
A Bio-Based Tackifier Synthesized by Room-Temperature Cationic Copolymerization of Isobutene and β-Pinene


Abstract

1. Introduction
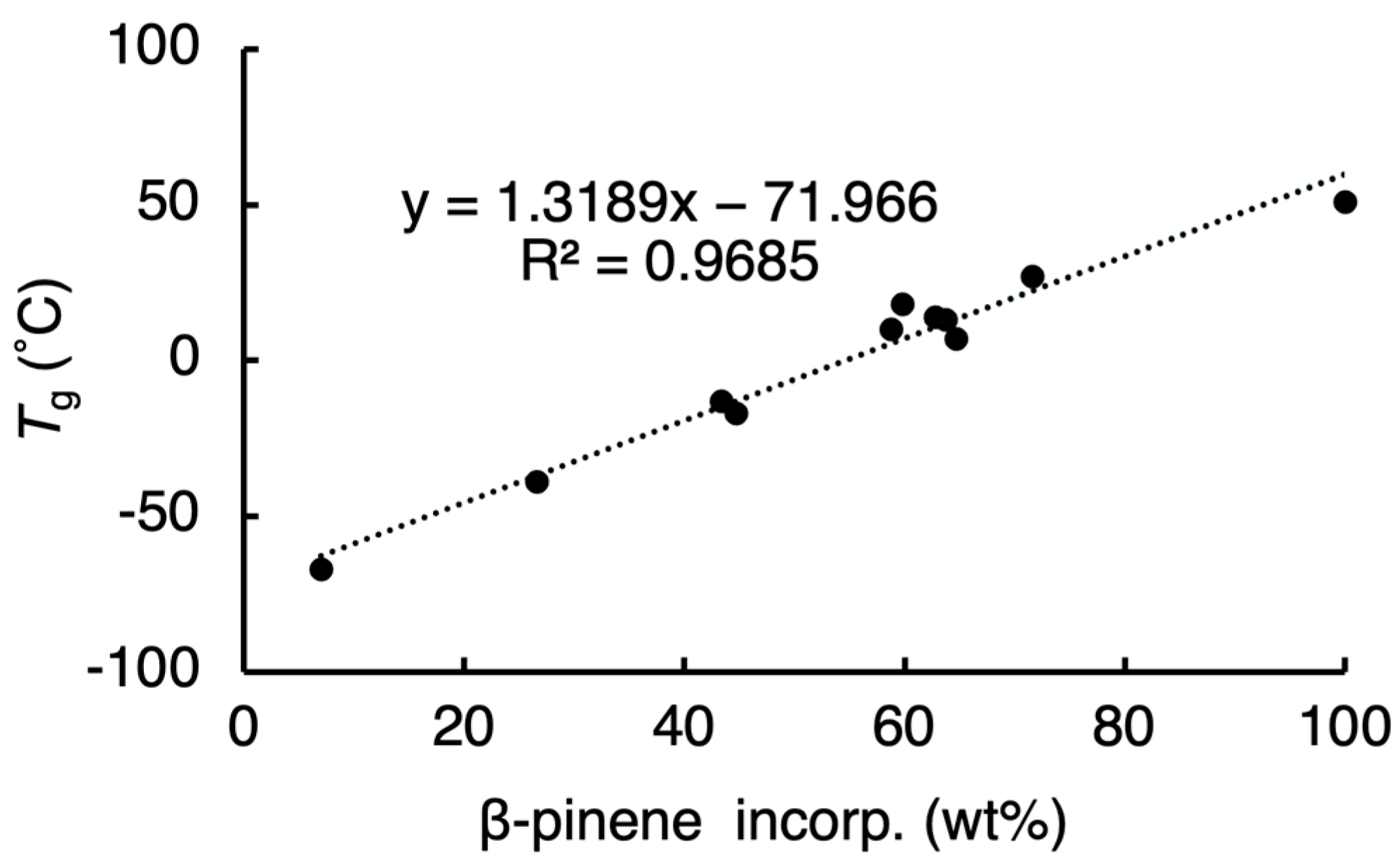
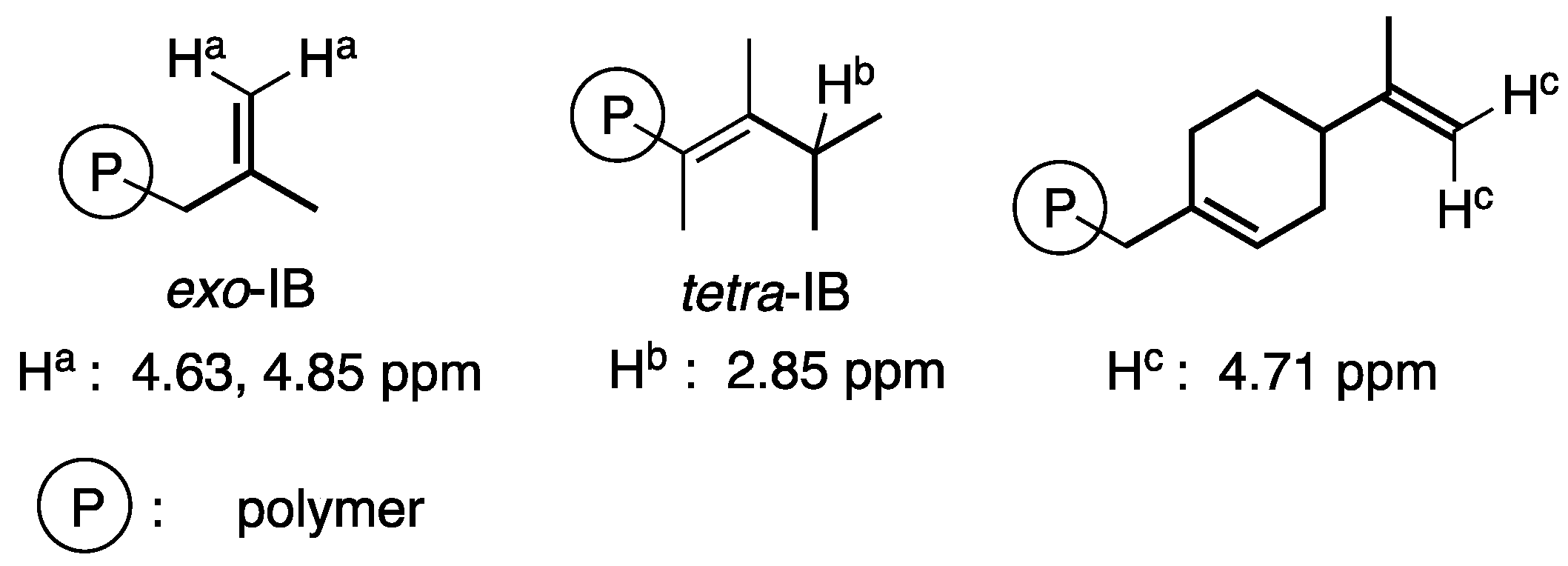
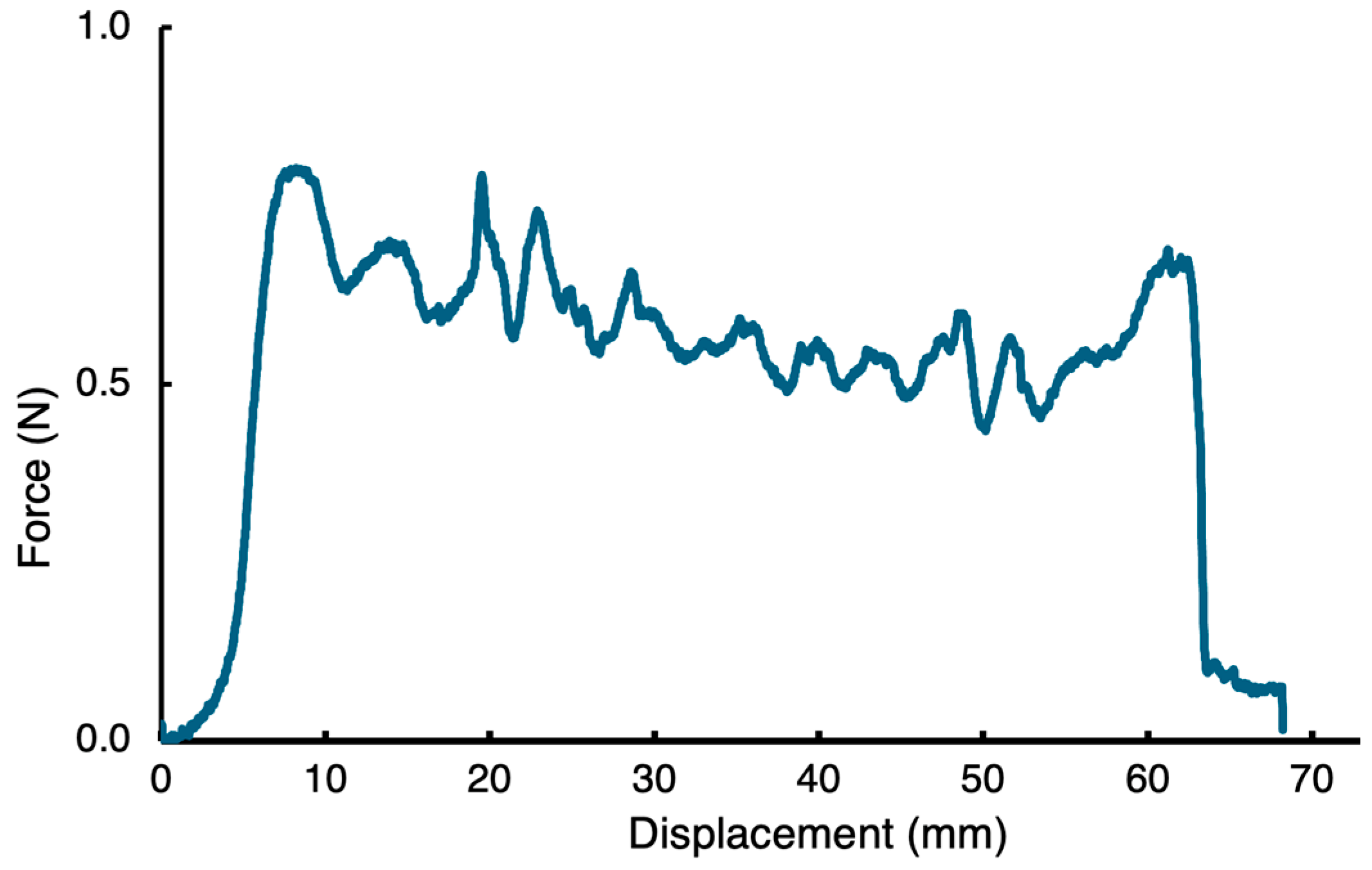
2. Results and Discussion
3. Experimental Section
3.1. Materials
3.2. Measurements
3.3. Copolymerization of β-Pinene and IB, Using AlCl3∙OiPr2 as a Catalyst
3.4. Copolymerization of β-Pinene and IB Using B(C6F5)3 as a Catalyst (Table 2, Run 13)
3.5. Sample Film Preparation for the Peeling Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilbon, P.A.; Chu, F.; Tang, C. Progress in Renewable Polymers from Natural Terpenes, Terpenoids, and Rosin. Macromol. Rapid Commun. 2013, 34, 8–37. [Google Scholar] [CrossRef] [PubMed]
- Winnacker, M. Pinenes: Abundant and Renewable Building Blocks for a Variety of Sustainable Polymers. Angew. Chem. Int. Ed. 2018, 57, 14362–14371. [Google Scholar] [CrossRef] [PubMed]
- Nyamwihura, R.J.; Ogungbe, I.V. The pinene scaffold: Its occurrence, chemistry, synthetic utility, and pharmacological importance. RSC Adv. 2022, 12, 11346. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living Cationic Isomerization Polymerization of β-Pinene. 1. Initiation with HCl-2-Chloroethyl Vinyl Ether Adduct/TiCl3(OiPr) in Conjunction with nBu4NCl. Macromolecules 1997, 30, 22–26. [Google Scholar] [CrossRef]
- Verebelyi, K.; Szabo, A.; Reti, Z.; Szarka, G.; Villanyi, A.; Ivan, B. Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions. Int. J. Mol. Sci. 2023, 24, 5170. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Sugiyama, H.; Kamigaito, M. Biomass-derived heat-resistant alicyclic hydrocarbon polymers: Poly(terpenes) and their hydrogenated derivatives. Green Chem. 2006, 8, 878–882. [Google Scholar] [CrossRef]
- Kukhta, N.A.; Vasilenko, I.V.; Kostjuk, S.V. Room temperature cationic polymerization of β-pinene using modified AlCl3 catalyst: Toward sustainable plastics from renewable biomass resources. Green Chem. 2011, 13, 2362–2364. [Google Scholar] [CrossRef]
- Satoh, K.; Nakahara, A.; Mukunoki, K.; Sugiyama, H.; Saito, H.; Kamigaito, M. Sustainable cycloolefin polymer from pine tree oil for optoelectronics material: Living cationic polymerization of β-pinene and catalytic hydrogenation of high-molecular-weight hydrogenated poly (β-pinene). Polym. Chem. 2014, 5, 3222–3230. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Liao, T.-P.; Guhaniyogi, S.; Chang, V.S.C. Poly(β-pinenes) carrying one, two, or three functional end groups. I. The effect of reaction conditions on the polymerization of β-pinene. J. Polym. Sci. Polym. Chem. 1982, 20, 3219–3227. [Google Scholar] [CrossRef]
- Destephen, A.; de San Román, E.G.; Martínez-Tong, D.E.; Ballard, N. Cationic Polymerization of β-Pinene Using B(C6F5)3 as a Lewis Acid for the Synthesis of Tackifiers in Pressure Sensitive Adhesives. Macromol. Mater. Eng. 2021, 306, 2100194. [Google Scholar] [CrossRef]
- Destephen, A.; Ballard, N. On the limitations of cationic polymerization of vinyl monomers in aqueous dispersed media. Polym. Chem. 2021, 12, 6444–6455. [Google Scholar] [CrossRef]
- Roberts, W.J.; Day, A.R. A Study of the Polymerization of α- and β-Pinene with Friedel-Crafts Type Catalysts. J. Am. Chem. Soc. 1950, 72, 1226–1230. [Google Scholar] [CrossRef]
- Karasawa, Y.; Kimura, M.; Kanazawa, A.; Kanaoka, S.; Aoshima, S. New initiating systems for cationic polymerization of plant-derived monomers: GaCl3/alkylbenzene-induced controlled cationic polymerization of β-pinene. Polym. J. 2015, 47, 152–157. [Google Scholar] [CrossRef]
- Hayatifar, M.; Marchetti, F.; Pampaloni, G.; Patil, Y.; Galletti, A.M.R. Room-temperature polymerization of β-pinene by niobium and tantalum halides. Catal. Today 2012, 192, 177–182. [Google Scholar] [CrossRef]
- Gaspar, A.S.; Cordeiro, J.B.; Simoes, P.A.; Gameiro, D.; Rocha, F.A.; Serra, A.C.; Coelho, J.F.J.; Fonseca, A.C. Poly(β-pinene) as an efficient biobased tackifier for metallocene poly(ethylene) based hotmelt adhesives. Int. J. Adh. Adh. 2022, 114, 103111. [Google Scholar] [CrossRef]
- Brandt, H.-D.; Nentwig, W.; Rooney, N.; LaFlair, R.T.; Wolf, U.U.; Duffy, J.; Puskas, J.E.; Kaszas, G.; Drewitt, M.; Glander, S. Rubber, 5. Solution Rubbers in Ullmann’s Encyclopedia of Industrial Chemitsry; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Faust, R.; Kennedy, J.P. Living Carbocationic Polymerization III. Demonstration of the Living Polymerization of Isobutylene. Polym. Bull. 1986, 15, 317–323. [Google Scholar] [CrossRef]
- Kaszas, G.; Puskas, J.E.; Chen, C.C.; Kennedy, J.P. Electron Pair Donors in Carbocationic Polymerization. 2. Mechanism of Living Carbocationic Polymerizations and the Role of in Situ and External Electron Pair Donors. Macromolecules 1990, 23, 3909–3915. [Google Scholar] [CrossRef]
- Storey, R.F.; Chisholm, B.J.; Brister, L.B. Kinetic Study of the Living Cationic Polymerization of Isobutylene Using a Dicumyl Chloride/TiCl4/Pyridine Initiating System. Macromolecules 1995, 28, 4055. [Google Scholar] [CrossRef]
- Fodor, Z.; Bae, Y.C.; Faust, R. Temperature effects on the living cationic polymerization of isobutylene: Determination of spontaneous chain-transfer constants in the presence of terminative chain transfer. Macromolecules 1998, 31, 4439–4446. [Google Scholar] [CrossRef]
- Storey, R.F.; Donnalley, A.B. Initiation effects in the living cationic polymerization of isobutylene. Macromolecules 1999, 32, 7003–7011. [Google Scholar] [CrossRef]
- Byrikhin, V.S.; Nesmelov, A.I.; Murachev, V.B.; Ezhova, E.A.; Orlinkov, A.V.; Akhrem, I.S. Cationic polymerization of hydrocarbon monomers under the action of acyl halide complexes with Lewis acids 4. Kinetics of cationic polymerization of isobutylene induced by the AcBr2AIBr3 complex. Russ. Chem. Bull. 1993, 42, 837–840. [Google Scholar] [CrossRef]
- Murachev, V.B.; Ezhova, E.A.; Nesmelov, A.I.; Byrikhin, V.S.; Orlinkov, A.V.; Akhrem, I.S. Cationic polymerization of hydrocarbon monomers induced by complexes of acyl halides with Lewis acids 8. The polymerization of isobutylene in the presence of superacids. Russ. Chem. Bull. 1997, 46, 934–938. [Google Scholar] [CrossRef]
- Harrison, J.J.; Mijares, C.M.; Cheng, M.T.; Hudson, J. Negative Ion Electrospray Ionization Mass Spectrum of Polyisobutenyl succinic Anhydride: Implications for Isobutylene Polymerization Mechanism. Macromolecules 2002, 35, 2494–2500. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, Y.X.; Zhang, Y.; Yan, P.F.; Xu, R.W. A cost-effective process for highly reactive polyisobutylenes via cationic polymerization coinitiated by AlCl3. Polymer 2010, 51, 5960–5969. [Google Scholar] [CrossRef]
- Vasilenko, I.V.; Shiman, D.I.; Kostjuk, S.V. Highly reactive polyisobutylenes via AlCl3·OBu2-coinitiated cationic polymerization of isobutylene: Effect of solvent polarity, temperature, and initiator. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 750–758. [Google Scholar] [CrossRef]
- Shiman, D.I.; Vasilenko, I.V.; Kostjuk, S.V. Cationic polymerization of isobutylene by complexes of alkylaluminum dichlorides with diisopropyl ether: An activating effect of water. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 2386–2393. [Google Scholar] [CrossRef]
- Vasilenko, I.V.; Nikishev, P.A.; Shiman, D.I.; Kostjuk, S.V. Cationic polymerization of isobutylene in toluene: Toward well-defined exo-olefin terminated medium molecular weight polyisobutylenes under mild conditions. Polym. Chem. 2017, 8, 1417–1425. [Google Scholar] [CrossRef]
- Rajasekhar, T.; Singh, G.; Kapur, G.S.; Ramakumar, S.S.V. Recent advances in catalytic chain transfer polymerization of isobutylene: A review. RSC Adv. 2020, 10, 18180. [Google Scholar] [CrossRef] [PubMed]
- Nifant’ev, I.E.; Korchagina, S.A.; Chinova, M.S.; Tavtorkin, A.N. Polyisobutylenes with Controlled Molecular Weight and Chain-End Structure: Synthesis and Actual Applications. Polymers 2023, 15, 3415. [Google Scholar] [CrossRef]
- Mitu, S.; Baird, M.C. Isobutene polymerization induced by adducts of B(C6F5)3 with carboxylic acids CH3(CH2)nCO2H (n = 2, 4, 6, 8, 10, 12, 14, 16, 18, 20); chain length effects on this strong acid, carbocationic polymerization initiating system. Eur. Polym. J. 2006, 42, 2039–2044. [Google Scholar] [CrossRef]
- Ott, E. Terpene Resins. U.S. Patent 2,373,706, 17 April 1945. [Google Scholar]
- Kennedy, J.P.; Chou, T. Poly(isobutylene-co-β-pinene). A new sulfur vulcanizable, ozone resistant elastomer by cationic isomerization copolymerization. Adv. Polym. Sci. 1976, 21, 1. [Google Scholar]
- Li, A.L.; Zhang, W.; Liang, H.; Lu, J. Living cationic random copolymerization of β-pinene and isobutylene with 1-phenylethyl chloride/TiCl4/Ti(OiPr)4/nBu4NCl. Polymer 2004, 45, 6533–6537. [Google Scholar] [CrossRef]
- Dimitrov, P.; Emert, J.; Faust, R. Polymerization of Isobutylene by AlCl3/Ether Complexes in Nonpolar Solvent. Macromolecules 2012, 45, 3318–3325. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) Polymer Handbook, 4th ed.; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Guo, A.; Yang, X.; Yan, P.; We, Y. Synthesis of Highly Reactive Polyisobutylenes with Exo-olefin Terminals via Controlled Cationic Polymerization with H2O/FeCl3/iPrOH Initiating System in Nonpolar Hydrocarbon Media. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4200–4212. [Google Scholar] [CrossRef]
- Shiman, D.I.; Vasilenko, I.V.; Kostjuk, S.V. Cationic polymerization of isobutylene by AlCl3/ether complexes in non-polar solvents: Effect of ether structure on the selectivity of β-H elimination. Polymer 2013, 54, 2235–2242. [Google Scholar] [CrossRef]
- Yang, X.; Guo, A.; Xu, H.; Wu, Y. Direct synthesis of highly reactive polyisobutylenes via cationic polymerization of isobutylene co-initiated with TiCl4 in nonpolar hydrocarbon media. J. Appl. Polym. Sci. 2015, 132, 42232. [Google Scholar] [CrossRef]
- Akiyama, S.; Kobori, Y.; Sugisaki, A.; Koyama, T.; Akiba, I. Phase behavior and pressure sensitive adhesive properties in blends of poly(styrene-b-isoprene-b-styrene) with tackifier resin. Polymer 2000, 41, 4021–4027. [Google Scholar] [CrossRef]
- Kamigaito, M.; Sawamoto, M.; Higashimura, T. Alkoxy-substituted titanium (IV) chlorides as Lewis acid activators for living cationic polymerization of isobutyl vinyl ether: Control of Lewis acidity in the design of initiating systems. Macromolecules 1995, 28, 5671–5675. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Catalyst (mmol) | Pinene Feed (mmol) | Yield | β-Pinene Incorp. a (mol%) | Mn b | Mw/Mn b | Tg c (°C) | |
|---|---|---|---|---|---|---|---|---|
| (g) | (wt%) | |||||||
| 1 | 0.20 | 0.50 | 0.23 | 40 | 3 | 1100 | 1.4 | −67 |
| 2 | 0.20 | 1.0 | 0.28 | 44 | 13 | 1000 | 1.4 | −39 |
| 3 d | 0.20 | 1.0 | 0.29 | 45 | 25 | 1000 | 1.3 | −17 |
| 4 | 0.20 | 2.0 | 0.37 | 48 | 24 | 1100 | 1.5 | −13 |
| 5 | 0.20 | 3.0 | 0.53 | 58 | 37 | 1400 | 2.0 | 10 |
| 6 | 0.10 | 3.0 | 0.42 | 46 | 42 | 1400 | 2.0 | 13 |
| 7 | 0.05 | 3.0 | 0.33 | 36 | 43 | 1200 | 1.7 | 7 |
| 8 e | 0.20 | 3.0 | 0.44 | 48 | 41 | 1400 | 2.0 | 14 |
| 9 f | 0.20 | 3.0 | 0.47 | 52 | 38 | 1300 | 1.9 | 18 |
| 10 | 0.20 | 5.0 | 0.83 | 70 | 51 | 1700 | 1.7 | 27 |
| 11 | 0.20 | 5.0 | 0.57 | 84 | 100 | 8100 | 2.1 | 51 |
| Run | Catalyst (mmol) | Yield (g) | β-pinene incorp. a (mmol) | Mn b | Mw/Mn b | Tg c (°C) |
|---|---|---|---|---|---|---|
| 5 | AlCl3∙OiPr2 | 0.53 | 36 | 1400 | 2.0 | 10 |
| 12 | AlCl3∙OBu2 | 0.52 | 33 | 1100 | 1.9 | −6 |
| 13 | AlCl3∙OEt2 | 0.59 | 41 | 1200 | 1.9 | 7 |
| 14 | B(C6F5)3 | 0.12 | 43 | 1400 | 3.4 | 16 |
| 15 | Ti(OiPr)Cl3 | trace | - d | - d | - d | - d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajala, O.A.; Nakayama, Y.; Shiono, T.; Tanaka, R. A Bio-Based Tackifier Synthesized by Room-Temperature Cationic Copolymerization of Isobutene and β-Pinene. Catalysts 2024, 14, 402. https://doi.org/10.3390/catal14070402
Ajala OA, Nakayama Y, Shiono T, Tanaka R. A Bio-Based Tackifier Synthesized by Room-Temperature Cationic Copolymerization of Isobutene and β-Pinene. Catalysts. 2024; 14(7):402. https://doi.org/10.3390/catal14070402
Chicago/Turabian StyleAjala, Oluwaseyi Aderemi, Yuushou Nakayama, Takeshi Shiono, and Ryo Tanaka. 2024. "A Bio-Based Tackifier Synthesized by Room-Temperature Cationic Copolymerization of Isobutene and β-Pinene" Catalysts 14, no. 7: 402. https://doi.org/10.3390/catal14070402
APA StyleAjala, O. A., Nakayama, Y., Shiono, T., & Tanaka, R. (2024). A Bio-Based Tackifier Synthesized by Room-Temperature Cationic Copolymerization of Isobutene and β-Pinene. Catalysts, 14(7), 402. https://doi.org/10.3390/catal14070402