 2022).
- Gasperi, J.; Wright, S.L.; Dris, R.; Collard, F.; Mandin, C.; Guerrouache, M.; Langlois, V.; Kelly, F.J.; Tassin, B. Microplastics in Air: Are We Breathing It In? Curr. Opin. Environ. Sci. Health 2018, 1, 1–5. [Google Scholar] [CrossRef]
- Suaria, G.; Achtypi, A.; Perold, V.; Lee, J.R.; Pierucci, A.; Bornman, T.G.; Aliani, S.; Ryan, P.G. Microfibers in Oceanic Surface Waters: A Global Characterization. Sci. Adv. 2020, 6, eaay8493. [Google Scholar] [CrossRef] [PubMed]
- Karami, A.; Golieskardi, A.; Keong Choo, C.; Larat, V.; Galloway, T.S.; Salamatinia, B. The Presence of Microplastics in Commercial Salts from Different Countries. Sci. Rep. 2017, 7, 46173. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, M.S. Effects of Microplastics on Fish and in Human Health. Front. Environ. Sci. 2022, 10, 827289. [Google Scholar] [CrossRef]
- Aguilar-Guzmán, J.C.; Bejtka, K.; Fontana, M.; Valsami-Jones, E.; Villezcas, A.M.; Vazquez-Duhalt, R.; Rodríguez-Hernández, A.G. Polyethylene Terephthalate Nanoparticles Effect on RAW 264.7 Macrophage Cells. Microplastics Nanoplastics 2022, 2, 9. [Google Scholar] [CrossRef]
- MacLeod, M.; Arp, H.P.H.; Tekman, M.B.; Jahnke, A. The Global Threat from Plastic Pollution. Science 2021, 373, 61–65. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Hecht, K.; Buller, R. Enzymatic PET Degradation. Chimia 2019, 73, 743–749. [Google Scholar] [CrossRef]
- Guo, B.; Vanga, S.R.; Lopez-Lorenzo, X.; Saenz-Mendez, P.; Ericsson, S.R.; Fang, Y.; Ye, X.; Schriever, K.; Bäckström, E.; Biundo, A.; et al. Conformational Selection in Biocatalytic Plastic Degradation by PETase. ACS Catal. 2022, 12, 3397–3409. [Google Scholar] [CrossRef]
- Thomsen, T.B.; Hunt, C.J.; Meyer, A.S. Influence of Substrate Crystallinity and Glass Transition Temperature on Enzymatic Degradation of Polyethylene Terephthalate (PET). New Biotechnol. 2022, 69, 28–35. [Google Scholar] [CrossRef]
- Zimmermann, W. Biocatalytic Recycling of Polyethylene Terephthalate Plastic: Biocatalytic Plastic Recycling. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2020, 378, 20190273. [Google Scholar] [CrossRef]
- Kawai, F.; Kawabata, T.; Oda, M. Current State and Perspectives Related to the Polyethylene Terephthalate Hydrolases Available for Biorecycling. ACS Sustain. Chem. Eng. 2020, 8, 8894–8908. [Google Scholar] [CrossRef]
- Carniel, A.; de Abreu Waldow, V.; de Castro, A.M. A Comprehensive and Critical Review on Key Elements to Implement Enzymatic PET Depolymerization for Recycling Purposes. Biotechnol. Adv. 2021, 52, 107811. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Breite, D.; Song, C.; Gräsing, D.; Ploss, T.; Hille, P.; Schwerdtfeger, R.; Matysik, J.; Schulze, A.; Zimmermann, W. Biocatalytic Degradation Efficiency of Postconsumer Polyethylene Terephthalate Packaging Determined by Their Polymer Microstructures. Adv. Sci. 2019, 6, 1900491. [Google Scholar] [CrossRef]
- Panowicz, R.; Konarzewski, M.; Durejko, T.; Szala, M.; Łazi, M. Properties of Polyethylene Terephthalate (PET) after Thermo-Oxidative Aging. Materials 2021, 14, 3833. [Google Scholar] [CrossRef] [PubMed]
- Lens-Pechakova, L.S. Recent Studies on Enzyme-Catalysed Recycling and Biodegradation of Synthetic Polymers. Adv. Ind. Eng. Polym. Res. 2021, 4, 151–158. [Google Scholar] [CrossRef]
- Kushwaha, A.; Goswami, L.; Singhvi, M.; Kim, B.S. Biodegradation of Poly(Ethylene Terephthalate): Mechanistic Insights, Advances, and Future Innovative Strategies. Chem. Eng. J. 2023, 457, 141230. [Google Scholar] [CrossRef]
- Ghosal, K.; Nayak, C. Recent Advances in Chemical Recycling of Polyethylene Terephthalate Waste into Value Added Products for Sustainable Coating Solutions—Hope vs. Hype. Mater. Adv. 2022, 3, 1974–1992. [Google Scholar] [CrossRef]
- Sovová, K.; Ferus, M.; Matulková, I.; Španěl, P.; Dryahina, K.; Dvořák, O.; Civiš, S. A Study of Thermal Decomposition and Combustion Products of Disposable Polyethylene Terephthalate (PET) Plastic Using High Resolution Fourier Transform Infrared Spectroscopy, Selected Ion Flow Tube Mass Spectrometry and Gas Chromatography Mass Spectrometr. Mol. Phys. 2008, 106, 1205–1214. [Google Scholar] [CrossRef]
- Jung, K.W.; Kim, J.H.; Choi, J.W. Synthesis of Magnetic Porous Carbon Composite Derived from Metal-Organic Framework Using Recovered Terephthalic Acid from Polyethylene Terephthalate (PET) Waste Bottles as Organic Ligand and Its Potential as Adsorbent for Antibiotic Tetracycline Hydrochlo. Compos. Part B Eng. 2020, 187, 107867. [Google Scholar] [CrossRef]
- Yang, Q.; Li, H.; Wang, D.; Zhang, X.; Guo, X.; Pu, S.; Guo, R.; Chen, J. Utilization of Chemical Wastewater for CO2 Emission Reduction: Purified Terephthalic Acid (PTA) Wastewater-Mediated Culture of Microalgae for CO2 Bio-Capture. Appl. Energy 2020, 276, 115502. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, J.; Ullah, M.; Wang, Q.; Zhang, Y.; Cao, G.; Chen, L.; Ullah, M.W.; Sun, S. Polyethylene Terephthalate Hydrolysate Increased Bacterial Cellulose Production. Carbohydr. Polym. 2023, 300, 120301. [Google Scholar] [CrossRef] [PubMed]
- Malafatti-Picca, L.; de Barros Chaves, M.R.; de Castro, A.M.; Valoni, É.; de Oliveira, V.M.; Marsaioli, A.J.; de Franceschi de Angelis, D.; Attili-Angelis, D. Hydrocarbon-Associated Substrates Reveal Promising Fungi for Poly (Ethylene Terephthalate) (PET) Depolymerization. Braz. J. Microbiol. 2019, 50, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A Bacterium That Degrades and Assimilates Poly(Ethylene Terephthalate). Science 2016, 353, 759. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Kong, T.; Li, Y.; Li, Q.; Li, Z.; Zhang, J. Biodegradation of Microplastic Derived from Poly(Ethylene Terephthalate) with Bacterial Whole-Cell Biocatalysts. Polymers 2018, 10, 1326. [Google Scholar] [CrossRef] [PubMed]
- Asmita, K.; Shubhamsingh, T.; Tejashree, S. Isolation of Plastic Degrading Micro-Organisms from Soil Samples Collected at Various Locations in Mumbai, India. Int. Res. J. Environ. Sci. 2015, 4, 77–85. [Google Scholar]
- Khoironi, A.; Anggoro, S.; Sudarno, S. Evaluation of the Interaction Among Microalgae Spirulina sp, Plastics Polyethylene Terephthalate and Polypropylene in Freshwater Environment. J. Ecol. Eng. 2019, 20, 161–173. [Google Scholar] [CrossRef]
- Dąbrowska, G.B.; Janczak, K.; Richert, A. Combined Use of Bacillus Strains and Miscanthus for Accelerating Biodegradation of Poly(Lactic Acid) and Poly(Ethylene Terephthalate). PeerJ 2021, 9, e10957. [Google Scholar] [CrossRef]
- Huang, Q.S.; Yan, Z.F.; Chen, X.Q.; Du, Y.Y.; Li, J.; Liu, Z.Z.; Xia, W.; Chen, S.; Wu, J. Accelerated Biodegradation of Polyethylene Terephthalate by Thermobifida fusca Cutinase Mediated by Stenotrophomonas pavanii. Sci. Total Environ. 2022, 808, 152107. [Google Scholar] [CrossRef]
- Dhaka, V.; Singh, S.; Ramamurthy, P.C.; Samuel, J.; Swamy Sunil Kumar Naik, T.; Khasnabis, S.; Prasad, R.; Singh, J. Biological Degradation of Polyethylene Terephthalate by Rhizobacteria. Environ. Sci. Pollut. Res. 2022. [Google Scholar] [CrossRef]
- Farzi, A.; Dehnad, A.; Fotouhi, A.F. Biodegradation of Polyethylene Terephthalate Waste Using Streptomyces Species and Kinetic Modeling of the Process. Biocatal. Agric. Biotechnol. 2019, 17, 25–31. [Google Scholar] [CrossRef]
- Qi, X.; Ma, Y.; Chang, H.; Li, B.; Ding, M.; Yuan, Y. Evaluation of PET Degradation Using Artificial Microbial Consortia. Front. Microbiol. 2021, 12, 778828. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Y.; Cheng, Y.; Wang, X.; Tong, S.; Yang, H.; Wang, Z. Efficient Biodegradation of Highly Crystallized Polyethylene Terephthalate through Cell Surface Display of Bacterial PETase. Sci. Total Environ. 2020, 709, 136138. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Duan, R.; Xiao, Y.; Wei, Y.; Zhang, H.; Sun, X.; Wang, S.; Cheng, Y.; Wang, X.; Tong, S.; et al. Biodegradation of Highly Crystallized Poly(Ethylene Terephthalate) through Cell Surface Codisplay of Bacterial PETase and Hydrophobin. Nat. Commun. 2022, 13, 7138. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ye, Q.; Seo, Y.; Wei, N. Enzymatic Degradation of Polyethylene Terephthalate Plastics by Bacterial Curli Display PETase. Environ. Sci. Technol. Lett. 2022, 9, 650–657. [Google Scholar] [CrossRef]
- Yan, F.; Wei, R.; Cui, Q.; Bornscheuer, U.T.; Liu, Y.J. Thermophilic Whole-Cell Degradation of Polyethylene Terephthalate Using Engineered Clostridium thermocellum. Microb. Biotechnol. 2021, 14, 374–385. [Google Scholar] [CrossRef]
- Kawai, F.; Kawabata, T.; Oda, M. Current Knowledge on Enzymatic PET Degradation and Its Possible Application to Waste Stream Management and Other Fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics, Preparation, and Application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef]
- Martínez, A.; Maicas, S. Cutinases: Characteristics and Insights in Industrial Production. Catalysts 2021, 11, 1194. [Google Scholar] [CrossRef]
- Xi, X.; Ni, K.; Hao, H.; Shang, Y.; Zhao, B.; Qian, Z. Secretory Expression in Bacillus subtilis and Biochemical Characterization of a Highly Thermostable Polyethylene Terephthalate Hydrolase from Bacterium HR29. Enzym. Microb. Technol. 2021, 143, 109715. [Google Scholar] [CrossRef]
- Austin, H.P.; Allen, M.D.; Donohoe, B.S.; Rorrer, N.A.; Kearns, F.L.; Silveira, R.L.; Pollard, B.C.; Dominick, G.; Duman, R.; Omari, K.E.; et al. Characterization and Engineering of a Plastic-Degrading Aromatic Polyesterase. Proc. Natl. Acad. Sci. USA 2018, 115, E4350–E4357. [Google Scholar] [CrossRef]
- Joo, S.; Cho, I.J.; Seo, H.; Son, H.F.; Sagong, H.Y.; Shin, T.J.; Choi, S.Y.; Lee, S.Y.; Kim, K.J. Structural Insight into Molecular Mechanism of Poly(Ethylene Terephthalate) Degradation. Nat. Commun. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Han, X.; Ko, T.P.; Liu, W.; Guo, R.T. Structural Studies Reveal the Molecular Mechanism of PETase. FEBS J. 2018, 285, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Han, X.; Li, X.; Jiang, P.; Niu, D.; Ma, L.; Liu, W.; Li, S.; Qu, Y.; Hu, H.; et al. General Features to Enhance Enzymatic Activity of Poly(Ethylene Terephthalate) Hydrolysis. Nat. Catal. 2021, 4, 425–430. [Google Scholar] [CrossRef]
- Müller, R.J.; Schrader, H.; Profe, J.; Dresler, K.; Deckwer, W.D. Enzymatic Degradation of Poly(Ethylene Terephthalate): Rapid Hydrolyse Using a Hydrolase from T. Fusca. Macromol. Rapid Commun. 2005, 26, 1400–1405. [Google Scholar] [CrossRef]
- Ronkvist, Å.M.; Xie, W.; Lu, W.; Gross, R.A. Cutinase-Catalyzed Hydrolysis of Poly(Ethylene Terephthalate). Macromolecules 2009, 42, 5128–5138. [Google Scholar] [CrossRef]
- Kawai, F.; Oda, M.; Tamashiro, T.; Waku, T.; Tanaka, N.; Yamamoto, M.; Mizushima, H.; Miyakawa, T.; Tanokura, M. A Novel Ca2+-Activated, Thermostabilized Polyesterase Capable of Hydrolyzing Polyethylene Terephthalate from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 98, 10053–10064. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a Novel Cutinase Homolog with Polyethylene Terephthalate-Degrading Activity from Leaf-Branch Compost by Using a Metagenomic Approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef]
- Sonnendecker, C.; Oeser, J.; Richter, P.K.; Hille, P.; Zhao, Z.; Fischer, C.; Lippold, H.; Blázquez-Sánchez, P.; Engelberger, F.; Ramírez-Sarmiento, C.A.; et al. Low Carbon Footprint Recycling of Post-Consumer PET Plastic with a Metagenomic Polyester Hydrolase. ChemSusChem 2022, 15, e202101062. [Google Scholar] [CrossRef]
- Pfaff, L.; Gao, J.; Li, Z.; Jäckering, A.; Weber, G.; Mican, J.; Chen, Y.; Dong, W.; Han, X.; Feiler, C.G.; et al. Multiple Substrate Binding Mode-Guided Engineering of a Thermophilic PET Hydrolase. ACS Catal. 2022, 12, 9790–9800. [Google Scholar] [CrossRef]
- Lu, H.; Diaz, D.J.; Czarnecki, N.J.; Zhu, C.; Kim, W.; Shroff, R.; Acosta, D.J.; Alexander, B.R.; Cole, H.O.; Zhang, Y.; et al. Machine Learning-Aided Engineering of Hydrolases for PET Depolymerization. Nature 2022, 604, 662–667. [Google Scholar] [CrossRef]
- Herrero Acero, E.; Ribitsch, D.; Steinkellner, G.; Gruber, K.; Greimel, K.; Eiteljoerg, I.; Trotscha, E.; Wei, R.; Zimmermann, W.; Zinn, M.; et al. Enzymatic Surface Hydrolysis of PET: Effect of Structural Diversity on Kinetic Properties of Cutinases from Thermobifida. Macromolecules 2011, 44, 4632–4640. [Google Scholar] [CrossRef]
- Arnling Bååth, J.; Novy, V.; Carneiro, L.V.; Guebitz, G.M.; Olsson, L.; Westh, P.; Ribitsch, D. Structure-Function Analysis of Two Closely Related Cutinases from Thermobifida cellulosilytica. Biotechnol. Bioeng. 2022, 119, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, S.; Cai, D.; Shao, C.; Zhang, C.; Zhou, J.; Cui, Z.; He, T.; Chen, C.; Chen, B.; et al. Depolymerization of Post-Consumer PET Bottles with Engineered Cutinase 1 from Thermobifida cellulosilytica. Green Chem. 2022, 24, 5998–6007. [Google Scholar] [CrossRef]
- Wyeth, N.C.; Mendenhall, P.; Roseveare, R.N. Biaxially Oriented Poly(Ethylene Terephthalate) Bottle. United States Patent US3733309A, 3 September 1985. [Google Scholar]
- Witt, U.; Müller, R.-J.; Augusta, J.; Widdecke, H.; Deckwer, W.-D. Synthesis, Properties and Biodegradability of Polyesters Based on 1,3-Propanediol. Macromol. Chem. Phys. 1994, 195, 793–802. [Google Scholar] [CrossRef]
- Han, X.; Liu, W.; Huang, J.W.; Ma, J.; Zheng, Y.; Ko, T.P.; Xu, L.; Cheng, Y.S.; Chen, C.C.; Guo, R.T. Structural Insight into Catalytic Mechanism of PET Hydrolase. Nat. Commun. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Araújo, R.; Silva, C.; O’Neill, A.; Micaelo, N.; Guebitz, G.; Soares, C.M.; Casal, M.; Cavaco-Paulo, A. Tailoring Cutinase Activity towards Polyethylene Terephthalate and Polyamide 6,6 Fibers. J. Biotechnol. 2007, 128, 849–857. [Google Scholar] [CrossRef]
- De Castro, A.M.; Carniel, A.; Stahelin, D.; Junior, L.S.C.; de Angeli Honorato, H.; de Menezes, S.M.C. High-Fold Improvement of Assorted Post-Consumer Poly(Ethylene Terephthalate) (PET) Packages Hydrolysis Using Humicola insolens Cutinase as a Single Biocatalyst. Process Biochem. 2019, 81, 85–91. [Google Scholar] [CrossRef]
- Bååth, J.A.; Borch, K.; Jensen, K.; Brask, J.; Westh, P. Comparative Biochemistry of Four Polyester (PET) Hydrolases**. ChemBioChem 2021, 22, 1627–1637. [Google Scholar] [CrossRef]
- Badino, S.F.; Bååth, J.A.; Borch, K.; Jensen, K.; Westh, P. Adsorption of Enzymes with Hydrolytic Activity on Polyethylene Terephthalate. Enzym. Microb. Technol. 2022, 152, 109937. [Google Scholar] [CrossRef]
- Vogel, K.; Wei, R.; Pfaff, L.; Breite, D.; Al-Fathi, H.; Ortmann, C.; Estrela-Lopis, I.; Venus, T.; Schulze, A.; Harms, H.; et al. Enzymatic Degradation of Polyethylene Terephthalate Nanoplastics Analyzed in Real Time by Isothermal Titration Calorimetry. Sci. Total Environ. 2021, 773, 145111. [Google Scholar] [CrossRef]
- Scandola, M.; Focarete, M.L.; Frisoni, G. Simple Kinetic Model for the Heterogeneous Enzymatic Hydrolysis of Natural Poly(3-Hydroxybutyrate). Macromolecules 1998, 31, 3846–3851. [Google Scholar] [CrossRef]
- Aer, L.; Jiang, Q.; Gul, I.; Qi, Z.; Feng, J.; Tang, L. Overexpression and Kinetic Analysis of Ideonella sakaiensis PETase for Polyethylene Terephthalate (PET) Degradation. Environ. Res. 2022, 212, 113472. [Google Scholar] [CrossRef] [PubMed]
- Reuveni, S.; Urbakh, M.; Klafter, J. Role of Substrate Unbinding in Michaelis-Menten Enzymatic Reactions. Proc. Natl. Acad. Sci. USA 2014, 111, 4391–4396. [Google Scholar] [CrossRef]
- Barth, M.; Oeser, T.; Wei, R.; Then, J.; Schmidt, J.; Zimmermann, W. Effect of Hydrolysis Products on the Enzymatic Degradation of Polyethylene Terephthalate Nanoparticles by a Polyester Hydrolase from Thermobifida fusca. Biochem. Eng. J. 2015, 93, 222–228. [Google Scholar] [CrossRef]
- Oda, M.; Yamagami, Y.; Inaba, S.; Oida, T.; Yamamoto, M.; Kitajima, S.; Kawai, F. Enzymatic Hydrolysis of PET: Functional Roles of Three Ca2+ Ions Bound to a Cutinase-like Enzyme, Cut190*, and Its Engineering for Improved Activity. Appl. Microbiol. Biotechnol. 2018, 102, 10067–10077. [Google Scholar] [CrossRef]
- Wei, R.; Oeser, T.; Schmidt, J.; Meier, R.; Barth, M.; Then, J.; Zimmermann, W. Engineered Bacterial Polyester Hydrolases Efficiently Degrade Polyethylene Terephthalate Due to Relieved Product Inhibition. Biotechnol. Bioeng. 2016, 113, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.; Honak, A.; Oeser, T.; Wei, R.; Belisário-Ferrari, M.R.; Then, J.; Schmidt, J.; Zimmermann, W. A Dual Enzyme System Composed of a Polyester Hydrolase and a Carboxylesterase Enhances the Biocatalytic Degradation of Polyethylene Terephthalate Films. Biotechnol. J. 2016, 11, 1082–1087. [Google Scholar] [CrossRef]
- Eugenio, E.D.Q.; Campisano, I.S.P.; de Castro, A.M.; Coelho, M.A.Z.; Langone, M.A.P. Kinetic Modeling of the Post-Consumer Poly(Ethylene Terephthalate) Hydrolysis Catalyzed by Cutinase from Humicola insolens. J. Polym. Environ. 2021, 30, 1627–1637. [Google Scholar] [CrossRef]
- Gamerith, C.; Zartl, B.; Pellis, A.; Guillamot, F.; Marty, A.; Acero, E.H.; Guebitz, G.M. Enzymatic Recovery of Polyester Building Blocks from Polymer Blends. Process Biochem. 2017, 59, 58–64. [Google Scholar] [CrossRef]
- Pirillo, V.; Pollegioni, L.; Molla, G. Analytical Methods for the Investigation of Enzyme-Catalyzed Degradation of Polyethylene Terephthalate. FEBS J. 2021, 288, 4730–4745. [Google Scholar] [CrossRef]
- Schubert, S.; Schaller, K.; Bååth, J.A.; Hunt, C.; Borch, K.; Jensen, K.; Brask, J.; Westh, P. Reaction Pathways for the Enzymatic Degradation of Poly(Ethylene Terephthalate): What Characterizes an Efficient PET-Hydrolase? ChemBioChem 2022, 24, e202200516. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Kawakami, N.; Oda, K.; Miyamoto, K. Acceleration of Enzymatic Degradation of Poly(Ethylene Terephthalate) by Surface Coating with Anionic Surfactants. ChemSusChem 2018, 11, 4018–4025. [Google Scholar] [CrossRef] [PubMed]
- Baath, J.A.; Jensen, K.; Borch, K.; Westh, P.; Kari, J. Sabatier Principle for Rationalizing Enzymatic Hydrolysis of a Synthetic Polyester. J. Am. Chem. Soc. 2022, 2, 1223–1231. [Google Scholar] [CrossRef]
- Gamerith, C.; Vastano, M.; Ghorbanpour, S.M.; Zitzenbacher, S.; Ribitsch, D.; Zumstein, M.T.; Sander, M.; Acero, E.H.; Pellis, A.; Guebitz, G.M. Enzymatic Degradation of Aromatic and Aliphatic Polyesters by P. Pastoris Expressed Cutinase 1 from Thermobifida cellulosilytica. Front. Microbiol. 2017, 8, 938. [Google Scholar] [CrossRef]
- Shirke, A.N.; White, C.; Englaender, J.A.; Zwarycz, A.; Butterfoss, G.L.; Linhardt, R.J.; Gross, R.A. Stabilizing Leaf and Branch Compost Cutinase (LCC) with Glycosylation: Mechanism and Effect on PET Hydrolysis. Biochemistry 2018, 57, 1190–1200. [Google Scholar] [CrossRef]
- Kawai, F.; Furushima, Y.; Mochizuki, N.; Muraki, N.; Yamashita, M.; Iida, A.; Mamoto, R.; Tosha, T.; Iizuka, R.; Kitajima, S. Efficient Depolymerization of Polyethylene Terephthalate (PET) and Polyethylene Furanoate by Engineered PET Hydrolase Cut190. AMB Express 2022, 12, 134. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, X.; Geng, Y.; Zhou, Q.; Lu, X.; Zhang, S. Deep Eutectic Solvents as Highly Active Catalysts for the Fast and Mild Glycolysis of Poly(Ethylene Terephthalate)(PET). Green Chem. 2015, 17, 2473–2479. [Google Scholar] [CrossRef]
- Tan, Y.; Henehan, G.T.; Kinsella, G.K.; Ryan, B.J. Cutinase from Amycolatopsis mediterannei: Marked Activation and Stabilisation in Deep Eutectic Solvents. Bioresour. Technol. Rep. 2021, 16, 100882. [Google Scholar] [CrossRef]
- Attallah, O.A.; Azeem, M.; Nikolaivits, E.; Topakas, E.; Fournet, M.B. Microwave-Assisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polymers 2022, 14, 109. [Google Scholar] [CrossRef]
- Kawabata, T.; Oda, M.; Kawai, F. Mutational Analysis of Cutinase-like Enzyme, Cut190, Based on the 3D Docking Structure with Model Compounds of Polyethylene Terephthalate. J. Biosci. Bioeng. 2017, 124, 28–35. [Google Scholar] [CrossRef]
- Numoto, N.; Kamiya, N.; Bekker, G.J.; Yamagami, Y.; Inaba, S.; Ishii, K.; Uchiyama, S.; Kawai, F.; Ito, N.; Oda, M. Structural Dynamics of the PET-Degrading Cutinase-like Enzyme from Saccharomonospora viridis AHK190 in Substrate-Bound States Elucidates the Ca2+-Driven Catalytic Cycle. Biochemistry 2018, 57, 5289–5300. [Google Scholar] [CrossRef] [PubMed]
- Inaba, S.; Kamiya, N.; Bekker, G.J.; Kawai, F.; Oda, M. Folding Thermodynamics of PET-Hydrolyzing Enzyme Cut190 Depending on Ca 2+ Concentration. J. Therm. Anal. Calorim. 2019, 135, 2655–2663. [Google Scholar] [CrossRef]
- Then, J.; Wei, R.; Oeser, T.; Barth, M.; Belisário-Ferrari, M.R.; Schmidt, J.; Zimmermann, W. Ca2+ and Mg2+ Binding Site Engineering Increases the Degradation of Polyethylene Terephthalate Films by Polyester Hydrolases from Thermobifida fusca. Biotechnol. J. 2015, 10, 592–598. [Google Scholar] [CrossRef]
- Then, J.; Wei, R.; Oeser, T.; Gerdts, A.; Schmidt, J.; Barth, M.; Zimmermann, W. A Disulfide Bridge in the Calcium Binding Site of a Polyester Hydrolase Increases Its Thermal Stability and Activity against Polyethylene Terephthalate. FEBS Open Bio 2016, 6, 425–432. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.-L.; Texier, H.; Gavalda, S.; et al. An Engineered PET Depolymerase to Break down and Recycle Plastic Bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Gavalda, S.; Borsenberger, V.; Duquesne, S.; Marty, A.; Tournier, V.; Lippens, G. An NMR Look at an Engineered PET Depolymerase. Biophys. J. 2022, 121, 2882–2894. [Google Scholar] [CrossRef] [PubMed]
- Brott, S.; Pfaff, L.; Schuricht, J.; Schwarz, J.-N.; Böttcher, D.; Badenhorst, C.P.S.; Wei, R.; Bornscheuer, U.T. Engineering and Evaluation of Thermostable IsPETase Variants for PET Degradation. Eng. Life Sci. 2022, 22, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Emori, M.; Numoto, N.; Senga, A.; Bekker, G.-J.; Kamiya, N.; Kobayashi, Y.; Ito, N.; Kawai, F.; Oda, M. Structural Basis of Mutants of PET-Degrading Enzyme from Saccharomonospora viridis AHK190 with High Activity and Thermal Stability. Proteins Struct. Funct. Bioinforma. 2021, 89, 502–511. [Google Scholar] [CrossRef]
- Sampedro, J.G.; Uribe, S. Trehalose-Enzyme Interactions Result in Structure Stabilization and Activity Inhibition. The Role of Viscosity. Mol. Cell. Biochem. 2004, 256, 319–327. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, Y.; Liu, X.; Dong, S.; Tian, Y.Y.; Qiao, Y.; Mitra, R.; Han, J.; Li, C.; Han, X.; et al. Computational Redesign of a PETase for Plastic Biodegradation under ambient condition by the GRAPE Strategy. ACS Catal. 2021, 11, 1340–1350. [Google Scholar] [CrossRef]
- Meng, X.; Yang, L.; Liu, H.; Li, Q.; Xu, G.; Zhang, Y.; Guan, F.; Zhang, Y.; Zhang, W.; Wu, N.; et al. Protein Engineering of Stable IsPETase for PET Plastic Degradation by Premuse. Int. J. Biol. Macromol. 2021, 180, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Rennison, A.; Winther, J.R.; Varrone, C. Rational Protein Engineering to Increase the Activity and Stability of Ispetase Using the Pross Algorithm. Polymers 2021, 13, 3884. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; You, S.; Zhang, J.; Qi, W.; Su, R. Enhancement of the Polyethylene Terephthalate and Mono-(2-Hydroxyethyl) Terephthalate Degradation Activity of Ideonella sakaiensis PETase by an Electrostatic Interaction-Based Strategy. Bioresour. Technol. 2022, 364, 128026. [Google Scholar] [CrossRef]
- Zeng, W.; Li, X.; Yang, Y.; Min, J.; Huang, J.W.; Liu, W.; Niu, D.; Yang, X.; Han, X.; Zhang, L.; et al. Substrate-Binding Mode of a Thermophilic PET Hydrolase and Engineering the Enzyme to Enhance the Hydrolytic Efficacy. ACS Catal. 2022, 12, 3033–3040. [Google Scholar] [CrossRef]
- Wu, B.; Cui, Y.; Chen, Y.; Sun, J.; Zhu, T.; Li, C.; Geng, W. Deep Learning-Aided Redesign of a Hydrolase for near 100% PET Depolymerization under Industrially Relevant Conditions. Research Square. 2023. (preprint). [Google Scholar] [CrossRef]
- Li, Z.; Zhao, Y.; Wu, P.; Wang, H.; Li, Q.; Gao, J.; Qin, H.M.; Wei, H.; Bornscheuer, U.T.; Han, X.; et al. Structural Insight and Engineering of a Plastic Degrading Hydrolase Ple629. Biochem. Biophys. Res. Commun. 2022, 626, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Song, C.; Gräsing, D.; Schneider, T.; Bielytskyi, P.; Böttcher, D.; Matysik, J.; Bornscheuer, U.T.; Zimmermann, W. Conformational Fitting of a Flexible Oligomeric Substrate Does Not Explain the Enzymatic PET Degradation. Nat. Commun. 2019, 10, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, P.; Wei, R.; Matysik, J.; Song, C. Mechanistic Investigation of Enzymatic Degradation of Polyethylene Terephthalate by Nuclear Magnetic Resonance. Methods Enzymol. 2021, 648, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. “The Entire Protein Universe”: AI Predicts Shape of Nearly Every Known Protein. Nature 2022, 608, 15–16. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold Models with Ligands and Cofactors. Nat. Methods 2022, 20, 205–213. [Google Scholar] [CrossRef]
- Furukawa, M.; Kawakami, N.; Tomizawa, A.; Miyamoto, K. Efficient Degradation of Poly(Ethylene Terephthalate) with Thermobifida fusca Cutinase Exhibiting Improved Catalytic Activity Generated Using Mutagenesis and Additive-Based Approaches. Sci. Rep. 2019, 9, 16038. [Google Scholar] [CrossRef]
- Herrero Acero, E.; Ribitsch, D.; Dellacher, A.; Zitzenbacher, S.; Marold, A.; Steinkellner, G.; Gruber, K.; Schwab, H.; Guebitz, G.M. Surface Engineering of a Cutinase from Thermobifida cellulosilytica for Improved Polyester Hydrolysis. Biotechnol. Bioeng. 2013, 110, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Son, H.F.; Cho, I.J.; Joo, S.; Seo, H.; Sagong, H.Y.; Choi, S.Y.; Lee, S.Y.; Kim, K.J. Rational Protein Engineering of Thermo-Stable PETase from Ideonella sakaiensis for Highly Efficient PET Degradation. ACS Catal. 2019, 9, 3519–3526. [Google Scholar] [CrossRef]
- Knott, B.C.; Erickson, E.; Allen, M.D.; Gado, J.E.; Graham, R.; Kearns, F.L.; Pardo, I.; Topuzlu, E.; Anderson, J.J.; Austin, H.P.; et al. Characterization and Engineering of a Two-Enzyme System for Plastics Depolymerization. Proc. Natl. Acad. Sci. USA 2020, 117, 25476–25485. [Google Scholar] [CrossRef]
- Wallerstein, J.; Ekberg, V.; Ignjatović, M.M.; Kumar, R.; Caldararu, O.; Peterson, K.; Wernersson, S.; Brath, U.; Leffler, H.; Oksanen, E.; et al. Entropy–Entropy Compensation between the Protein, Ligand, and Solvent Degrees of Freedom Fine-Tunes Affinity in Ligand Binding to Galectin-3C. JACS Au 2021, 1, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.A.; Okur, H.I.; Yan, C.; Yang, T.; Heyda, J.; Cremer, P.S. Weakly Hydrated Anions Bind to Polymers but Not Monomers in Aqueous Solutions. Nat. Chem. 2022, 14, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Q.; Guo, Z.-Y.; Wang, L.; Yan, Z.-F.; Jin, C.-X.; Huang, Q.-S.; Kong, D.-M.; Rao, D.-M.; Wu, J. Directional-Path Modification Strategy Enhances PET Hydrolase Catalysis of Plastic Degradation. J. Hazard. Mater. 2022, 433, 128816. [Google Scholar] [CrossRef]
- Brizendine, R.K.; Erickson, E.; Haugen, S.J.; Ramirez, K.J.; Miscall, J.; Salvachúa, D.; Pickford, A.R.; Sobkowicz, M.J.; Mcgeehan, J.E.; Beckham, G.T. Particle Size Reduction of Poly(Ethylene Terephthalate) Increases the Rate of Enzymatic Depolymerization But Does Not Increase the Overall Conversion Extent. ACS Sustain. Chem. Eng. 2022, 10, 9131–9140. [Google Scholar] [CrossRef]
- Erickson, E.; Gado, J.E.; Avilán, L.; Bratti, F.; Brizendine, R.K.; Cox, P.A.; Gill, R.; Graham, R.; Kim, D.-J.; König, G.; et al. Sourcing Thermotolerant Poly(Ethylene Terephthalate) Hydrolase Scaffolds from Natural Diversity. Nat. Commun. 2022, 13, 7850. [Google Scholar] [CrossRef]
- Sulaiman, S.; You, D.-J.; Kanaya, E.; Koga, Y.; Kanaya, S. Crystal Structure and Thermodynamic and Kinetic Stability of Metagenome-Derived LC-Cutinase. Biochemistry 2014, 53, 1858–1869. [Google Scholar] [CrossRef]
- Graham, R.; Erickson, E.; Brizendine, R.K.; Salvachúa, D.; Michener, W.E.; Li, Y.; Tan, Z.; Beckham, G.T.; McGeehan, J.E.; Pickford, A.R. The Role of Binding Modules in Enzymatic Poly(Ethylene Terephthalate) Hydrolysis at High-Solids Loadings. Chem Catal. 2022, 2, 2644–2657. [Google Scholar] [CrossRef]
- De Castro, A.M.; Carniel, A.; Nicomedes, J., Jr.; da Conceição Gomes, A.; Valoni, É. Screening of Commercial Enzymes for Poly(Ethylene Terephthalate) (PET) Hydrolysis and Synergy Studies on Different Substrate Sources. J. Ind. Microbiol. Biotechnol. 2017, 44, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, S.; Long, L.; Cao, Y.; Ding, S. Engineering a Chimeric Lipase-Cutinase (Lip-Cut) for Efficient Enzymatic Deinking of Waste Paper. BioResources 2018, 13, 981–996. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, T.; Long, L.; Zhang, R.; Ding, S. Efficient Enzymatic Degradation of Poly (ɛ-Caprolactone) by an Engineered Bifunctional Lipase-Cutinase. Polym. Degrad. Stab. 2019, 160, 120–125. [Google Scholar] [CrossRef]
- Ribitsch, D.; Yebra, A.O.; Zitzenbacher, S.; Wu, J.; Nowitsch, S.; Steinkellner, G.; Greimel, K.; Doliska, A.; Oberdorfer, G.; Gruber, C.C.; et al. Fusion of Binding Domains to Thermobifida cellulosilytica Cutinase to Tune Sorption Characteristics and Enhancing PET Hydrolysis. Biomacromolecules 2013, 14, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Petrović, D.; Strodel, B.; Smits, S.H.J.; Kolkenbrock, S.; Leggewie, C.; Jaeger, K.E. Interaction of Carbohydrate-Binding Modules with Poly(Ethylene Terephthalate). Appl. Microbiol. Biotechnol. 2019, 103, 4801–4812. [Google Scholar] [CrossRef]
- Xue, R.; Chen, Y.; Rong, H.; Wei, R.; Cui, Z.; Zhou, J.; Dong, W.; Jiang, M. Fusion of Chitin-Binding Domain From Chitinolyticbacter meiyuanensis SYBC-H1 to the Leaf-Branch Compost Cutinase for Enhanced PET Hydrolysis. Front. Bioeng. Biotechnol. 2021, 9, 1315. [Google Scholar] [CrossRef]
- Palm, G.J.; Reisky, L.; Böttcher, D.; Müller, H.; Michels, E.A.P.; Walczak, M.C.; Berndt, L.; Weiss, M.S.; Bornscheuer, U.T.; Weber, G. Structure of the Plastic-Degrading Ideonella sakaiensis MHETase Bound to a Substrate. Nat. Commun. 2019, 10, 1717. [Google Scholar] [CrossRef]
- Von Haugwitz, G.; Han, X.; Pfaff, L.; Li, Q.; Wei, H.; Gao, J.; Methling, K.; Ao, Y.; Brack, Y.; Mican, J.; et al. Structural Insights into (Tere)Phthalate-Ester Hydrolysis by a Carboxylesterase and Its Role in Promoting PET Depolymerization. ACS Catal. 2022, 12, 15259–15270. [Google Scholar] [CrossRef]
- Puspitasari, N.; Tsai, S.L.; Lee, C.K. Fungal Hydrophobin RolA Enhanced PETase Hydrolysis of Polyethylene Terephthalate. Appl. Biochem. Biotechnol. 2021, 193, 1284–1295. [Google Scholar] [CrossRef]
- Chaires, J.B. Calorimetry and Thermodynamics in Drug Design. Annu. Rev. Biophys. 2008, 37, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Markova, N. Isothermal Titration Calorimetry: Theory and Practice. 2015. Available online: https://www.malvernpanalytical.com/en/learn/knowledge-center/whitepapers/wp150318itctheoryandpractice (accessed on 2 November 2022).
- Tafoukt, D.; Soric, A.; Sigoillot, J.-C.; Ferrasse, J.-H. Determination of Kinetics and Heat of Hydrolysis for Non-Homogenous Substrate by Isothermal Calorimetry. Bioprocess Biosyst. Eng. 2017, 40, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; Ko, C.-H.; Chang, K.-L.; Leu, S.-Y. Construction of a Structural Enzyme Adsorption/Kinetics Model to Elucidate Additives Associated Lignin–Cellulase Interactions in Complex Bioconversion System. Biotechnol. Bioeng. 2021, 118, 4065–4075. [Google Scholar] [CrossRef]
- Thomsen, T.B.; Hunt, C.J.; Meyer, A.S. Standardized Method for Controlled Modification of Poly (Ethylene Terephthalate) (PET) Crystallinity for Assaying PET Degrading Enzymes. MethodsX 2022, 9, 101815. [Google Scholar] [CrossRef] [PubMed]
- Badia, J.D.; Strömberg, E.; Karlsson, S.; Ribes-Greus, A. The Role of Crystalline, Mobile Amorphous and Rigid Amorphous Fractions in the Performance of Recycled Poly (Ethylene Terephthalate) (PET). Polym. Degrad. Stab. 2012, 97, 98–107. [Google Scholar] [CrossRef]
- Carniel, A.; Valoni, É.; Junior, J.N.; da Conceição Gomes, A.; de Castro, A.M. Lipase from Candida antarctica (CALB) and Cutinase from Humicola insolens Act Synergistically for PET Hydrolysis to Terephthalic Acid. Process Biochem. 2017, 59, 84–90. [Google Scholar] [CrossRef]
- Pellis, A.; Gamerith, C.; Ghazaryan, G.; Ortner, A.; Herrero Acero, E.; Guebitz, G.M. Ultrasound-Enhanced Enzymatic Hydrolysis of Poly(Ethylene Terephthalate). Bioresour. Technol. 2016, 218, 1298–1302. [Google Scholar] [CrossRef]
- Morshed, M.N.; Bouazizi, N.; Behary, N.; Guan, J.; Nierstrasz, V. Stabilization of Zero Valent Iron (Fe0) on Plasma/Dendrimer Functionalized Polyester Fabrics for Fenton-like Removal of Hazardous Water Pollutants. Chem. Eng. J. 2019, 374, 658–673. [Google Scholar] [CrossRef]
- Farhadi, A.R.K.; Rahemi, N.; Allahyari, S.; Tasbihi, M. Metal-Doped Perovskite BiFeO3/RGO Nanocomposites towards the Degradation of Acetaminophen in Aqueous Phase Using Plasma-Photocatalytic Hybrid Technology. J. Taiwan Inst. Chem. Eng. 2021, 120, 77–92. [Google Scholar] [CrossRef]
- Schmidt, J.; Wei, R.; Oeser, T.; Belisário-Ferrari, M.R.; Barth, M.; Then, J.; Zimmermann, W. Effect of Tris, MOPS, and Phosphate Buffers on the Hydrolysis of Polyethylene Terephthalate Films by Polyester Hydrolases. FEBS Open Bio 2016, 6, 919–927. [Google Scholar] [CrossRef]
- Barth, M.; Wei, R.; Oeser, T.; Then, J.; Schmidt, J.; Wohlgemuth, F.; Zimmermann, W. Enzymatic Hydrolysis of Polyethylene Terephthalate Films in an Ultrafiltration Membrane Reactor. J. Memb. Sci. 2015, 494, 182–187. [Google Scholar] [CrossRef]
- Carniel, A.; Gomes, A.D.C.; Coelho, M.A.Z.; de Castro, A.M. Process Strategies to Improve Biocatalytic Depolymerization of Post-Consumer PET Packages in Bioreactors, and Investigation on Consumables Cost Reduction. Bioprocess Biosyst. Eng. 2021, 44, 507–516. [Google Scholar] [CrossRef] [PubMed]
- De Queiros Eugenio, E.; Campisano, I.S.P.; de Castro, A.M.; Coelho, M.A.Z.; Langone, M.A.P. Experimental and Mathematical Modeling Approaches for Biocatalytic Post-Consumer Poly(Ethylene Terephthalate) Hydrolysis. J. Biotechnol. 2021, 341, 76–85. [Google Scholar] [CrossRef] [PubMed]
- CARBIOS Investor Day 2022 Presentation. 2022. Available online: https://www.carbios.com/en/regulated-information/page/2/ (accessed on 2 March 2023).
- Singh, A.; Rorrer, N.A.; Nicholson, S.R.; Erickson, E.; DesVeaux, J.S.; Avelino, A.F.T.; Lamers, P.; Bhatt, A.; Zhang, Y.; Avery, G.; et al. Techno-Economic, Life-Cycle, and Socioeconomic Impact Analysis of Enzymatic Recycling of Poly(Ethylene Terephthalate). Joule 2021, 5, 2479–2503. [Google Scholar] [CrossRef]
- Kawai, F. Emerging Strategies in Polyethylene Terephthalate Hydrolase Research for Biorecycling. ChemSusChem 2021, 14, 4115–4122. [Google Scholar] [CrossRef]
- Kawai, F. The Current State of Research on PET Hydrolyzing Enzymes Available for Biorecycling. Catalysts 2021, 11, 206. [Google Scholar] [CrossRef]
- Anishchenko, I.; Pellock, S.J.; Chidyausiku, T.M.; Ramelot, T.A.; Ovchinnikov, S.; Hao, J.; Bafna, K.; Norn, C.; Kang, A.; Bera, A.K.; et al. De Novo Protein Design by Deep Network Hallucination. Nature 2021, 600, 547–552. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lisanza, S.; Juergens, D.; Tischer, D.; Watson, J.L.; Castro, K.M.; Ragotte, R.; Saragovi, A.; Milles, L.F.; Baek, M.; et al. Scaffolding Protein Functional Sites Using Deep Learning. Science 2022, 377, 387–394. [Google Scholar] [CrossRef]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.J.; Milles, L.F.; Wicky, B.I.M.; Courbet, A.; de Haas, R.J.; Bethel, N.; et al. Robust Deep Learning–Based Protein Sequence Design Using ProteinMPNN. Science 2022, 378, 49–56. [Google Scholar] [CrossRef]
- Ferruz, N.; Heinzinger, M.; Akdel, M.; Goncearenco, A.; Naef, L.; Dallago, C. From Sequence to Function through Structure: Deep Learning for Protein Design. bioRxiv 2022, 21, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wicky, B.I.M.; Milles, L.F.; Courbet, A.; Ragotte, R.J.; Dauparas, J.; Kinfu, E.; Tipps, S.; Kibler, R.D.; Baek, M.; DiMaio, F.; et al. Hallucinating Symmetric Protein Assemblies. Science 2022, 378, 56–61. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Enzyme | Substrate | Reaction Conditions | TPA (mM) | Productivity (mgTPA L−1 h−1) | Reference |
|---|---|---|---|---|---|---|
| Idonella sakaiensis | PETase | PET film C c | T d, B d, pH 7.2, 50 nM, 96 h time reaction | 0.511 | 0.88 | [41] |
| PETase-W159H/S238F | 0.421 | 0.42 | ||||
| Chimera IsMHETase/IsPETase-MP20 | GF-PET C d | T d, B d, pH 7.5, 10% DMSO, 96 h reaction time | 1.5 | 2.6 | [106] | |
| IsPETase-S121E/D186H/R280A/R224Q/N233K | 19 gGF-PET L−1 C d | T c, B c, pH 8, 200 nM, 96 h reaction time | 33.8 a | 58.49 a | [51] | |
| Humicola insolens | Novozym® 51032 | 20 gPET L−1 C c | T b, B b, A c, pH 7, 10 mgprotein gPET−1, 336 h reaction time | ~45 | ~22.25 | [114] |
| 20 gPET L−1 C b | ~0.450 | ~0.22 | ||||
| 81 gPC-PET L−1 C b | T b, B b, A b, pH 8.95, 65 mgprotein gPET−1, 96 h reaction time | 64 | 110.75 | [59] | ||
| 81 gPC-PET L−1 C b | T c, B b, A b, pH 8.95, 32 mgprotein gPET−1, 96 h reaction time | 96.54 | 47.73 | [135] | ||
| 220 gPC-PET L−1 C b | T b, B b, A b, pH 7, 1 mgprotein mL−1, 96 h reaction time | 105.6 | 182.74 | [70] | ||
| Metagenome from leaf branch compost | LCC | 25 gPET L−1 | T b, B c, pH 8, 5 µg enzyme, 24 h reaction time | 28 | 193.82 | [112,139] |
| LCC-F243I/D238C/S283C/Y127G | 200 gPC-PET kg−1 C c | T b, B c, A b, pH 8, 3 mgenzyme gPET−1, 9.3 h reaction time | 935 a | 16,702.32 a | [87] | |
| LCC-F243I/D238C/S283C/Y127G | 0.6 gGF-PET L−1 C d | T b, B c, A b, pH 8, 0.5 μmol enzyme, 12 h reaction time | 0.76 | 10.52 | [119] | |
| LCC-F243I/D238C/S283C/Y127G-CBM | 1.23 | 17.03 | ||||
| LCC-F243I/D238C/S283C/Y127G | 100 gLC-PET L−1 C d | T b, B c, A b, pH 8, 3 mgICCG gPET−1, 144 h reaction time | 475 | 548 | [110] | |
| 100 gWC-PET L−1 C c | 124.9 | 144.09 | ||||
| 100 gHC-PET L−1 C b | 100 | 115.37 | ||||
| LCC-F243Y/D238C/S283C/Y127G | 200 gGF-PET L−1 C d | T b, B c, A b, pH 7.5, 12.5 mM enzyme, 96 h reaction time | 953 | 1649 | [113] | |
| LCC-F243Y/D238C/S283C/Y127G-TrCBM1 | 829 | 1435 | ||||
| Metagenome compost | PHL7 | 83 gGF-PET L−1 C d | T b, B b, A c, pH 8, 0.6 mgenzyme gPET−1, 16 h reaction time | ~95 | ~986.4 | [49] |
| PES-H1 | PET powder C c | T b, B b, A c, pH 8, 1 mgenzyme gPET−1, 24 h reaction time | ~75 | ~519.16 | [50] | |
| PES-H1-L92F/Q94Y | ~200 | ~1384.42 | ||||
| Thermobifida fusca | TfCut2 | PET nanoparticle | T b, B b, A c, pH 8.5, 50 µg mL−1 enzyme, 24 h reaction time | 0.6 | 4.15 | [66] |
| GF-PET C d | T b, B b, A b, pH 8, 18 µg mL−1 enzyme, 24 h reaction time | 12 | 83.07 | [134] | ||
| Thermobifida cellulosilytica | Thc_Cut2 | 20 gPET L−1 C b | T b, B c, A c, pH 8, 18 µg mL−1 enzyme, 30 h reaction time | ~0.018 | ~0.1 | [53] |
| Thc_Cut1 | 50 gPET L−1 C b | T b, B b, A c, pH 8, 5 µM enzyme, 96 h reaction time | 55 | 95.18 | [76] | |
| Thc_Cut1_koST | 62 | 107.29 | ||||
| Thc_Cut1-G63A/F210I/D205C/E254C/Q93G | 8.3 gGF-PET L−1 C d | T b, B c, A c, pH 8, 75 µg enzyme, 96 h reaction time | 34.7 | 60.05 | [54] | |
| Saccharomonospora viridis | Cut190Q138A/D250C-E296C/Q123H/N202H | GF-PET C d | T b, B c, pH 8.2, 2 µM enzyme, 72 h reaction time | 15 a | 34.61 a | [67,140] |
| Cut190**SS/L136F/Q138G/F255I * | 20 gPET/L C d | T b, B c, A c, pH 9, 2 µM enzyme, 72 h reaction time | 94.9 a | 218.97 a | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Rodríguez, J.A.; Rodríguez-Sotres, R.; Farrés, A. Determinants for an Efficient Enzymatic Catalysis in Poly(Ethylene Terephthalate) Degradation. Catalysts 2023, 13, 591. https://doi.org/10.3390/catal13030591
Castro-Rodríguez JA, Rodríguez-Sotres R, Farrés A. Determinants for an Efficient Enzymatic Catalysis in Poly(Ethylene Terephthalate) Degradation. Catalysts. 2023; 13(3):591. https://doi.org/10.3390/catal13030591
Chicago/Turabian StyleCastro-Rodríguez, José Augusto, Rogelio Rodríguez-Sotres, and Amelia Farrés. 2023. "Determinants for an Efficient Enzymatic Catalysis in Poly(Ethylene Terephthalate) Degradation" Catalysts 13, no. 3: 591. https://doi.org/10.3390/catal13030591