
Investigation of an Ethanol Electroreforming Cell Based on a Pt1Ru1/C Catalyst at the Anode





Abstract
1. Introduction
2. Results
2.1. Physicochemical Characterization
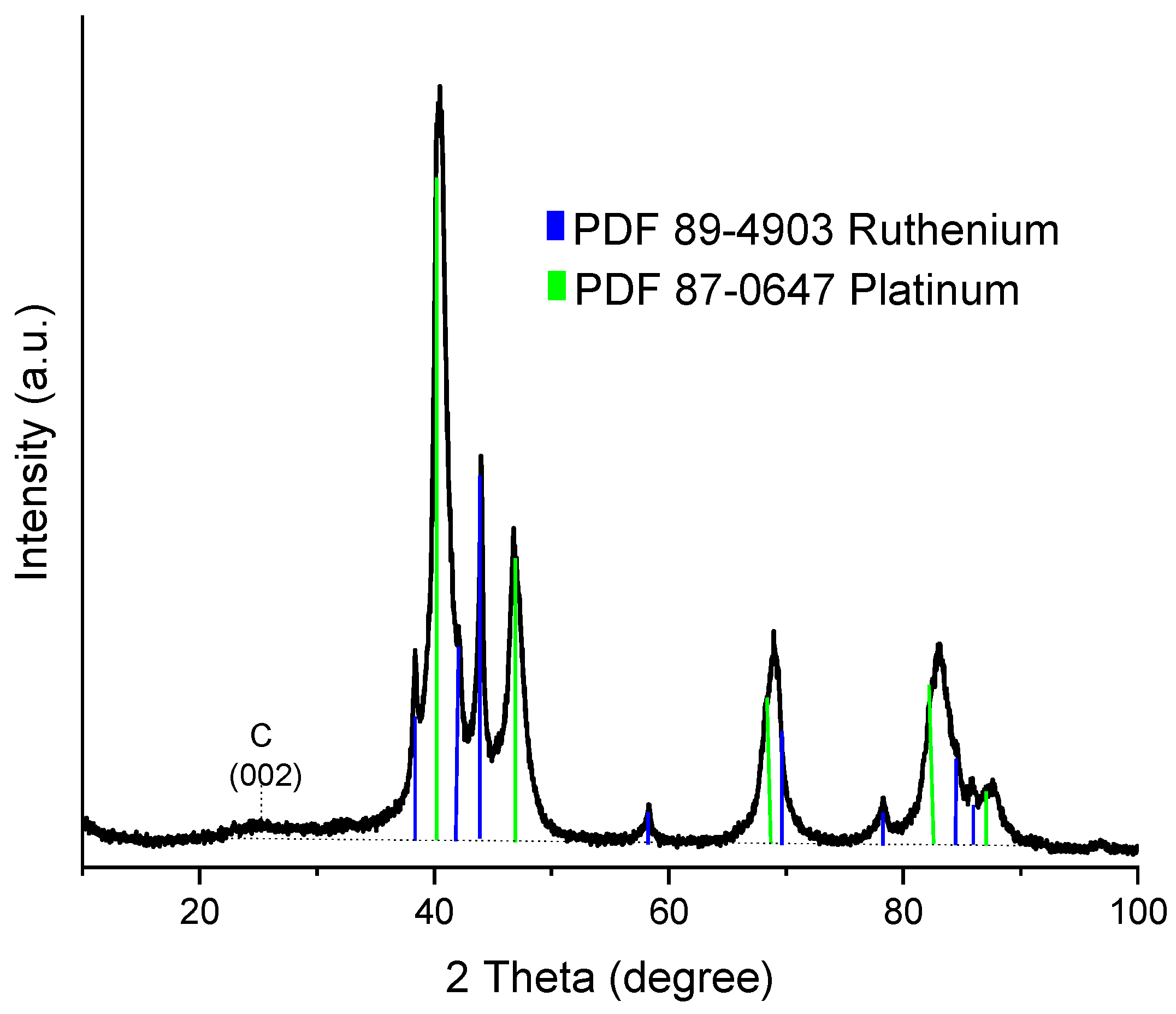
2.1.1. X-ray Diffraction (XRD) Analysis
2.1.2. Scanning Transmission Electron Microscopy–Energy-Dispersive X-ray Spectroscopy (S/TEM-EDX) Investigations
2.2. Electrochemical Characterization
2.2.1. Half-Cell Electrochemical Measurements
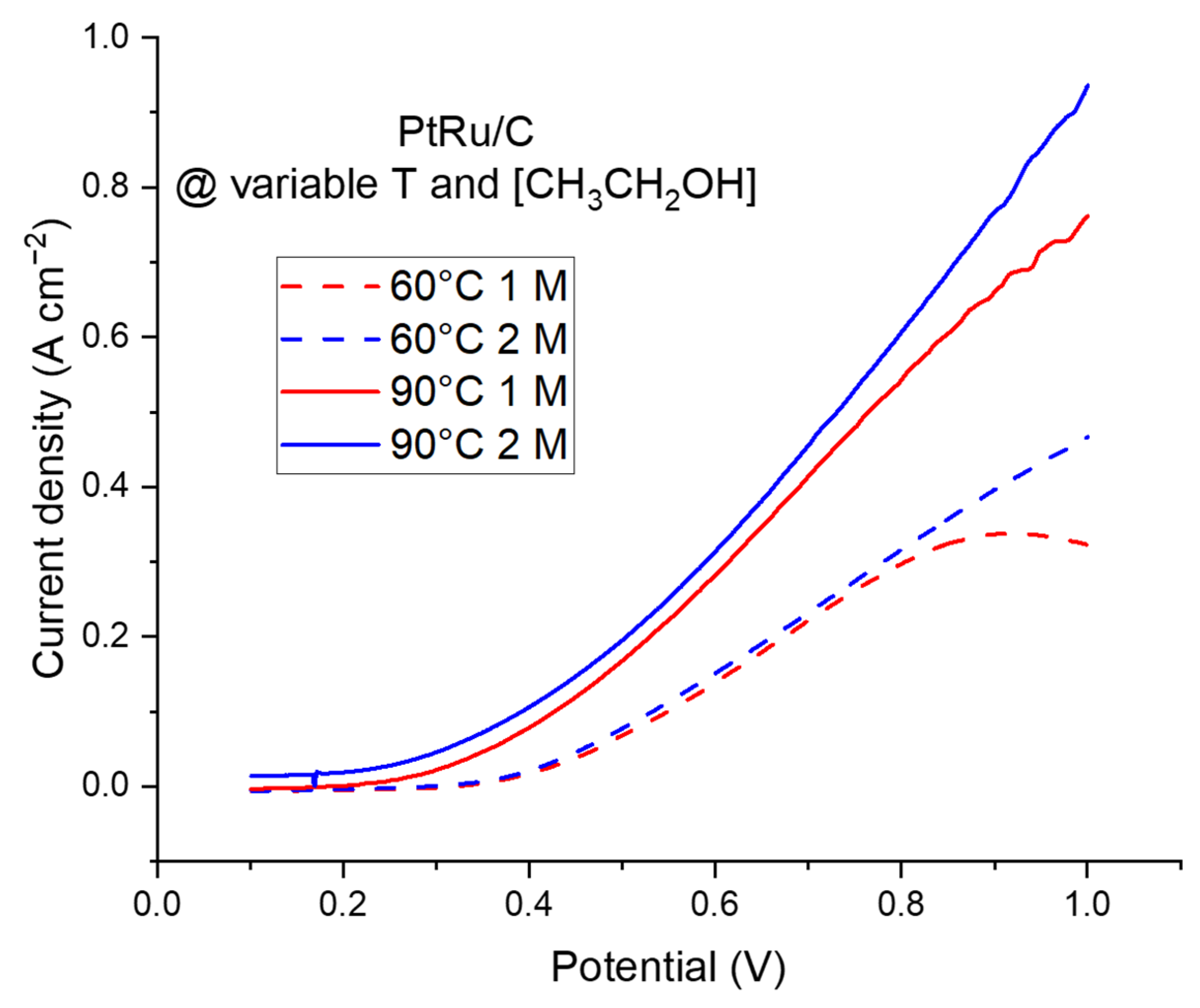
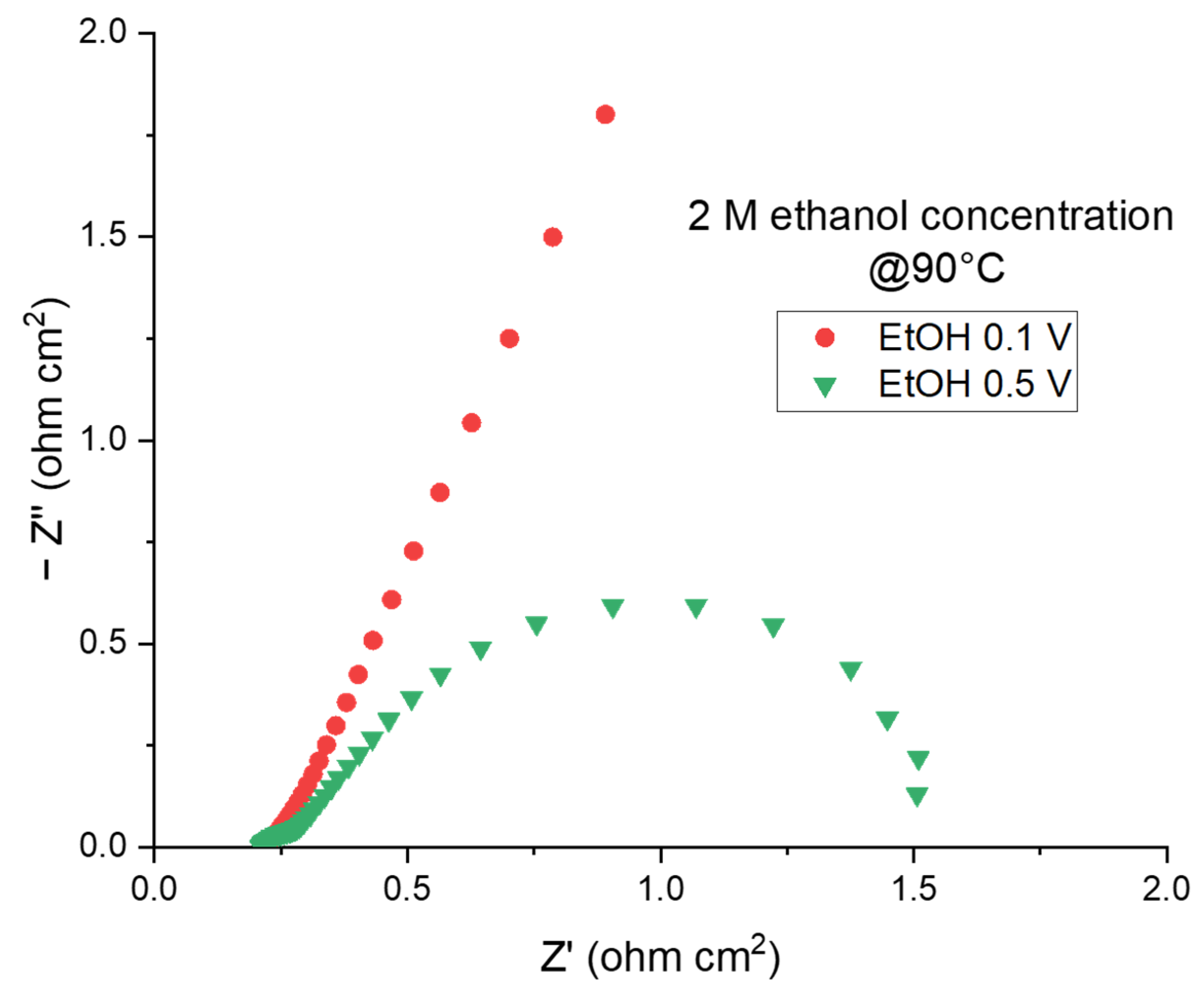
2.2.2. The Electroreforming (ER) Process in a Complete Cell
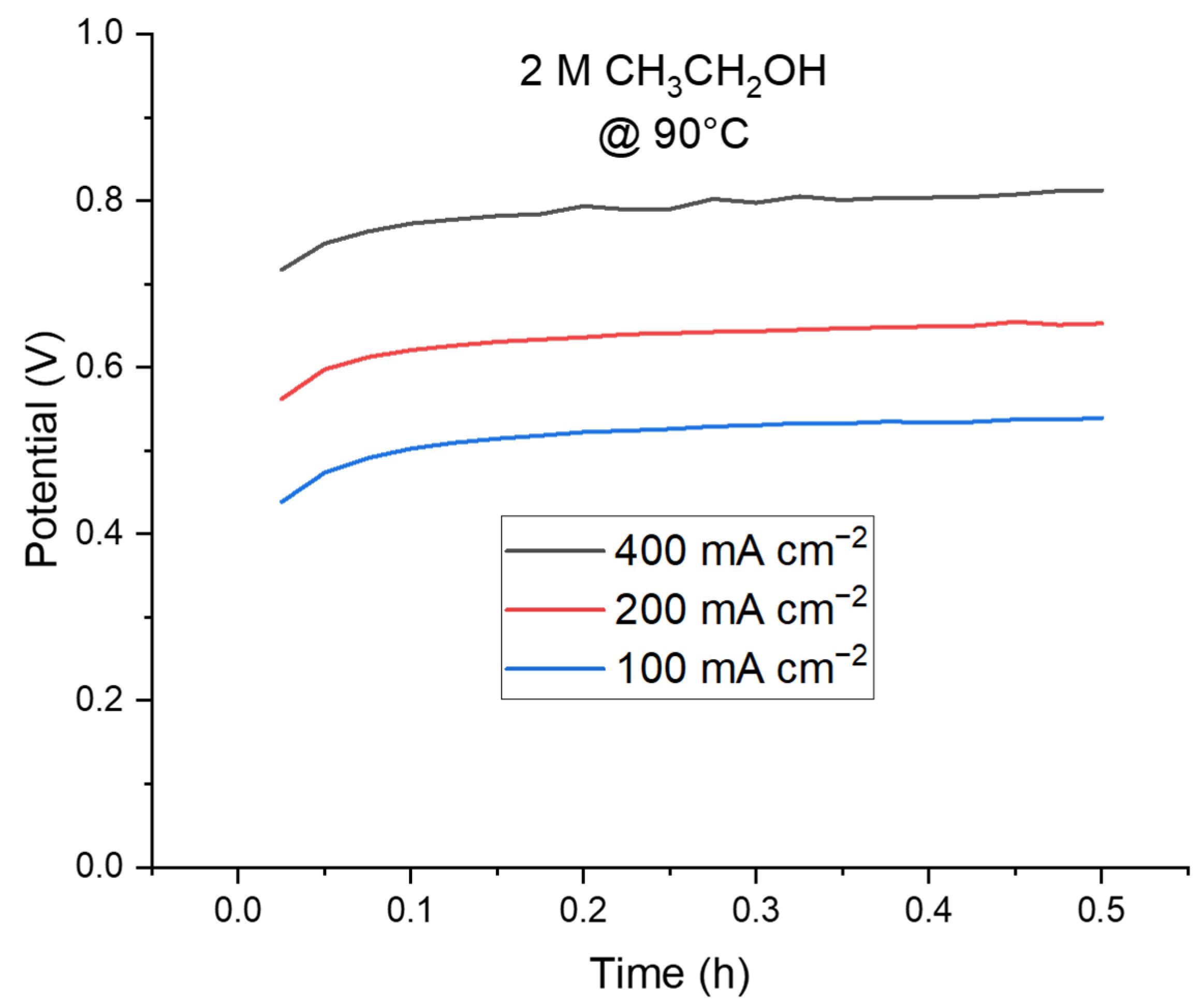
2.2.3. Stability and Hydrogen Detection in a Complete Cell
2.2.4. Physicochemical Characterization after the Stability Test
3. Materials and Methods
3.1. Synthesis of 50 wt.% Pt1Ru1/KB
3.2. Physicochemical Analysis
3.3. Electrochemical Investigation
3.3.1. Half-Cell Analyses
3.3.2. Electroreforming in a Full Cell
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Yan, Q.; Zhang, M. Ecosystem Carbon Storage Considering Combined Environmental and Land-Use Changes in the Future and Pathways to Carbon Neutrality in Developed Regions. Sci. Total Environ. 2023, 903, 166204. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Ambati, M.S.K.; Chakraborty, A.K.; Chakrabortty, S.; Biring, S.; Ramakrishna, S.; Wong, T.K.S.; Kumar, A.; Lawaniya, R.; Dalapati, G.K. Photovoltaic/Photo-Electrocatalysis Integration for Green Hydrogen: A Review. Energy Convers. Manag. 2022, 261, 115648–115685. [Google Scholar] [CrossRef]
- Rabaia, M.K.H.; Abdelkareem, M.A.; Sayed, E.T.; Elsaid, K.; Chae, K.J.; Wilberforce, T.; Olabi, A.G. Environmental Impacts of Solar Energy Systems: A Review. Sci. Total Environ. 2021, 754, 141989. [Google Scholar] [CrossRef] [PubMed]
- Sadorsky, P. Wind Energy for Sustainable Development: Driving Factors and Future Outlook. J. Clean. Prod. 2021, 289, 125779. [Google Scholar] [CrossRef]
- Dhar, A.; Naeth, M.A.; Jennings, P.D.; Gamal El-Din, M. Perspectives on Environmental Impacts and a Land Reclamation Strategy for Solar and Wind Energy Systems. Sci. Total Environ. 2020, 718, 134602. [Google Scholar] [CrossRef] [PubMed]
- Züttel, A.; Remhof, A.; Borgschulte, A.; Friedrichs, O. Hydrogen: The Future Energy Carrier. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 3329–3342. [Google Scholar] [CrossRef] [PubMed]
- Sazali, N. Emerging Technologies by Hydrogen: A Review. Int. J. Hydrogen Energy 2020, 45, 18753–18771. [Google Scholar] [CrossRef]
- Benghanem, M.; Mellit, A.; Almohamadi, H.; Haddad, S.; Chettibi, N.; Alanazi, A.M.; Dasalla, D.; Alzahrani, A. Hydrogen Production Methods Based on Solar and Wind Energy: A Review. Energies 2023, 16, 757. [Google Scholar] [CrossRef]
- Serov, A.; Kwak, C. Review of Non-Platinum Anode Catalysts for DMFC and PEMFC Application. Appl. Catal. B 2009, 90, 313–320. [Google Scholar] [CrossRef]
- Yang, D.; Yan, Z.; Li, B.; Higgins, D.C.; Wang, J.; Lv, H.; Chen, Z.; Zhang, C. Highly Active and Durable Pt–Co Nanowire Networks Catalyst for the Oxygen Reduction Reaction in PEMFCs. Int. J. Hydrogen Energy 2016, 41, 18592–18601. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Y.; Nairan, A.; Khan, U.; Hu, Y.; Wu, B.; Sun, L.; Zeng, L.; Gao, J. Constructing FeNiPt@C Trifunctional Catalyst by High Spin-Induced Water Oxidation Activity for Zn-Air Battery and Anion Exchange Membrane Water Electrolyzer. Adv. Sci. 2024, 11, 2308205. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Lavacchi, A.; Miller, H.A.; Bevilacqua, M.; Filippi, J.; Innocenti, M.; Marchionni, A.; Oberhauser, W.; Wang, L.; Vizza, F. Nanotechnology Makes Biomass Electrolysis More Energy Efficient than Water Electrolysis. Nat. Commun. 2014, 5, 4036. [Google Scholar] [CrossRef] [PubMed]
- Dolle, C.; Neha, N.; Coutanceau, C. Electrochemical Hydrogen Production from Biomass. Curr. Opin. Electrochem. 2022, 31, 100841. [Google Scholar] [CrossRef]
- Sebastián, D.; Serov, A.; Matanovic, I.; Artyushkova, K.; Atanassov, P.; Aricò, A.S.; Baglio, V. Insights on the Extraordinary Tolerance to Alcohols of Fe-N-C Cathode Catalysts in Highly Performing Direct Alcohol Fuel Cells. Nano Energy 2017, 34, 195–204. [Google Scholar] [CrossRef]
- Berretti, E.; Longhi, M.; Atanassov, P.; Sebastián, D.; Lo Vecchio, C.; Baglio, V.; Serov, A.; Marchionni, A.; Vizza, F.; Santoro, C.; et al. Platinum Group Metal-Free Fe-Based (Fe–N–C) Oxygen Reduction Electrocatalysts for Direct Alcohol Fuel Cells. Curr. Opin. Electrochem. 2021, 29, 100756. [Google Scholar] [CrossRef]
- Gupta, U.K.; Pramanik, H. Electrooxidation Study of Pure Ethanol/Methanol and Their Mixture for the Application in Direct Alcohol Alkaline Fuel Cells (DAAFCs). Int. J. Hydrogen Energy 2019, 44, 421–435. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Aricò, A.S.; Baglio, V. Application of Low-Cost Me-N-C (Me = Fe or Co) Electrocatalysts Derived from Edta in Direct Methanol Fuel Cells (DMFCs). Materials 2018, 11, 1193. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Ren, J.; Wang, Y.; Xiong, Y.; Wang, Q.; Li, Q.; Zhao, X.; Zhan, L.; Zheng, L.; Tang, Y.; et al. One-Stone, Two Birds: Alloying Effect and Surface Defects Induced by Pt on Cu2−xSe Nanowires to Boost C-C Bond Cleavage for Electrocatalytic Ethanol Oxidation. Nano Energy 2021, 88, 106307. [Google Scholar] [CrossRef]
- Yaqoob, L.; Noor, T.; Iqbal, N. A Comprehensive and Critical Review of the Recent Progress in Electrocatalysts for the Ethanol Oxidation Reaction. RSC Adv. 2021, 11, 16768–16804. [Google Scholar] [CrossRef]
- Miller, H.A.; Bouzek, K.; Hnat, J.; Loos, S.; Bernäcker, C.I.; Weißgärber, T.; Röntzsch, L.; Meier-Haack, J. Green Hydrogen from Anion Exchange Membrane Water Electrolysis: A Review of Recent Developments in Critical Materials and Operating Conditions. Sustain. Energy Fuels 2020, 4, 2114–2133. [Google Scholar] [CrossRef]
- Gatto, I.; Caprì, A.; Lo Vecchio, C.; Zignani, S.; Patti, A.; Baglio, V. Optimal Operating Conditions Evaluation of an Anion-Exchange-Membrane Electrolyzer Based on FUMASEP® FAA3-50 Membrane. Int. J. Hydrogen Energy 2023, 48, 11914–11921. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, A.; Lepre, E.; Sánchez-Silva, L.; López-Salas, N.; de la Osa, A.R. PtRu Nanoparticles Supported on Noble Carbons for Ethanol Electrooxidation. J. Energy Chem. 2022, 66, 168–180. [Google Scholar] [CrossRef]
- Sudachom, N.; Warakulwit, C.; Parpainainar, P. The Effect of Ternary Catalyst Atomic Ratios (PtRuSn/C and PtRuNi/C) on Ethanol Electrooxidation for Direct Ethanol Fuel Cell. In Key Engineering Materials; Trans Tech Publications Ltd.: Zurich, Switzerland, 2015; Volume 659, pp. 247–251. [Google Scholar]
- Lamy, C.; Jaubert, T.; Baranton, S.; Coutanceau, C. Clean Hydrogen Generation through the Electrocatalytic Oxidation of Ethanol in a Proton Exchange Membrane Electrolysis Cell (PEMEC): Effect of the Nature and Structure of the Catalytic Anode. J. Power Sources 2014, 245, 927–936. [Google Scholar] [CrossRef]
- Sebastián, D.; Stassi, A.; Siracusano, S.; Lo Vecchio, C.; Aricò, A.S.; Baglio, V. Influence of Metal Oxide Additives on the Activity and Stability of PtRu/C for Methanol Electro-Oxidation. J. Electrochem. Soc. 2015, 162, F713. [Google Scholar] [CrossRef]
- Caravaca, A.; Sapountzi, F.M.; De Lucas-Consuegra, A.; Molina-Mora, C.; Dorado, F.; Valverde, J.L. Electrochemical Reforming of Ethanol-Water Solutions for Pure H2 Production in a PEM Electrolysis Cell. Int. J. Hydrogen Energy 2012, 37, 9504–9513. [Google Scholar] [CrossRef]
- Pagliaro, M.V.; Bellini, M.; Bevilacqua, M.; Filippi, J.; Folliero, M.G.; Marchionni, A.; Miller, H.A.; Oberhauser, W.; Caporali, S.; Innocenti, M.; et al. Carbon Supported Rh Nanoparticles for the Production of Hydrogen and Chemicals by the Electroreforming of Biomass-Derived Alcohols. RSC Adv. 2017, 7, 13971–13978. [Google Scholar] [CrossRef]
- Miller, H.A.; Bellini, M.; Vizza, F.; Hasenöhrl, C.; Tilley, R.D. Carbon Supported Au-Pd Core-Shell Nanoparticles for Hydrogen Production by Alcohol Electroreforming. Catal. Sci. Technol. 2016, 6, 6870–6878. [Google Scholar] [CrossRef]
- Bellini, M.; Pagliaro, M.V.; Marchionni, A.; Filippi, J.; Miller, H.A.; Bevilacqua, M.; Lavacchi, A.; Oberhauser, W.; Mahmoudian, J.; Innocenti, M.; et al. Hydrogen and Chemicals from Alcohols through Electrochemical Reforming by Pd-CeO2/C Electrocatalyst. Inorganica Chim. Acta 2021, 518, 120245. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Sebastián, D.; Alegre, C.; Aricò, A.S.; Baglio, V. Carbon-Supported Pd and Pd-Co Cathode Catalysts for Direct Methanol Fuel Cells (DMFCs) Operating with High Methanol Concentration. J. Electroanal. Chem. 2017, 808, 464–473. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Anode | Membrane | Cathode | Oxidant | T (°C) | Jmax @1.0 V (A cm−2) |
|---|---|---|---|---|---|---|
| This work | 50% PtRu/KB | Nafion 115 | 40% Pt/C | 2 M EtOH | 90 | 0.94 |
| This work | 50% PtRu/KB | Nafion 115 | 40% Pt/C | 1 M EtOH | 90 | 0.76 |
| This work | 50% PtRu/KB | Nafion 115 | 40% Pt/C | 2 M EtOH | 60 | 0.47 |
| This work | 50% PtRu/KB | Nafion 115 | 40% Pt/C | 1 M EtOH | 60 | 0.34 @0.9V |
| [26] | 40% Pt-20% Ru/C | Sterion PEM | 20% Pt/C | 6M EtOH | 80 | 0.24 |
| [26] | 40% Pt-20% Ru/C | Sterion PEM | 20% Pt/C | 6M EtOH | 60 | 0.14 |
| [24] | 30% PtSnRu | Nafion 117 PEM | 30% Pt/C | 2M EtOH + 0.5M H2SO4 | 20 | 0.13 |
| [27] | Rh/C | Tokuyama A-201 AEM | 40% Pt/C | 2M EtOH + 2M KOH | 60 | 0.5 @0.7V |
| [28] | AuPd/C | Tokuyama A-201 AEM | 40% Pt/C | 2M EtOH + 2M KOH | 60 | 0.41 @0.9V |
| [12] | Pd/TNTA-web | Tokuyama A-201 AEM | 40% Pt/C | 2M EtOH + 2M NaOH | 80 | 1.95 |
| [12] | Pd/TNTA-web | Tokuyama A-201 AEM | 40% Pt/C | 2M EtOH + 2M NaOH | 50 | 1.25 @0.9V |
| [29] | Pd-CeO2/C | Tokuyama A-201 AEM | 40% Pt/C | 2M EtOH + 2M KOH | 60 | 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Vecchio, C.; Mosca, E.; Trocino, S.; Baglio, V. Investigation of an Ethanol Electroreforming Cell Based on a Pt1Ru1/C Catalyst at the Anode. Catalysts 2024, 14, 415. https://doi.org/10.3390/catal14070415
Lo Vecchio C, Mosca E, Trocino S, Baglio V. Investigation of an Ethanol Electroreforming Cell Based on a Pt1Ru1/C Catalyst at the Anode. Catalysts. 2024; 14(7):415. https://doi.org/10.3390/catal14070415
Chicago/Turabian StyleLo Vecchio, Carmelo, Erminia Mosca, Stefano Trocino, and Vincenzo Baglio. 2024. "Investigation of an Ethanol Electroreforming Cell Based on a Pt1Ru1/C Catalyst at the Anode" Catalysts 14, no. 7: 415. https://doi.org/10.3390/catal14070415
APA StyleLo Vecchio, C., Mosca, E., Trocino, S., & Baglio, V. (2024). Investigation of an Ethanol Electroreforming Cell Based on a Pt1Ru1/C Catalyst at the Anode. Catalysts, 14(7), 415. https://doi.org/10.3390/catal14070415