Abstract
In this study, Pt1Sn1 intermetallic nanoparticles (NPs) on SiO2/CeO2@SiO2 composites were located either on SiO2 or on CeO2@SiO2, thereby varying the average distance (intimacy) between metal sites and CeOx sites from “closest” to “nanoscale”. The catalytic performance of these catalysts was compared to dual-bed mixtures of Pt1Sn1@SiO2 and CeO2@SiO2 powders, which provided a “milliscale” distance between sites. Several beneficial effects on the catalytic performance of CO2-assisted oxidative dehydrogenation of C5-paraffins were observed when Pt1Sn1 nanoparticles were located on SiO2 in nanoscale proximity to the CeO2 sites, as opposed to Pt and Sn species located on CeO2@SiO2 with the closest proximity and milliscale intimacy between Pt1Sn1 and CeO2. The former catalysts exhibited the highest C5-paraffin conversion of 32.8%, with a C5 total olefin selectivity of 68.7%, while the closest-proximity sample had a lower conversion of 17.4%, with a C5 total olefin selectivity of 20.9%. The FT-IR (Fourier transform infrared spectroscopy) spectroscopic study of the CO adsorption and X-ray photoelectron spectroscopy results revealed that the closest proximity between Pt and Ce inhibited PtSn alloy formation due to their strong interaction. However, for the nanoscale-proximity sample, neighboring CeO2@SiO2 did not disturb Pt1Sn1 intermetallic formation. This strategy can be applied to other CO2 activation catalysts, instead of CeO2@SiO2. This paper aims to provide insights into the influence of metal–CeOx intimacy in bi-functional catalysts.
1. Introduction
CO2-assisted oxidative dehydrogenation (CO2-ODH) of alkane to the corresponding olefins (ethylene, propylene, and styrene) involves the use of CO2 as a soft oxidant, which serves as one of the strategies for CO2 utilization [1,2,3,4,5]. The CO2-ODH process can offer an alternative to the catalytic non-oxidative dehydrogenation process used in various industries. This method can lead to an enhanced thermodynamic yield of olefins by removing H2 through reverse water–gas shift reactions (i.e., H2 + CO2 → H2O + CO) [2,5]. Furthermore, CO2 can improve the catalyst lifetime by facilitating coke removal via the reverse Boudouard reaction [2,6].
Naphtha steam cracking is a process that involves heating naphtha in the presence of steam at high temperatures (750–950 °C) to produce light olefins, which serve as feedstocks for polyethylene and polypropylene. Typically, ethylene constitutes around 57% of the cracked products, while propylene accounts for approximately 39%, although these percentages may vary based on the specific process conditions and naphtha composition [7]. In addition to these light olefins, naphtha cracking also generates a mixture of saturated hydrocarbons, including pentane and hexane. Typically, these heavier hydrocarbons (mostly C5 and C6) are either recycled back into the naphtha cracking process or added to the gasoline pool, both of which have relatively low value. In this regard, there is a high demand for converting these heavier hydrocarbons into value-added products.
C5 olefins (n-pentene and its isomers) are of immense importance in the petrochemical industry as raw materials in manufacturing a vast array of products comprising rubber, elastomers (polyisoprene, butyl rubber, etc.), fine chemicals, hydrocarbons, and resins [8]. ODH of C5-paraffin to olefin is a cost-effective process that can overcome the thermodynamic limitations of endothermic dehydrogenation. However, only a limited number of articles have been reported on C5-paraffin ODH. This is due to the observation that the presence of pure oxygen negatively affects the selectivity and product distribution [8,9,10]. To address the limitations of using oxygen as an oxidant in ODH, CO2 has been identified as a more viable alternative. However, CO2 activation remains a concern. This is because dehydrogenation of C5-paraffin requires a reaction temperature of approximately 500 °C to attain high selectivity, which is significantly lower than the typical ODH reaction temperature (~600 °C) when using ethane and propane as reactants. Consequently, under these conditions, the extent of CO2 activation is remarkably reduced compared to the light-alkane dehydrogenation process.
Over the past two decades, Pt-based bimetallic catalysts have been extensively studied in the dehydrogenation of lower alkanes. Pt-based bimetallic catalysts featuring rare-earth elements (REEs) as secondary metals have also been utilized very recently by our groups [2,11]. Among them, supported Pt-Sn alloys are extensively reported catalysts in ethane and propane dehydrogenation, exhibiting high catalytic activity, olefin selectivity, and improved catalyst durability [12,13,14,15,16,17,18]. Based on these observations, we explored the combination of CeO2 and silica-supported Pt-Sn (Pt-Sn@SiO2) as a bi-functional catalyst at lower temperatures (450–500 °C) for C5 ODH with CO2. We chose cerium oxide as the CO2 activator because various studies have reported that CeO2, having lattice oxygen and oxygen defects, can facilitate CO2 reduction during the dehydrogenation reaction [2,14]. However, at lower reaction temperatures, the significantly reduced activation of CO2 presents a challenge. To balance CO2 activation with alkane dehydrogenation, the cerium content should be increased, as its surface lattice oxygen can facilitate CO2 activation. Nonetheless, an excess of cerium can also lead to CO2 dry reforming reactions, which favor C–C bond cleavage. To address this limitation, the proximity between CeO2 and Pt-Sn@SiO2 was investigated. Scheme 1 presents the various integration methods used, including physical mixing, co-impregnation, ball milling, and a dual-bed catalyst (details in the experimental section), in order to achieve superior catalytic activity and high C5 olefin selectivity.
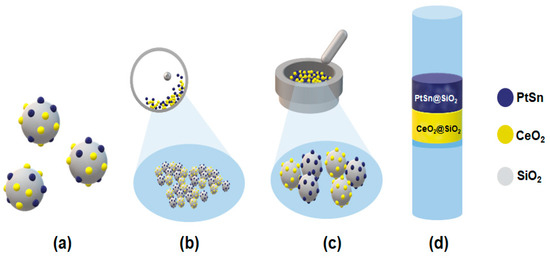
Scheme 1.
Illustration of different arrangements of the proximity between Pt1Sn1 and CeO2 in a bi-functional catalyst: (a) co-impregnation; (b) ball milling; (c) physical mixing; (d) dual-bed catalyst.
2. Results and Discussion
2.1. Texture and Structural Properties of 1Pt-0.6Sn@SiO2 Catalyst
Figure S1a shows the XRD (X-ray diffraction) pattern for the SiO2 support (CARiACT Grade G-6), which exhibited a broad peak at around a 2θ of 22.6°, indicating the amorphous nature of SiO2 [13]. The N2 adsorption–desorption isotherm result (Figure S1b) shows that the SiO2 support exhibited an increase in N2 adsorption in the region of 0.45 < P/P0 < 0.8, which demonstrates the mesoporous nature of SiO2, resulting in a high specific surface area of 500 m2 g−1, with a pore volume of 0.77 cm3 g−1. On this silica support, 1 wt% Pt and 0.6 wt% Sn were supported using the incipient wetness method and subsequently activated in O2 at 350 °C, followed by H2 reduction at 550 °C, according to the previous literature [12]. The resulting material was designated 1Pt-0.6Sn@SiO2 (details in experimental Section 3.2.1). N2 adsorption–desorption isotherm analysis was also employed to verify the textural characteristics of the 1Pt-0.6Sn@SiO2 catalyst. As shown in Figure 1a, similar to pristine SiO2, the region of 0.45 < P/P0 < 0.8 showed an increase in N2 adsorption, resulting from the available mesoporous architecture, which exhibited a typical type IV pattern. 1Pt-0.6Sn@SiO2 had a large surface area of 486 m2 g−1, with a pore volume of 0.77 cm3 g−1. For the reduced 1Pt-0.6Sn@SiO2 catalyst (Figure 1b), the XRD results with a deep scan in the range of a 2θ of 35–47° show the characteristic peaks centered at a 2θ of ~41.8° and 44.1°, which represent the intermetallic compounds of Pt1Sn1 [12]. Except for this, no XRD peaks corresponding to Pt and Sn were detected. The particle size calculated from the peak centered at a 2θ of 41.8° using the Scherrer equation (D = Kλ/β.cosθ) was ~2 nm, reflecting the presence of intermetallic NPs composed of Pt and Sn [12]. Additionally, STEM (scanning transmission electron spectroscopy) analysis was conducted to investigate the dispersion of the Pt1Sn1 intermetallic NPs. Figure 1c shows uniformly dispersed NPs throughout the SiO2 support. The particle size distribution was obtained by selecting more than 50 particles (Figure S2), and the average particle size was ~2 nm in diameter, which is well-matched to the XRD result. The reduction properties of the catalysts were measured using H2-TPR (H2 temperature-programmed reduction) analysis (Figure S3). The peak observed at 176 °C is related to the co-reduction of Pt and Sn species supported on SiO2. According to the previous literature, when Pt and Sn were loaded on MCM-41 silica with a Sn/Pt weight ratio of less than 0.6, a hydrogen consumption peak was observed at approximately ~180 °C, attributed to the co-reduction of Pt and Sn [19]. This co-reduction process is facilitated by the reduction of Sn species, aided by the spillover of hydrogen from Pt metal nanoparticles. Furthermore, the XPS (X-ray photoelectron spectroscopy) analysis results were obtained to understand the oxidation state of Pt and Sn species. The binding energies around 71.4 eV for 4f7/2 and 74.7 eV for 4f5/2 for the 1Pt-0.6Sn@SiO2 catalyst revealed the presence of metallic Pt0 (Figure 1d) [13,14,20]. On the other hand, the Sn 3d XPS spectra were deconvoluted to study the oxidation state of Sn. An XPS peak centered at 485.7 eV with a small peak at 486.7 eV was observed, corresponding to Sn0 and Sn2+/4+ species, respectively (Figure 1d) [13]. The quantitative ratio between Sn0 and Sn2+/4+ was calculated as 2.6.
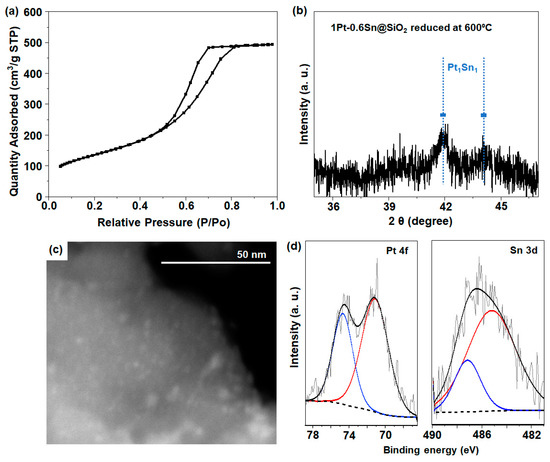
Figure 1.
(a) N2 isotherm, (b) XRD pattern, (c) STEM image, and (d) XPS spectra for 1Pt-0.6Sn@SiO2.
The XRD analysis of the 75CeO2@SiO2 sample returned characteristic peaks at a 2θ of 28.9°, 33.1°, 47.8°, and 56.3°, which were assigned to the (111), (199), (220), and (311) crystal planes of the cubic fluorite structure of CeO2, respectively (Figure 2a) [13]. The CeO2 particle size calculated from the XRD peak 2θ = 28.9° was ~4 nm. The N2 adsorption–desorption isotherm of 75CeO2@SiO2 showed an increase in N2 adsorption in the 0.45 < P/P0 < 0.8 region, indicating the presence of mesopores (Figure 2b). The catalyst had a comparatively lower surface area of 113.3 m2 g−1, with a pore volume of 0.1 cm3 g−1. Moreover, H2-TPR analysis was performed (Figure S3), where the reduction peak (appearing as an edge) at 410 °C was attributed to the removal of surface oxygen from small-sized ceria NPs situated on the SiO2 surface [21]. The broad peaks of H2 consumption at ~530 °C and above 760 °C indicated the reduction of lattice oxygen from the surface and bulk ceria.
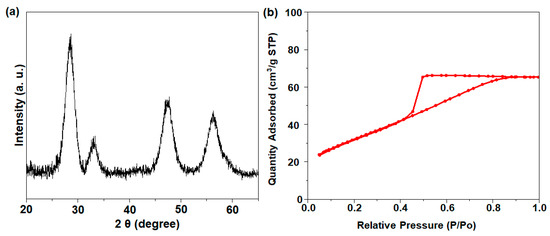
Figure 2.
(a) XRD pattern and (b) N2 isotherm result for 75CeO2@SiO2.
2.2. Effect of Proximity between Ceox and Pt/Sn Species on Intermetallic NP Formation
We studied the effect of the proximity between Pt/Sn and CeO2 in the bi-functional catalyst on the formation of intermetallic compounds between Pt and Sn. This study is based on the assumption that Pt species are known to strongly interact with CeO2−x via strong metal–support interactions [13,14]. Given this premise, when 1 wt% PtOx, 0.6 wt% SnOx, and 75 wt% CeO2 are simultaneously supported on SiO2, PtOx might preferentially be located on CeO2 without direct contact with SnOx. This could potentially result in the absence of intermetallic compound formation. To this end, we prepared two catalysts: one involving the co-impregnation of Pt, Sn, and Ce precursors together on a silica support, and another consisting of a physical mixture of equal quantities of 1Pt-0.6Sn@SiO2 and 75CeO2@SiO2 (Scheme 1a–c). Accordingly, the resulting catalysts were designated co-impregnated catalysts, and physically mixed catalysts, respectively.
The co-impregnated and physically mixed catalysts were characterized using various techniques. The XRD patterns obtained for both catalysts showed characteristic peaks related to cerium oxide (Figure S4). From the XRD results, the CeO2 particle size was calculated for the representative peak (2θ = 28.9°), where the co-impregnated catalyst had a particle size of 3.01 nm, and the physically mixed sample had a particle size of 4 nm. Based on the STEM analysis images (Figure S5), the regions of high intensity correspond to areas with a high concentration of CeO2. Distinguishing Pt and Sn from CeO2 particles is challenging due to the dominance of CeO2 particles. For the co-impregnated catalyst, the CeO2 particles appeared to be homogeneously dispersed; however, it is difficult to determine the particle size of Pt-based nanoparticles owing to the high content of cerium and its random arrangement. A similar situation was observed for the physically mixed catalysts.
The electronic properties of both the physically mixed and co-impregnated catalysts were studied using XPS analysis, as shown in Figure 3a,b. Figure 3a displays the XPS spectra for Pt 4f, indicating that Pt species were present in metallic form in both catalyst types. However, in the case of Sn 3d, we observed a notable difference between the two catalysts. As compared to 1Pt-0.6Sn@SiO2, for the co-impregnated catalyst, the Sn 3d XPS spectra after deconvolution showed that the intensity of the peak related to Sn0 decreased, and that for Sn2+/4+ at a higher binding energy increased significantly (Figure 3b). The quantitative ratio between Sn0 and Sn2+/4+ was 0.432, which was about 6 times lower than that of the 1Pt-0.6Sn@SiO2 catalyst. This indicates that the reduction of SnOx was prevented by the addition of CeOx. This observation could be explained by the stabilization effect of CeO2 on Pt or Sn species. Pt NPs could strongly interact with CeO2 through a strong metal–support interaction, preventing direct contact between Pt NPs and SnOx. Under this situation, facilitation of the reduction of SnOx by spillover hydrogen might also be inhibited [13]. In addition to this, tin oxides might be effectively stabilized by CeO2 via mixed metal oxide formation. In contrast to the co-impregnated catalysts, the XPS result obtained for the physically mixed sample was similar to that of 1Pt-0.6Sn@SiO2. The calculated Sn0-to-Sn2+/4+ ratio was 3.5, indicating that microscale intimacy between Pt/Sn and CeO2 does not interfere with the formation of intermetallic compounds.
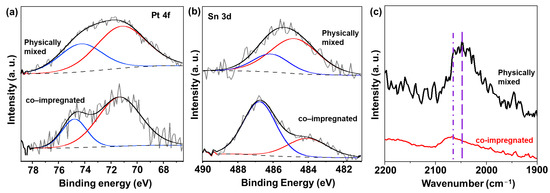
Figure 3.
XPS spectra of (a) Pt 4f and (b) Sn 3d, and (c) CO-FTIR results of physically mixed and co-impregnated catalysts.
To investigate the surface arrangement of Pt atoms on metal nanoparticles for the two catalysts, in situ CO-FTIR analysis was performed. For the co-impregnated catalyst, the peak observed at 2066 cm−1 was attributed to highly coordinated, medium-sized ensembles of Pt atoms (Figure 3c) [22,23]. Additionally, the small peak at ~2080 cm−1 pertains to a collection of Pt atoms arranged in a similar manner to unobstructed, large ensembles [23,24]. This can be explained as follows: With the addition of Ce in the co-impregnation method, the strong interaction between Pt and Ce hindered the formation of the Pt-Sn alloy. As a result, pure Pt nanoparticles were formed. On the other hand, the physically mixed catalyst exhibited a peak centered at 2046 cm−1 (related to Pt-Sn intermetallic NPs) [13,14]. The Pt-Sn NPs are expected to exhibit electron transfer from Sn to Pt due to their electronegativity difference, which leads to a higher degree of Pt electron back-donation into the 2π* antibonding orbital of the CO molecules. This phenomenon accounts for the peak shift towards a lower wavenumber, compared to the pure Pt NPs. In the physically mixed catalyst, in contrast to the co-impregnated catalyst, CeO2 could not impede the formation of Pt-Sn NPs due to the long distance between CeO2 and PtSn. Consequently, the physically mixed catalyst enabled the formation of Pt-Sn NPs.
2.3. C5 Oxidative Dehydrogenation with CO2: Effect of Nanoscale Intimacy between Ce and PtSn Catalysts
Before studying the effect of the special arrangement of the bi-functional catalysts, different parameters including the Ce content (Figure S6), CO2 partial pressure (Figure S7), and reaction temperature (Figure S8) effects were inspected to optimize the reaction conditions. These studies were performed using granules of both catalysts, physically mixed. It was observed that mixed 1Pt-0.6Sn@SiO2 and 75CeO2@SiO2, with a feed flow of Ar:CO2:C5 = 4:4:1 at 500 °C, had better activity with high C5 olefin (mono-olefins + dienes) selectivity. These conditions were applied in the following study.
The effect of the proximity of the silica-supported Pt-Sn and CeO2 catalysts was investigated in the C5 ODH reaction. For the catalytic activity, the conditions were fixed as follows: T = 500 °C; Ar: CO2: C5 molar ratio = 4:4:1 with a total flow rate of 18 mL/min. In the case of the 1Pt-0.6Sn@SiO2 catalyst, C5 conversion of 22.9% (1 h time on stream) with 71.4% C5 total olefin selectivity and 16.3% yield was obtained. However, after the addition of CeO2 in a physically mixed manner, excellent catalytic activity with 32.8% C5 conversion, 68.7% C5 total olefin selectivity, and 22.5% yield was obtained (Figure 4). The introduction of cerium enhanced the C5 ODH, but the selectivity was slightly decreased. The increase in C5 conversion can be attributed to the increase in CO2 conversion (Figure 4d), with CO2 performing as a soft oxidant activated by CeO2. On the other hand, the co-impregnated catalyst had a minimal conversion of 17.4% and C5 total olefin selectivity of 20.9%, with a 3.6% yield, which was even lower than that of 1Pt-0.6Sn@SiO2. For all studied catalysts, the by-products formed included CH4, CnH2n+2, and CnH2n (n = 2, 3, 4) with a collective selectivity of ~7%, as well as other unknown products. Considering the overall catalytic activity, an increasing trend of co-impregnated << 1Pt-0.6Sn@SiO2 < physically mixed was observed.
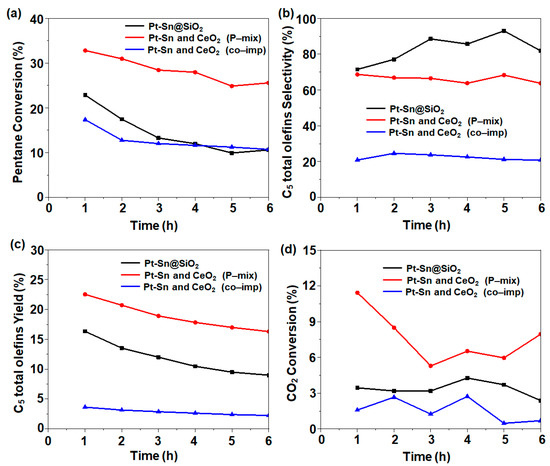
Figure 4.
(a) C5 conversion, (b) C5 total olefin selectivity, (c) C5 total olefin yield, and (d) CO2 conversion obtained using different integration methods. Reaction conditions: temperature = 500 °C; molar flow ratio of CO2:Ar:C5 = 4:4:1; catalyst amount, 0.2 g.
To determine the reason for the significant difference in the catalytic performance, particularly between the physically mixed and co-impregnated samples, the catalysts were characterized using XPS and CO-FTIR (see Section 2.2). The XPS analysis results of the physically mixed granules showed that the Pt-Sn intermetallic NPs were not disturbed (Figure 3a,b), which was further confirmed using CO-FTIR (Figure 3c), where a peak related to CO chemisorbed on Pt1Sn1 was observed. It is well-known that Pt-Sn intermetallic NPs usually result in C–H bond breaking in alkane dehydrogenation with the concomitant generation of H2. The addition of CeO2@SiO2 can catalyze the reverse water–gas shift reactions, where CO2 reacts with the generated H2 to produce syngas. The removal of H2 might facilitate the C5-paraffin dehydrogenation reaction, leading to a dramatic increase in C5 conversion. In the case of the co-impregnated 1Pt-0.6Sn/75CeO2@SiO2 catalyst, the comparatively strong interaction between Ce and Pt, which formed a Pt–O–Ce bond instead [14], restricted the Pt and Sn interaction to form intermetallic NPs. This resulted in SnO2 formation, as confirmed in the XPS analysis (Figure 3b). The CO-FTIR results also showed the formation of large Pt ensembles, which prefer C–C bond cleavage over C–H bond cleavage (Figure 3c) [2]. Therefore, the co-impregnated samples exhibited a lower olefin selectivity as compared to 1Pt0.6Sn@SiO2.
We further controlled the nanoscale intimacy between PtSn and CeO2 by shortening the distance between the two components from milliscale to nanoscale. To this end, we followed different treatments including a dual-bed combination, physical mixing, and ball milling (Scheme 1). Considering other possible distributions of the bi-functional catalyst, the dual-bed combination and ball milling were also investigated for the CO2-ODH of pentane, which returned a C5 conversion of 25.9% and 24.7% with a C5 total olefin selectivity of 62.3% and 62%, respectively (Figure 5). These activities were lower compared to those of the physically mixed catalyst. The dual-bed combination had slightly higher activity compared with 1Pt-0.6Sn@SiO2, which could be due to the limited interaction between the two bed surfaces inside the reactor. On the other hand, the ball milling process damaged the pore structure of the 1Pt-0.6Sn@SiO2 and 75CeO2@SiO2 samples, leading to a decrease in the specific surface area from ~486 m2 g−1 to 147 m2 g−1 (see below). Due to the collapse of the structure, the ball milling catalysts might exhibit lower activity than the physically mixed catalysts.
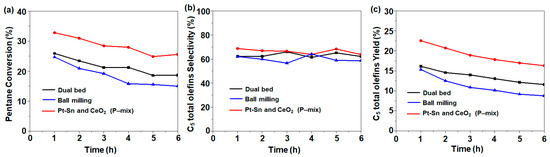
Figure 5.
(a) C5 conversion, (b) C5 total olefin selectivity, and (c) C5 total olefin yield following different catalyst arrangements. Reaction conditions: temperature = 500 °C; molar flow ratio of CO2:Ar:C5 = 4:4:1; catalyst amount, 0.2 g.
It is well-known that Cu supported on alumina (Cu/Al) promotes reverse water–gas shift reactions [25,26]. In this study, we also examined the effect of Cu/Al in the C5 ODH reaction. Zinc (Zn) was added to enhance the Cu dispersion. For comparison, Ce-Zn/Al was also prepared. These catalysts were individually mixed with 1Pt-0.6Sn@SiO2 and were analyzed for C5 ODH (Figure 6). The catalytic results show that with the addition of Ce-Zn/Al, the initial activity (1 h time on stream) was almost doubled, with a C5 conversion of 44%, but continuously decreased with the time on stream. On the other hand, the bi-functional catalyst composed of 1Pt-0.6Sn@SiO2 and Cu-Zn/Al returned an initial conversion of 22% (1 h time on stream), where slow deactivation was observed after 2 h of time on stream. These results indicate that the addition of a metal, which has the ability to actively participate in the reverse water–gas shift (RWGS) reaction, drives the equilibrium concentration towards the products, leading to an enhanced yield of C5 olefins.
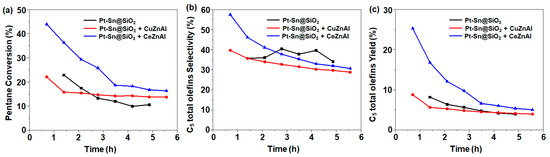
Figure 6.
(a) C5 conversion, (b) C5 total olefin selectivity, and (c) C5 total olefin yield. Reaction conditions: temperature = 500 °C; molar flow ratio of CO2:Ar:C5 = 4:4:1; catalyst amount, 0.2 g.
3. Materials and Methods
3.1. Materials
The following materials were used: Chloroplatinic acid hexahydrate (H2PtCl6·6H2O, ACS reagent, ≥37.50% Pt basis, Aldrich, St. Louis, MO, USA), Tin(II) chloride dihydrate (SnCl2·2H2O, ACS reagent, 98% trace metal basis Thermoscientific, Shanghai, China), Ammonium Cerium (IV) nitrate (Ce(NH4)2(NO3)6, ACS reagent, 99%), LUDOX-AS-40 (Aldrich, St. Louis, MO, USA), CARiACT Grade G-6 (denoted as SiO2) (size: 75–250 µm, Fuji Silysia Chemical LTD, Japan), and Cerium oxide (99.99%) 10 nm, CAS NO: 1306-38-3 (Avention, Siheung, Republic of Korea).
3.2. Catalyst Preparation
3.2.1. Preparation of Pt-Sn Supported on SiO2 (1Pt-0.6Sn@SiO2)
The silica-supported Pt-Sn catalyst was prepared using the incipient wetness impregnation method reported by Motagamwala et al. [12]. Firstly, H2PtCl6·6H2O and SnCl2·2H2O sources were stirred in 2 mL of 0.1 M HCl solution for 3 h, followed by the addition of 1 mL deionized water. The solution obtained was loaded on the SiO2 support. This sample was collected vacuum-dried for 12 h at room temperature, and calcined in air for 2 h at 350 °C. The calcined catalyst was labeled as 1Pt-0.6Sn@SiO2 and stored in a desiccator. The Pt content was 1 wt%, and that of Sn was 0.6 wt%; this ratio was maintained constant throughout the study.
3.2.2. Preparation of CeO2@SiO2 Catalyst
CeO2@SiO2 catalysts were prepared following a simple two-step procedure. Generally, an appropriate amount of the cerium source (Ce(NH4)2(NO3)6) was stirred in 5 g of H2O. After 20 min, 1 g of LUDOX-AS-40 was added in a dropwise manner. After 30 min of stirring, diluted ammonia solution (35%) was added dropwise to the reaction mixture to attain a pH of ~8–10. After filtration and washing with distilled water, the sample was dried at 100 °C for 12 h, followed by calcination at 500 °C for 4 h. Samples with different cerium contents were prepared and labeled as xCeO2@SiO2 (x = 5, 25, 50, 75, 100), characterized, and stored for further use.
3.2.3. Preparation of Pt-Sn/CeO2@SiO2 Catalyst (Co-Impregnated)
For the co-impregnated sample, Pt-Sn solution was prepared following the procedure discussed in Section 3.2.1 and subsequently applied to 75CeO2@SiO2 via incipient wetness impregnation. The remaining protocol was the same as that discussed in Section 3.2.1. The catalyst prepared was labeled as a co-impregnated catalyst.
3.3. Characterization
N2 adsorption–desorption was measured using a Micrometrics Tristar II 3020 instrument (Norcross, GA, USA) at −196 °C. All samples were degassed at 250 °C for 3 h before analysis. The Brunauer–Emmett–Teller (BET) equation was used for the surface area calculation using an adsorption branch obtained within a relative pressure (P/P0) range of 0.05 to 0.3. The powder X-ray diffraction (XRD) patterns were investigated using a Rigaku Miniflex instrument (Rigaku corporations, Tokyo, Japan) at 30 kV and 15 mA with a Cu Kα source (λ = 0.154 nm). STEM images were obtained using a FEI Titan environmental transmission electron microscope (ETEM) G2 (Hillsboro, OR, USA) at an acceleration voltage of 300 kV. X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Scientific (NEXSA) instrument (Waltham, MA, USA)with an Al–Kα source (1486.6 eV). The in situ CO-FTIR was analyzed using a Shimadzu IR Tracer-100 spectrometer (Kyoto, Japan), with an in situ IR cell. H2-TPR analysis was performed on catalysts using a BELCAT-M instrument (MicrotracBEL, Osaka, Japan). The catalyst was pretreated in an Ar flow at 300 °C for 1 h and cooled, and the analysis was carried out in a 10%H2/Ar flow at 50mL/min. The temperature was increased to 800 °C with a ramping rate of 5 °C/min.
3.4. Catalytic Test
The C5 ODH was performed at 500 °C using a fixed-bed downflow quartz reactor (L = 45 cm, i.d. = 1 cm). Before the activity measurement, 0.2 g of catalyst (0.1 g 1Pt-0.6Sn@SiO2 and 0.1 g 75CeO2@SiO2) was exposed to 10 vol.%H2/Ar at 550 °C. After holding for 2 h, the 10 vol.%H2/Ar flow was switched with Ar, and the reactor was cooled to 500 °C. A laboratory-designed Pyrex bubbler was used for C5-alkane. The C5 saturator was set to −16 °C to obtain a weight hour space velocity (WHSV) = 3.88 h−1. A feed of Ar and CO2 with a total of 16 mL/min (8 mL each) was passed through the bubbler followed by the catalyst bed, and fixed in the center of the reactor with the feed composition of Ar:CO2:C5 = 4:4:1. The reactor outlet line was directly connected to an online GC (gas chromatography) system (Agilent: 7890B, Santa Clara, CA, USA) equipped with flame ionization and thermal conductivity detectors. For the hydrocarbon separation, an HP-Al/S column was used, and for the CO2 and CO separation, an Rt-U Bond capillary column from Restek (Bellefonte, PA, USA) was used.
The C5-alkane and CO2 conversion and the product selectivity were obtained using the equations below:
C5 conversion:
C5 olefin selectivity:
where represents different products formed in pentane oxidative dehydrogenation with CO2 (10 − x = number of hydrogens, and x = 0, 2).
The CO2 conversion was calculated using the equation below:
4. Conclusions
Mesoporous silica (SiO2)-supported Pt-Sn and CeO2 catalysts (1Pt-0.6Sn@SiO2 and 75CeO2@SiO2) were prepared and utilized for C5 ODH using different integration patterns to obtain better activity with high C5 olefin selectivity. Among all the catalysts, the physically mixed bi-functional catalyst returned a higher C5 conversion of 32.8%, with a C5 total olefin selectivity of 68.7%, while for the co-impregnated catalyst, the activity was even lower than that of 1Pt-0.6Sn@SiO2. The XPS and CO-FTIR results confirmed that for the physically mixed catalyst, the Pt-Sn intermetallic NPs were not disturbed. Additionally, the existence of CeO2@SiO2 in the vicinity effectively enhanced the activity, which was also attributed to the rise in CO2 conversion, which enhanced the C5 ODH. On the other hand, in the case of the co-impregnated catalyst, the strong Pt–Ce interaction hindered the formation of Pt-Sn intermetallic NPs. Moreover, the addition of Cu-Zn/Al supplemented the RWGS reaction, which elevated the catalyst stability by pushing the equilibrium towards the products.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal13060933/s1, Figure S1. (a) XRD patterns and (b) N2-isotherm result for SiO2 support, Figure S2. Particle size distribution for 1Pt-0.6Sn@SiO2, Figure S3. H2-TPR for 1Pt-0.6Sn@SiO2, 75CeO2@SiO2 catalysts, Figure S4. XRD pattern of (a) co-impregnated catalyst and (b) physically mixed catalyst, Figure S5. STEM images of (a) co-impregnated catalyst and (b) physically mixed catalyst, Figure S6. C5 conversion, based on cerium content changed. Reaction conditions: temperature = 500 °C, CO2:Ar:C5 = 4:4:1, catalyst amount: 0.2 g, Figure S7. (a) C5 conversion, (b) C5 mono-olefins selectivity, and (c) C5 mono-olefins yield based on varying flow composition. Reaction conditions: temperature = 500 °C, molar flow ratio of CO2:Ar:C5 = x:y:1, (x = 1,2,4 and y = 7,6,4), catalyst amount: 0.2 g, Figure S8. (a) C5 conversion, (b) C5 mono-olefins selectivity, and (c) C5 mono-olefins yield, by varying reaction temperature. Reaction conditions: molar flow ratio of CO2:Ar:C5 = 4:4:1, catalyst amount: 0.2 g.
Author Contributions
Writing—original draft, figures, data collection, validation, and analysis, M.N.; data collection, analysis, and validation, G.L. and E.E.; STEM analysis, J.W.S.; data interpretation, study design, D.-H.C. and C.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research work was supported by the INHA University Research Grant (No. INHA-68949).
Data Availability Statement
All research data in this work is available from the corresponding author upon reasonable request.
Acknowledgments
This research work was supported by the INHA University Research Grant (No. INHA-68949).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gambo, Y.; Adamu, S.; Tanimu, G.; Abdullahi, I.M.; Lucky, R.A.; Ba-Shammakh, M.S.; Hossain, M.M. CO2-mediated oxidative dehydrogenation of light alkanes to olefins: Advances and perspectives in catalyst design and process improvement. Appl. Catal. A Gen. 2021, 623, 118273. [Google Scholar] [CrossRef]
- Numan, M.; Eom, E.; Li, A.; Mazur, M.; Cha, H.W.; Ham, H.C.; Jo, C.; Park, S.E. Oxidative dehydrogenation of ethane with CO2 as a soft oxidant over a PtCe bimetallic catalyst. ACS Catal. 2021, 11, 9221–9232. [Google Scholar] [CrossRef]
- Amghizar, I.; Vandewalle, L.A.; van Geem, K.M.; Marin, G.B. New Trends in Olefin Production. Engineering 2017, 3, 171–178. [Google Scholar] [CrossRef]
- Najari, S.; Saeidi, S.; Concepcion, P.; Dionysiou, D.D.; Bhargava, S.K.; Lee, A.F.; Wilson, K. Oxidative dehydrogenation of ethane: Catalytic and mechanistic aspects and future trends. Chem. Soc. Rev. 2021, 50, 4564–4605. [Google Scholar] [CrossRef] [PubMed]
- Numan, M.; Kim, T.; Jo, C.; Park, S.E. Ethane Dehydrogenation with CO2 as a soft oxidant over a Cr-TUD-1 catalyst. J. CO2 Util. 2020, 39, 101184. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Loizidis, C.; Heracleous, E.; Lemonidou, A.A. CO2-oxidative ethane dehydrogenation over highly efficient carbon-resistant Fe-catalysts. J. Catal. 2020, 388, 52–65. [Google Scholar] [CrossRef]
- Zacharopoulou, V.; Lemonidou, A.A. Olefins from biomass intermediates: A review. Catalysts 2017, 8, 2. [Google Scholar] [CrossRef]
- Valenzuela, R.X.; Muñoz Asperilla, J.M.; Corberán, V.C. Isoprene and C5 olefins production by oxidative dehydrogenation of isopentane. Ind. Eng. Chem.Res. 2008, 47, 8037–8042. [Google Scholar] [CrossRef]
- Ezinkwo, G.O.; Tretjakov, V.F.; Talyshinky, R.M.; Ilolov, A.M.; Mutombo, T.A. Overview of the Catalytic Production of Isoprene from different raw materials; Prospects of Isoprene production from bio-ethanol. Catal. Sustain. Energy 2013, 1, 100–111. [Google Scholar] [CrossRef]
- Huang, R.; Liang, C.H.; Su, D.S.; Zong, B.; Rong, J. The difference between borate and phosphate modified carbon nanotubes in isopentane oxidative dehydrogenation. Catal. Today 2015, 249, 161–166. [Google Scholar] [CrossRef]
- Ryoo, R.; Kim, J.; Jo, C.; Han, S.W.; Kim, J.C.; Park, H.; Han, J.; Shin, H.S.; Shin, J.W. Rare-earth–platinum intermetallic NPs NPs in mesoporous zeolite for catalysis. Nature 2020, 585, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Motagamwala, A.H.; Almallahi, R.; Wortman, J.; Igenegbai, V.O.; Linic, S. Stable and selective catalysts for propane dehydrogenation operating at thermodynamic limit. Science 2021, 373, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, G.Q.; Ren, X.; Liu, Z.W. CeO2-promoted PtSn/SiO2 as a high-performance catalyst for the oxidative dehydrogenation of propane with carbon dioxide. Nanomaterials 2022, 12, 417. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Q.; Ren, X.; Kondratenko, V.A.; Zhang, H.B.; Kondratenko, E.V.; Liu, Z.W. Promotional nature of Sn on Pt/CeO2 for the oxidative dehydrogenation of propane with carbon dioxide. Nano Res. 2023, 16, 6237–6250. [Google Scholar] [CrossRef]
- Wang, X.; Cui, J.; Zhang, N.; Song, J.; Fan, X.; Zhao, Z.; Kong, L.; Xiao, X.; Xie, Z. Propane Dehydrogenation over PtSn/Al2O3 Catalysts: Influence of Urea to Al(NO3)3·9H2O Ratio. Catalysts 2022, 12, 157. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, K.; Wang, C.; Ma, L.; Zhang, Q.; Liu, Q.; Chen, L.; Chen, L.; Zhang, Q.; Tian, Z. The properties and catalytic performance of PtSn/Mg(x-Ga)AlO catalysts for ethane dehydrogenation. RSC Adv. 2017, 7, 22836–22844. [Google Scholar] [CrossRef]
- Pham, H.N.; Sattler, J.J.; Weckhuysen, B.M.; Datye, A.K. Role of Sn in the regeneration of Pt/γ-Al2O3 light alkane dehydrogenation catalysts. ACS Catal. 2016, 6, 2257–2264. [Google Scholar] [CrossRef]
- Xiong, H.; Lin, S.; Goetze, J.; Pletcher, P.; Guo, H.; Kovarik, L.; Artyushkova, K.; Weckhuysen, B.M.; Datye, A.K. Thermally stable and regenerable platinum–tin clusters for propane dehydrogenation prepared by atom trapping on ceria. Angew. Chem. Int. Ed. 2017, 129, 9114–9119. [Google Scholar] [CrossRef]
- Coman, S.N.; Parvulescu, V.I.; De Bruyn, M.; De Vos, D.E.; Jacobs, P.A. Reduction of Prostaglandin Unsaturated Ketones to Secondary Allylic Alcohols by Hydrogen Transfer over Mesoporous-Supported PtSn Catalysts. J. Catal. 2002, 206, 218–229. [Google Scholar] [CrossRef]
- Ono, L.K.; Croy, J.R.; Heinrich, H.; Roldan Cuenya, B. Oxygen chemisorption, formation, and thermal stability of Pt oxides on Pt NPs supported on SiO2/Si (001): Size effects. J. Phys. Chem. C 2011, 115, 16856–16866. [Google Scholar] [CrossRef]
- Peng, H.; Dong, T.; Zhang, L.; Wang, C.; Liu, W.; Bao, J.; Wang, X.; Zhang, N.; Wang, Z.; Wu, P.; et al. Active and stable Pt-Ceria nanowires@ silica shell catalyst: Design, formation mechanism and total oxidation of CO and toluene. Appl. Catal. B Environ. 2019, 256, 117807. [Google Scholar] [CrossRef]
- Elgayyar, T.; Atwi, R.; Tuel, A.; Meunier, F.C. Contributions and limitations of IR spectroscopy of CO adsorption to the characterization of bimetallic and nanointermetallic NPs catalysts. Catal. Today 2021, 373, 59–68. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, J.; Wang, S.; Zhao, Y.; Ma, X. A well fabricated PtSn/SiO2 catalyst with enhanced synergy between Pt and Sn for acetic acid hydrogenation to ethanol. RSC Adv. 2016, 6, 51005–51013. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, H.; Ma, H.; Ying, W.; Fang, D. The effect of preparation method on the performance of PtSn/AlO catalysts for acetic acid hydrogenation. Pol. J. Chem. Technol. 2015, 17, 11–17. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Héroguel, F.; Kılıç, M.; Baranowski, C.J.; Artiglia, L.; Röthlisberger, U.; Luterbacher, J.S.; Kröcher, O. Cu–Al spinel as a highly active and stable catalyst for the reverse water gas shift reaction. ACS Catal. 2019, 9, 6243–6251. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Le Monnier, B.P.; Du, Y.P.; Héroguel, F.; Luterbacher, J.S.; Kröcher, O. Increasing the activity of the Cu/CuAl2O4/Al2O3 catalyst for the RWGS through preserving the Cu2+ ions. Chem. Commun. 2021, 57, 1153–1156. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).