
Autocatalyzed Kinetics of 6-Electron Electroreduction of Iodic Acid Studied by Rotating Disk Electrode Technique


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
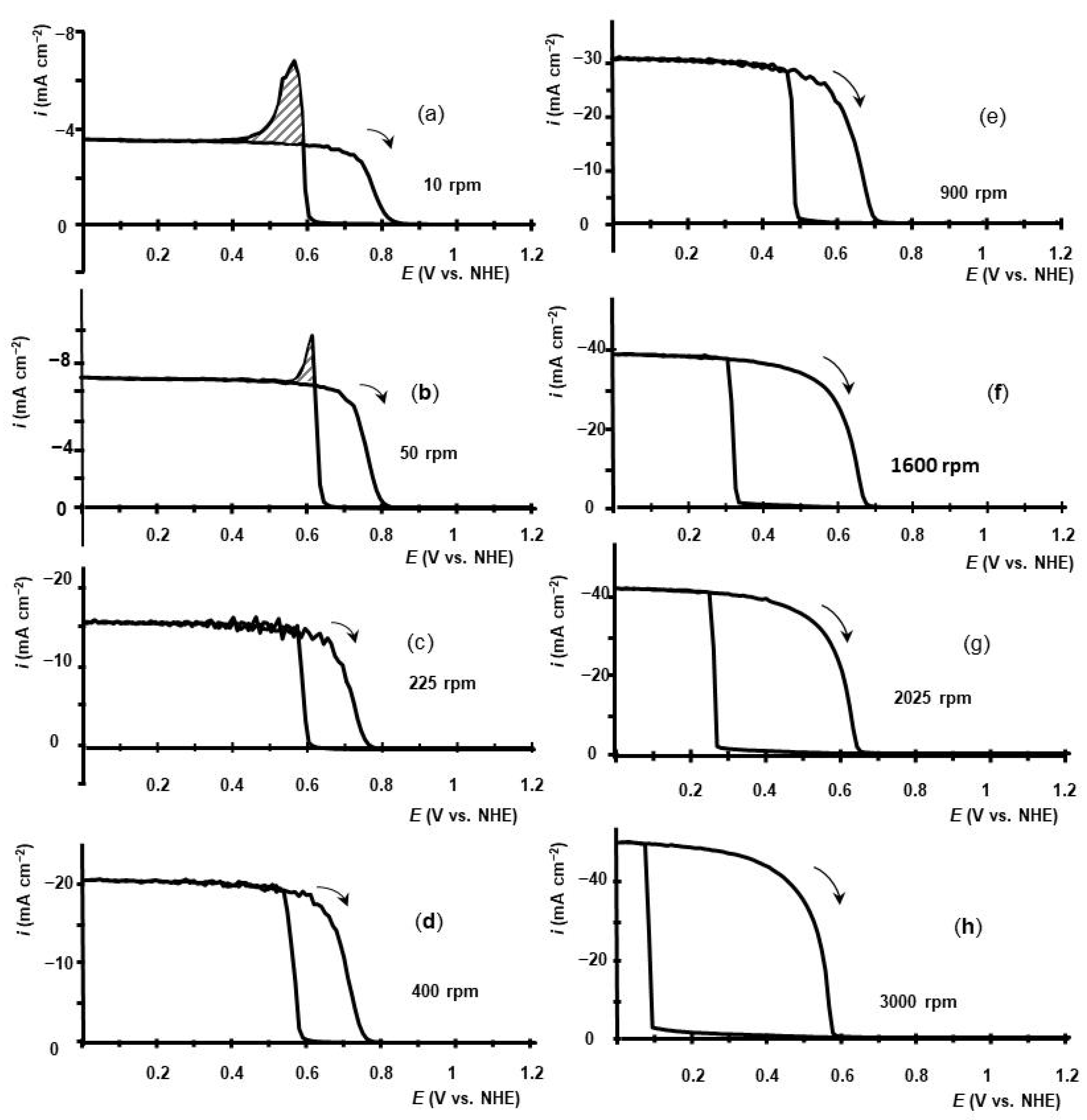
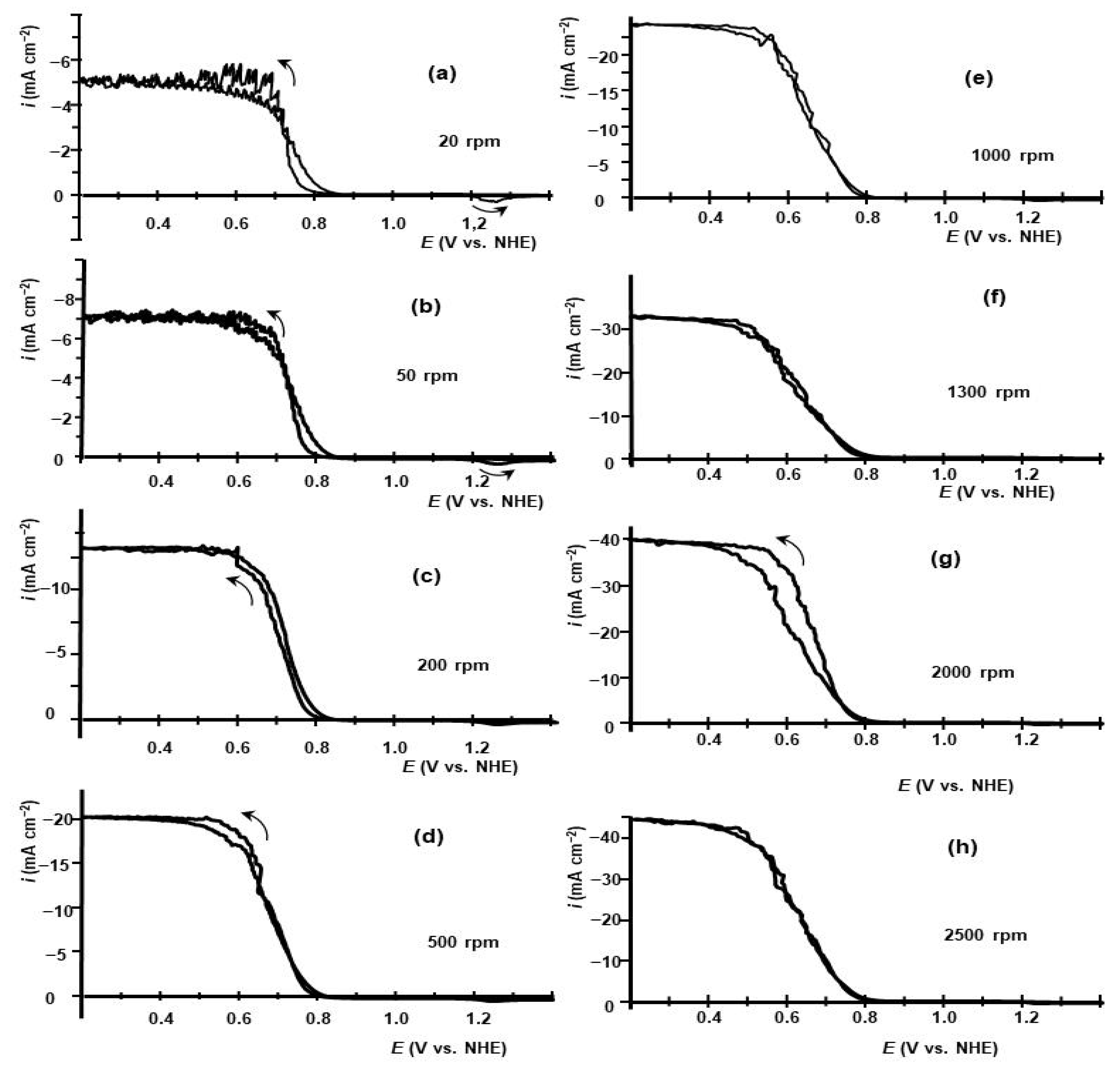
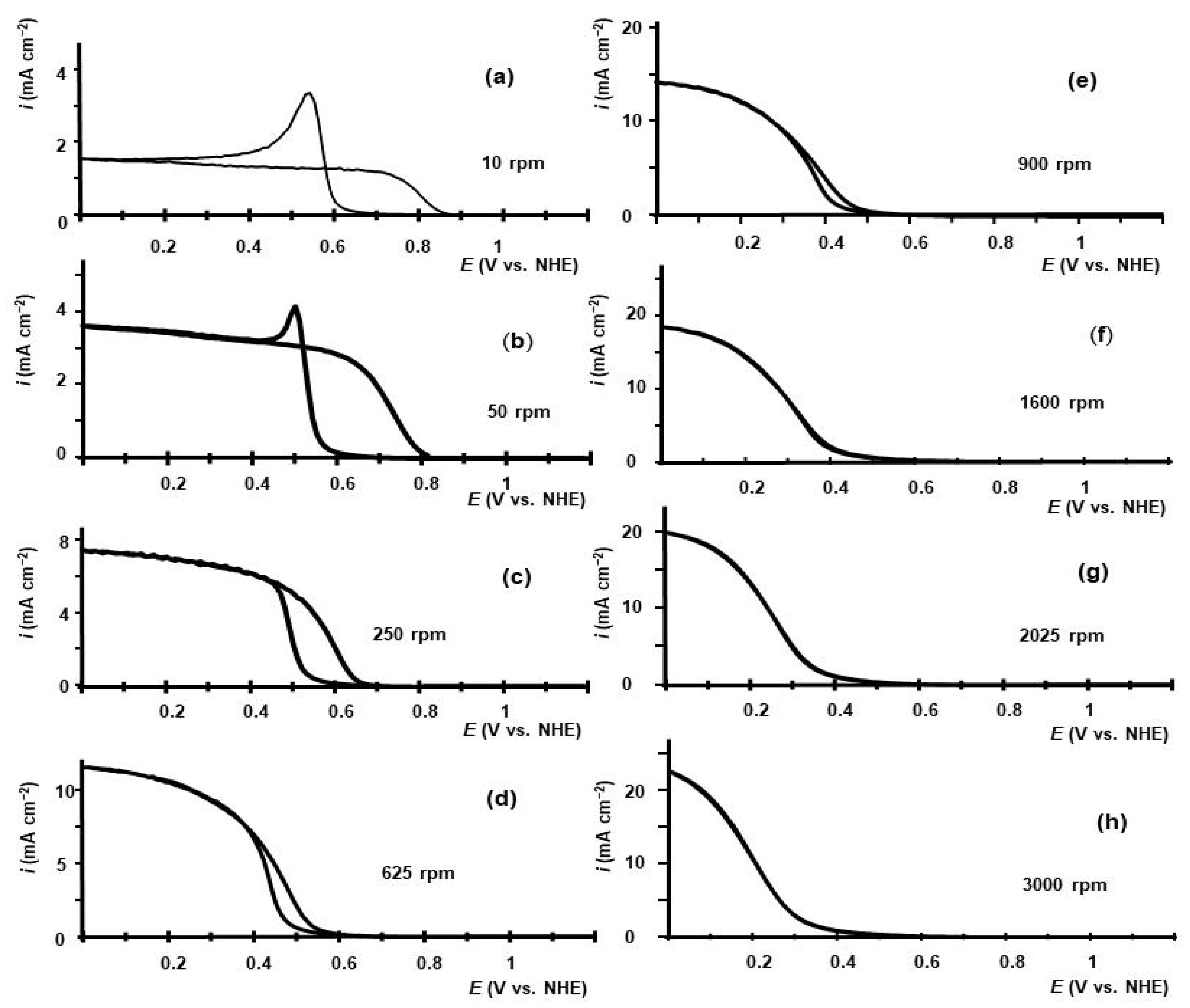
2.1. Voltammetry of HIO3 Reduction on GC RDE in Aqueous Electrolyte
2.2. Hypoiodic Acid
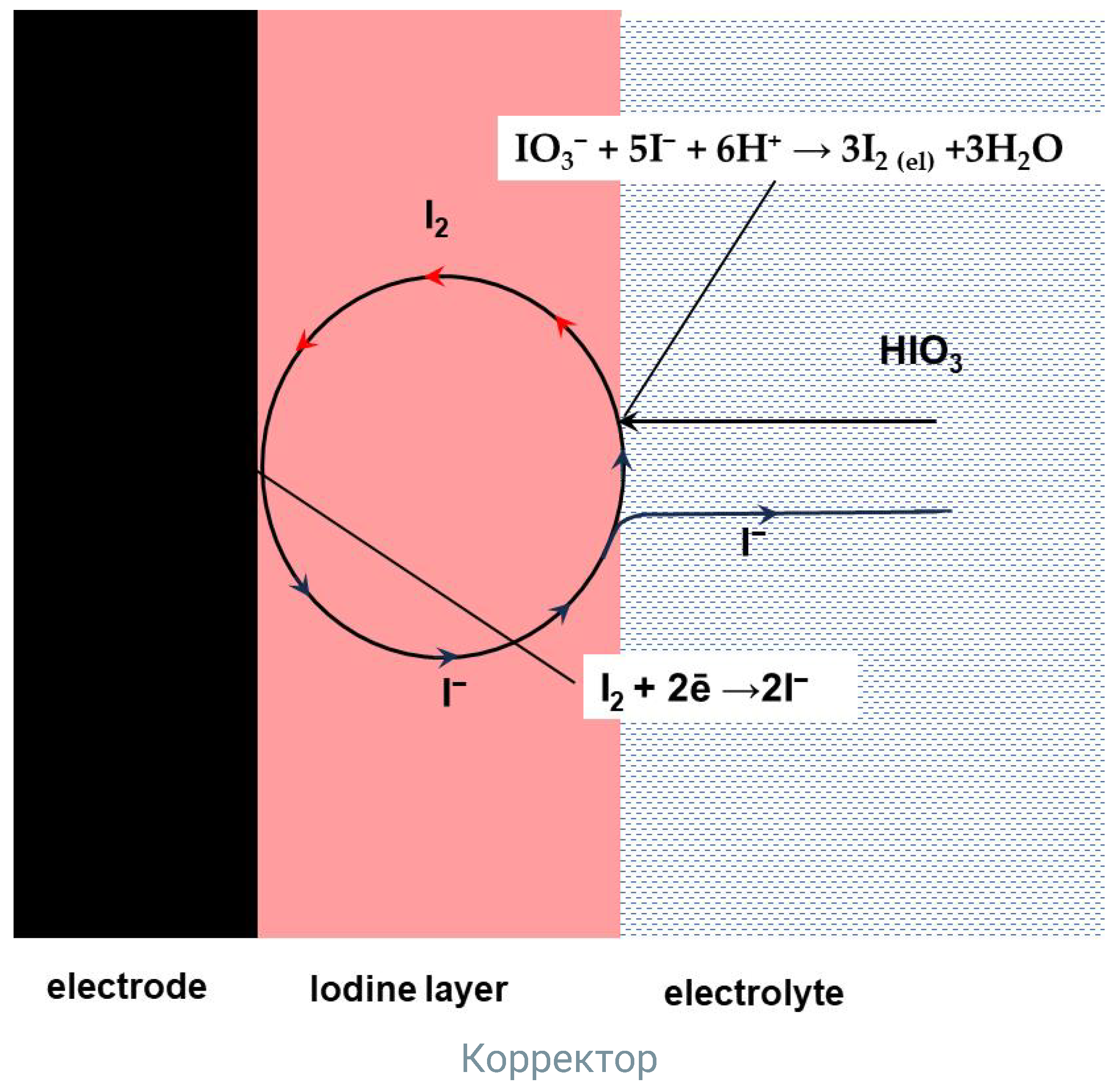
2.3. Iodine Layer on GC Electrode
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tolmachev, Y.V.; Piatkivskyi, A.; Ryzhov, V.V.; Konev, D.V.; Vorotyntsev, M.A. Energy cycle based on a high specific energy aqueous flow battery and its potential use for fully electric vehicles and for direct solar-to-chemical energy conversion. J. Solid State Electr. 2015, 19, 2711–2722. [Google Scholar] [CrossRef]
- Konev, D.V.; Istakova, O.I.; Ruban, E.A.; Glazkov, A.T.; Vorotyntsev, M.A. Hydrogen-chlorate electric power source: Feasibility of the device, discharge characteristics and modes of operation. Molecules 2022, 27, 5638. [Google Scholar] [CrossRef]
- Modestov, A.D.; Konev, D.V.; Tripachev, O.V.; Antipov, A.E.; Tolmachev, Y.V. A hydrogen–bromate flow battery for air-deficient environments. Energy Technol. 2018, 6, 242–245. [Google Scholar] [CrossRef]
- Vanysek, P. Electrochemical series. In CRC Handbook of Chemistry and Physics, 95th ed.; Haynes, W.M., Ed.; CRC PRESS: Boca Raton, FL, USA, 2014; pp. 5–80. [Google Scholar]
- Anson, F.C.; Lingane, J.J. Anodic chronopotentiometry with platinum and gold electrodes. The iodide-iodine-iodate system. J. Am. Chem. Soc. 1957, 79, 1015–1020. [Google Scholar] [CrossRef]
- Badea, G.E. Autocatalytic reduction of iodate at the platinum electrode in 0.5 M H2SO4 solutions. Rev. Roum. Chim. 2007, 52, 491–498. [Google Scholar]
- Beran, P.; Bruckenstein, S. Voltammetry of iodine (I) chloride, iodine, and iodate at rotated platinum disk and ring disk electrodes. Anal. Chem. 1968, 40, 1044–1051. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Wu, B. Determination of the adsorbed intermediate of iodide electro-oxidation on a platinum electrode by rotating disk electrode potential step coulometry (RDE-PSC). J. Electroanal. Chem. 1992, 335, 321–326. [Google Scholar] [CrossRef]
- Desideri, P.G. Reduction of iodate in sulphuric medium I. The reduction mechanism. J. Electroanal. Chem. 1965, 9, 218–228. [Google Scholar]
- Desideri, P.G. Electrochemical behaviour of iodate in hydrochloric acid solutions. J. Electroanal. Chem. 1968, 17, 129–136. [Google Scholar] [CrossRef]
- Grgur, B.N.; Gvozdenovic, M.M.; Stevanovic, J.S.; Jugovic, B.Z.; Trisovic, L.T. Electrochemical oxidation of iodide in aqueous solution. Chem. Eng. J. 2006, 124, 47–54. [Google Scholar] [CrossRef]
- Wels, B.R.; Austin-Harrison, D.S.; Johnson, D.C. Electrocatalytic reduction of iodate at platinum electrodes in 0.5 M H2SO4. Langmuir 1991, 7, 559–566. [Google Scholar] [CrossRef]
- Modestov, A.; Andreev, V.; Antipov, A.; Petrov, M.M. Aluminum/bromate and aluminum/iodate mechanically rechargeable batteries. Batteries 2022, 8, 270. [Google Scholar] [CrossRef]
- Jones, J.L.; Arranaga, A.B. A New zinc-lodate primary battery. J. Electrochem. Soc. 1958, 105, 435–439. [Google Scholar] [CrossRef]
- Modestov, A.; Andreev, V.; Antipov, A. Novel aqueous zinc–halogenate flow batteries as an offspring of zinc–air fuel cells for use in oxygen-deficient environment. Energy Technol. 2021, 9, 2100233. [Google Scholar] [CrossRef]
- Ma, W.; Liu, T.; Xu, C.; Lei, C.; Jiang, P.; He, X.; Liang, X. A twelve-electron conversion iodine cathode enabled by interhalogen chemistry in aqueous solution. Nat. Commun. 2023, 14, 5508. [Google Scholar] [CrossRef]
- Xie, C.; Wang, C.; Xu, Y.; Li, T.; Fu, Q.; Li, X. Reversible Multielectron Transfer I−/IO3− Cathode Enabled by a Hetero-Halogen Electrolyte for High-Energy-Density Aqueous Batteries. Nat. Energy 2024, 9, 714–724. [Google Scholar] [CrossRef]
- Vetter, K.J. Electrochemical Kinetics. In Theoretical and Experimental Aspects; Academic Press: New York, NY, USA, 1967; pp. 471–477. [Google Scholar]
- Schmitz, G. Kinetics and mechanism of the iodate–iodide reaction and other related reactions. Phys. Chem. Chem. Phys. 1999, 1, 1909–1914. [Google Scholar] [CrossRef]
- Perrin, D.D. Dissociation Constants of Organic Bases in Aqueous Solution; Butterworth & Co., Ltd.: London, UK, 1965; pp. 111–200. [Google Scholar]
- Spitz, R.D.; Liefbafsky, H.A. The Iodate-Iodine Electrode Mechanism, Standard Potentials, Related Thermodynamic Data. J. Electrochem. Soc. 1975, 122, 363–367. [Google Scholar] [CrossRef]
- Bichsel, Y.; Von Gunten, U. Hypoiodous acid: Kinetics of the buffer-catalyzed disproportionation. Wat. Res. 2000, 34, 3197–3203. [Google Scholar] [CrossRef]
- Myers, O.E.; Kennedy, J.W. The kinetics of iodine-iodate isotopic exchange reaction. J. Am. Chem. Soc. 1950, 72, 897–906. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Antipov, A.E.; Konev, D.V. Bromate anion reduction: Novel autocatalytic (EC″) mechanism of electrochemical processes. Its implication for redox flow batteries of high energy and power densities. Pure Appl. Chem. 2017, 89, 0306. [Google Scholar] [CrossRef]
- Mastragostino, M.; Gramellini, C. Kinetic study of the electrochemical processes of the bromine/bromide aqueous system on vitreous carbon electrodes. Electrochim. Acta 1985, 30, 373–380. [Google Scholar] [CrossRef]
- Modestov, A.D.; Konev, D.V.; Antipov, A.E.; Petrov, M.M.; Pichugov, R.D.; Vorotyntsev, M.A. Bromate electroreduction from sulfuric acid solution at rotating disk electrode: Experimental study. Electrochim. Acta 2018, 259, 655–663. [Google Scholar] [CrossRef]
- Faulkner, L.R.; Bard, A.J. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley and Sons: New York, NY, USA, 2001; p. 339. [Google Scholar]
- Varlamova, T.M.; Rubtsova, E.M.; Mushtakova, S.P. Solubility Diagrams of the Potassium Iodide–Water–Ethanol and Iodine–Water–Ethanol Ternary Systems. Rus. J. Phys. Chem. A 2009, 83, 1896–1899. [Google Scholar] [CrossRef]
- Bunerjee, P.; Bagchi, B. Ions’ motion in water. J. Chem. Phys. 2019, 150, 190901. [Google Scholar] [CrossRef]
- Joshi, B.K.; Kandapal, N.D. Volumetric and transport properties of aqueous sulphuric acid. Phys. Chem. Liq. 2007, 45, 463–470. [Google Scholar] [CrossRef]
- Bejerano, T.; Gileadi, E. Formation of thick layers of iodine during the anodic oxidation of iodide on a RDE Part I. The precipitation-dissolution mechanism. J. Electroanal. Chem. 1977, 82, 209—225. [Google Scholar] [CrossRef]
- Bejerano, T.; Gileadi, E. Formation of Thick Layers of Iodine During the Anodic Oxidation of Iodide on a RDE. II. Open-Circuit Behavior. J. Electrochem. Soc. 1977, 124, 1720–1723. [Google Scholar] [CrossRef]
- Jang, W.J.; Cha, J.S.; Kim, H.; Yang, J.H. Effect of an iodine film on charge-transfer resistance during the electro-oxidation of iodide in redox flow batteries. ACS Appl. Mater. Inter. 2021, 13, 6385−6393. [Google Scholar] [CrossRef]
- Sorti, L.; Vitulano, F.; Cappellini, E.; Uggeri, F.; Morelli, C.F.; Sello, G.; Minguzzi, A.; Vertova, A. Electrochemical iodination through the in situ generation of iodinating agents: A promising green approach. Molecules 2023, 28, 5555. [Google Scholar] [CrossRef]
- Allen, T.L.; Keeper, R.M. The formation of hypoiodous acid and hydrated iodine cation by the hydrolysis of iodine. J. Am. Chem. Soc. 1955, 77, 2957–2960. [Google Scholar] [CrossRef]
- Khattab, S.K.; Bandarkar, F.; Fakhree, M.A.A.; Jouyban, A. Density, viscosity, and surface tension of water+ethanol mixtures from 293 to 323 K. Korean J. Chem. Eng. 2012, 29, 812–817. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antipova, L.; Tripachev, O.; Rybakova, A.; Andreev, V.; Pichugov, R.; Sudarev, G.; Antipov, A.; Modestov, A. Autocatalyzed Kinetics of 6-Electron Electroreduction of Iodic Acid Studied by Rotating Disk Electrode Technique. Catalysts 2024, 14, 437. https://doi.org/10.3390/catal14070437
Antipova L, Tripachev O, Rybakova A, Andreev V, Pichugov R, Sudarev G, Antipov A, Modestov A. Autocatalyzed Kinetics of 6-Electron Electroreduction of Iodic Acid Studied by Rotating Disk Electrode Technique. Catalysts. 2024; 14(7):437. https://doi.org/10.3390/catal14070437
Chicago/Turabian StyleAntipova, Liliya, Oleg Tripachev, Alexandra Rybakova, Vladimir Andreev, Roman Pichugov, George Sudarev, Anatoly Antipov, and Alexander Modestov. 2024. "Autocatalyzed Kinetics of 6-Electron Electroreduction of Iodic Acid Studied by Rotating Disk Electrode Technique" Catalysts 14, no. 7: 437. https://doi.org/10.3390/catal14070437