


Are Rh Catalysts a Suitable Choice for Bio-Oil Reforming? The Case of a Commercial Rh Catalyst in the Combined H2O and CO2 Reforming of Bio-Oil
 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
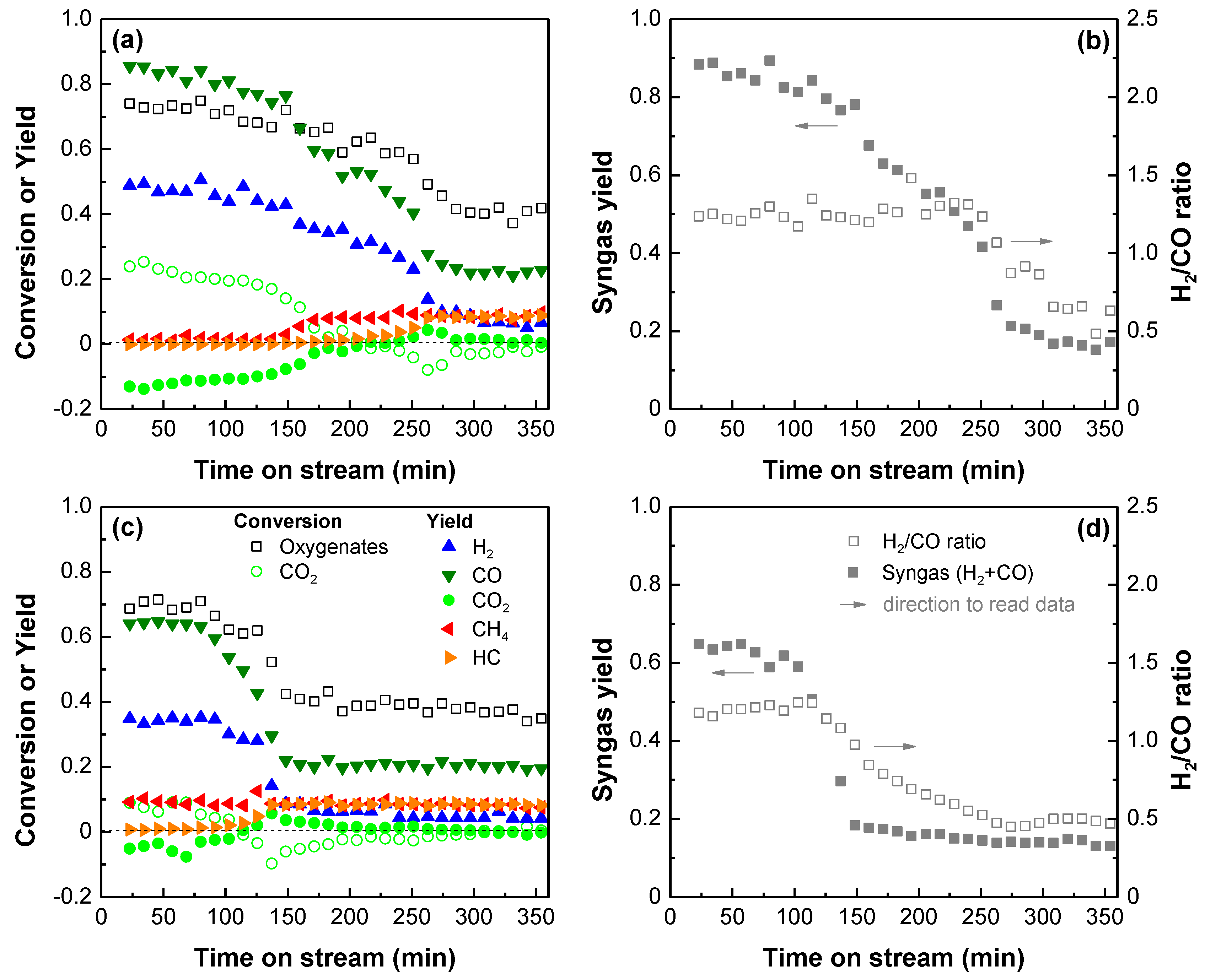
2.1. Performance of the Rh Catalyst in Bio-Oil Reforming
2.2. Chacaractarization of Deactivated Catalyst Samples
2.2.1. Coke Formation
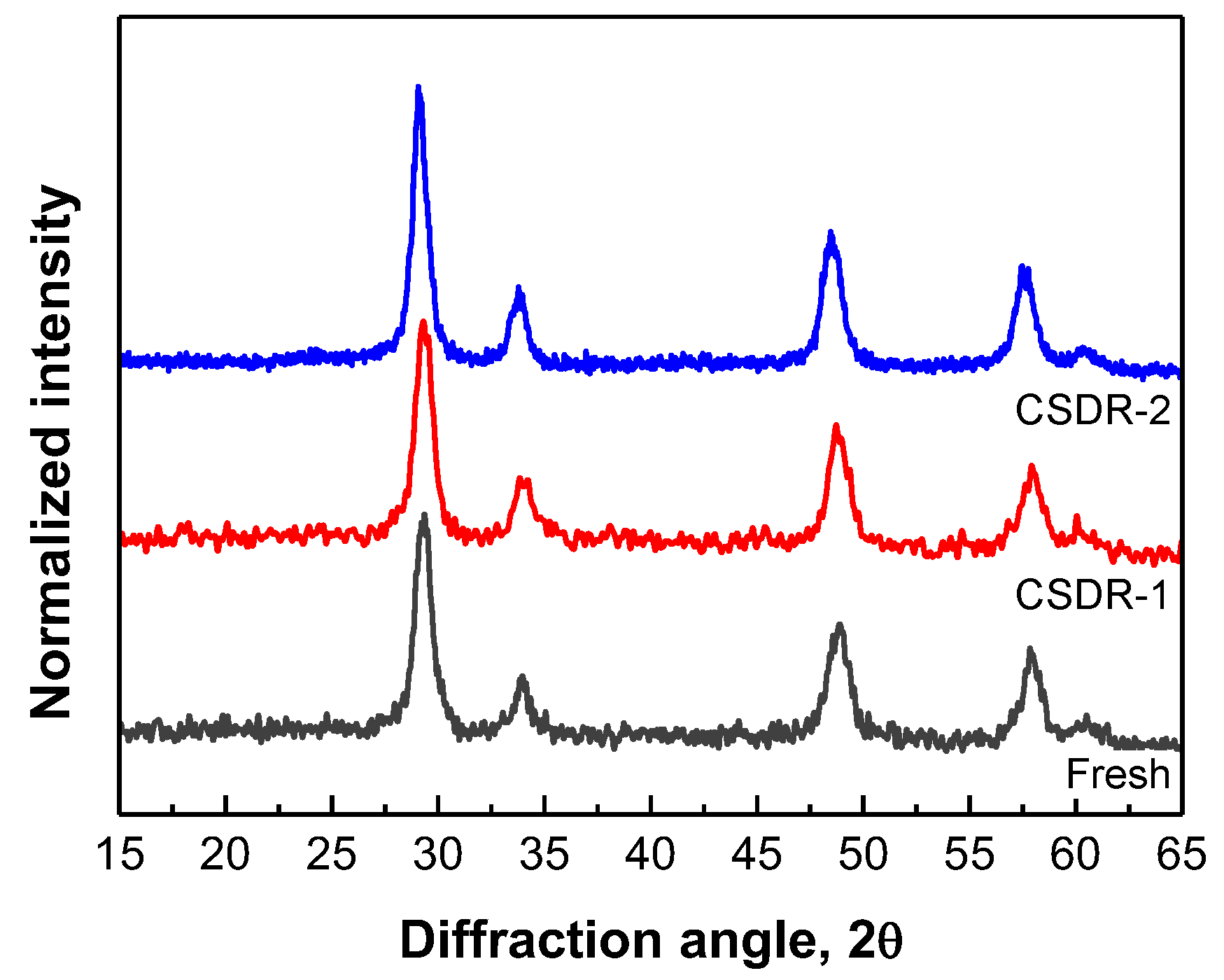
2.2.2. Characterization of Catalyst Structure and Rh Sites
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bachmann, M.; Völker, S.; Kleinekorte, J.; Bardow, A. Syngas from What? Comparative Life-Cycle Assessment for Syngas Production from Biomass, CO2, and Steel Mill Off-Gases. ACS Sustain. Chem. Eng. 2023, 11, 5356–5366. [Google Scholar] [CrossRef]
- García-Gómez, N.; Valecillos, J.; Valle, B.; Remiro, A.; Bilbao, J.; Gayubo, A.G. Combined effect of bio-oil composition and temperature on the stability of Ni spinel derived catalyst for hydrogen production by steam reforming. Fuel 2022, 326, 124966. [Google Scholar] [CrossRef]
- Baloch, H.A.; Nizamuddin, S.; Siddiqui, M.T.H.; Riaz, S.; Jatoi, A.S.; Dumbre, D.K.; Mubarak, N.M.; Srinivasan, M.P.; Griffin, G.J. Recent advances in production and upgrading of bio-oil from biomass: A critical overview. J. Environ. Chem. Eng. 2018, 6, 5101–5118. [Google Scholar] [CrossRef]
- Landa, L.; Remiro, A.; De Torre, R.; Aguado, R.; Bilbao, J.; Gayubo, G. Global vision from the thermodynamics of the effect of the bio-oil composition and the reforming strategies in the H 2 production and the energy requirement. Energy Convers. Manag. 2021, 239, 114181. [Google Scholar] [CrossRef]
- Landa, L.; Remiro, A.; Valecillos, J.; Bilbao, J.; Gayubo, A.G. Thermodynamic study of the CO2 valorization in the combined steam-dry reforming of bio-oil into syngas. J. CO2 Util. 2023, 72, 102503. [Google Scholar] [CrossRef]
- Zhao, Z.; Situmorang, Y.A.; An, P.; Chaihad, N.; Wang, J.; Hao, X.; Xu, G.; Abudula, A.; Guan, G. Hydrogen production from catalytic steam reforming of bio-oils: A critical review. Chem. Eng. Technol. 2020, 43, 625–640. [Google Scholar] [CrossRef]
- Silva, J.; Rocha, C.; Soria, M.A.; Madeira, L.M. Catalytic Steam Reforming of Biomass-Derived Oxygenates for H2 Production: A Review on Ni-Based Catalysts. ChemEngineering 2022, 6, 39. [Google Scholar] [CrossRef]
- Pafili, A.; Charisiou, N.D.; Douvartzides, S.L.; Siakavelas, G.I.; Wang, W.; Liu, G.; Papadakis, V.G.; Goula, M.A. Recent progress in the steam reforming of bio-oil for hydrogen production: A review of operating parameters, catalytic systems and technological innovations. Catalysts 2021, 11, 1526. [Google Scholar] [CrossRef]
- Setiabudi, H.D.; Aziz, M.A.A.; Abdullah, S.; Teh, L.P.; Jusoh, R. Hydrogen production from catalytic steam reforming of biomass pyrolysis oil or bio-oil derivatives: A review. Int. J. Hydrogen Energy 2020, 45, 18376–18397. [Google Scholar] [CrossRef]
- Zhang, C. Review of catalytic reforming of biomass pyrolysis oil for hydrogen production. Front. Chem. 2022, 10, 1–6. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V. Thermochemical production of bio-oil: A review of downstream processing technologies for bio-oil upgrading, production of hydrogen and high value-added products Oil in Water. Renew. Sustain. Energy Rev. 2021, 135, 110152. [Google Scholar] [CrossRef]
- Landa, L.; Remiro, A.; Valecillos, J.; Bilbao, J.; Gayubo, A.G. Syngas production through combined steam-dry reforming of raw bio-oil over a NiAl2O4 spinel derived catalyst. J. CO2 Util. 2023, 78, 102637. [Google Scholar] [CrossRef]
- Hu, X.; Lu, G. Bio-oil steam reforming, partial oxidation or oxidative steam reforming coupled with bio-oil dry reforming to eliminate CO2 emission. Int. J. Hydrogen Energy 2010, 35, 7169–7176. [Google Scholar] [CrossRef]
- Qingli, X.; Peng, F.; Wei, Q.; Kai, H.; Shanzhi, X.; Yongjie, Y. Catalyst deactivation and regeneration during CO2 reforming of bio-oil. Int. J. Hydrogen Energy 2019, 44, 10277–10285. [Google Scholar] [CrossRef]
- Rioche, C.; Kulkarni, S.; Meunier, F.C.; Breen, J.P.; Burch, R. Steam reforming of model compounds and fast pyrolysis bio-oil on supported noble metal catalysts. Appl. Catal. B Environ. 2005, 61, 130–139. [Google Scholar] [CrossRef]
- Rennard, D.; French, R.; Czernik, S.; Josephson, T.; Schmidt, L. Production of synthesis gas by partial oxidation and steam reforming of biomass pyrolysis oils. Int. J. Hydrogen Energy 2010, 35, 4048–4059. [Google Scholar] [CrossRef]
- Remiro, A.; Arandia, A.; Bilbao, J.; Gayubo, A.G. Comparison of Ni based and Rh based catalyst performance in the oxidative steam reforming of raw bio-oil. Energy Fuels 2017, 31, 7147–7156. [Google Scholar] [CrossRef]
- Arandia, A.; Remiro, A.; Oar-Arteta, L.; Bilbao, J.; Gayubo, A.G. Reaction conditions effect and pathways in the oxidative steam reforming of raw bio-oil on a Rh/CeO2-ZrO2 catalyst in a fluidized bed reactor. Int. J. Hydrogen Energy 2017, 42, 29175–29185. [Google Scholar] [CrossRef]
- Remiro, A.; Arandia, A.; Oar-Arteta, L.; Bilbao, J.; Gayubo, A.G. Stability of a Rh/CeO2-ZrO2 Catalyst in the Oxidative Steam Reforming of Raw Bio-oil. Energy Fuels 2018, 32, 3588–3598. [Google Scholar] [CrossRef]
- Iojoiu, E.E.; Domine, M.E.; Davidian, T.; Guilhaume, N.; Mirodatos, C. Hydrogen production by sequential cracking of biomass-derived pyrolysis oil over noble metal catalysts supported on ceria-zirconia. Appl. Catal. Gen. 2007, 323, 147–161. [Google Scholar] [CrossRef]
- Domine, M.E.; Iojoiu, E.E.; Davidian, T.; Guilhaume, N.; Mirodatos, C. Hydrogen production from biomass-derived oil over monolithic Pt- and Rh-based catalysts using steam reforming and sequential cracking processes. Catal. Today 2008, 133–135, 565–573. [Google Scholar] [CrossRef]
- Ranjekar, A.M.; Yadav, G.D. Dry reforming of methane for syngas production: A review and assessment of catalyst development and efficacy. J. Indian Chem. Soc. 2021, 98, 100002. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Chen, M.; Liang, D.; Yang, Z.; Cheng, W.; Tang, Z.; Wang, J.; Zhang, H. Recent advances during CH4 dry reforming for syngas production: A mini review. Int. J. Hydrogen Energy 2021, 46, 5852–5874. [Google Scholar] [CrossRef]
- Fan, M.S.; Abdullah, A.Z.; Bhatia, S. Catalytic technology for carbon dioxide reforming of methane to synthesis gas. ChemCatChem 2009, 1, 192–208. [Google Scholar] [CrossRef]
- da Silva, A.M.; de Souza, K.R.; Jacobs, G.; Graham, U.M.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam and CO2 reforming of ethanol over Rh/CeO2 catalyst. Appl. Catal. B Environ. 2011, 102, 94–109. [Google Scholar] [CrossRef]
- Díaz López, E.; Comas-Vives, A. CO2 activation dominating the dry reforming of methane catalyzed by Rh(111) based on multiscale modelling. Catal. Sci. Technol. 2023, 13, 7162–7171. [Google Scholar] [CrossRef]
- Kartavova, K.E.; Mashkin, M.Y.; Kostin, M.Y.; Finashina, E.D.; Kalmykov, K.B.; Kapustin, G.I.; Pribytkov, P.V.; Tkachenko, O.P.; Mishin, I.V.; Kustov, L.M.; et al. Rhodium-Based Catalysts: An Impact of the Support Nature on the Catalytic Cyclohexane Ring Opening. Nanomaterials 2023, 13, 936. [Google Scholar] [CrossRef]
- Mashkin, M.Y.; Tedeeva, M.A.; Fedorova, A.A.; Fatula, E.R.; Egorov, A.V.; Dvoryak, S.V.; Maslakov, K.I.; Knotko, A.V.; Baranchikov, A.E.; Kapustin, G.I.; et al. Synthesis of CexZr1-xO2/SiO2 supports for chromium oxide catalysts of oxidative dehydrogenation of propane with carbon dioxide. J. Chem. Technol. Biotechnol. 2023, 98, 1247–1259. [Google Scholar] [CrossRef]
- Remiro, A.; Ochoa, A.; Arandia, A.; Castaño, P.; Bilbao, J.; Gayubo, A.G. On the dynamics and reversibility of the deactivation of a Rh/CeO2–ZrO2 catalyst in raw bio-oil steam reforming. Int. J. Hydrogen Energy 2019, 44, 2620–2632. [Google Scholar] [CrossRef]
- García-Gómez, N.; Valle, B.; Remiro, A.; Bilbao, J.; Gayubo, A.G. The effect of the support in the deactivation and regeneration of Rh catalysts used in the steam reforming of bio-oil. In Proceedings of the 3rd International Congress Chemistry & Environment ANQUE-ICCE3, Santander, Spain, 19–21 June 2019. [Google Scholar]
- Machida, M.; Yoshida, H.; Kamiuchi, N.; Fujino, Y.; Miki, T.; Haneda, M.; Tsurunari, Y.; Iwashita, S.; Ohta, R.; Yoshida, H.; et al. Thermal Aging of Rh/ZrO2-CeO2 Three-Way Catalysts under Dynamic Lean/Rich Perturbation Accelerates Deactivation via an Encapsulation Mechanism. ACS Catal. 2023, 13, 3806–3814. [Google Scholar] [CrossRef]
- Machida, M.; Yoshida, H.; Kamiuchi, N.; Fujino, Y.; Miki, T.; Haneda, M.; Tsurunari, Y.; Iwashita, S.; Ohta, R.; Yoshida, H.; et al. Rh Nanoparticles Dispersed on ZrO2-CeO2 Migrate to Al2O3 Supports to Mitigate Thermal Deactivation via Encapsulation. ACS Appl. Nano Mater. 2023, 6, 9805–9815. [Google Scholar] [CrossRef]
- Craciun, R.; Daniell, W.; Knözinger, H. The effect of CeO2 structure on the activity of supported Pd catalysts used for methane steam reforming. Appl. Catal. A Gen. 2002, 230, 153–168. [Google Scholar] [CrossRef]
- He, L.; Liao, G.; Li, H.; Ren, Q.; Hu, S.; Han, H.; Xu, J.; Jiang, L.; Su, S.; Wang, Y.; et al. Evolution characteristics of different types of coke deposition during catalytic removal of biomass tar. J. Energy Inst. 2020, 93, 2497–2504. [Google Scholar] [CrossRef]
- García-Gómez, N.; Valecillos, J.; Remiro, A.; Valle, B.; Bilbao, J.; Gayubo, A.G. Effect of reaction conditions on the deactivation by coke of a NiAl2O4 spinel derived catalyst in the steam reforming of bio-oil. Appl. Catal. B Environ. 2021, 297, 120445. [Google Scholar] [CrossRef]
- Gong, X.; Guo, Z.; Wang, Z. Reactivity of pulverized coals during combustion catalyzed by CeO2 and Fe2O3. Combust. Flame 2010, 157, 351–356. [Google Scholar] [CrossRef]
- Arandia, A.; Remiro, A.; García, V.; Castaño, P.; Bilbao, J.; Gayubo, A. Oxidative Steam Reforming of Raw Bio-Oil over Supported and Bulk Ni Catalysts for Hydrogen Production. Catalysis 2018, 8, 322. [Google Scholar] [CrossRef]
- Sagar Vijay Kumar, P.; Suresh, L.; Vinodkumar, T.; Reddy, B.M.; Chandramouli, G.V.P. Zirconium Doped Ceria Nanoparticles: An Efficient and Reusable Catalyst for a Green Multicomponent Synthesis of Novel Phenyldiazenyl-Chromene Derivatives Using Aqueous Medium. ACS Sustain. Chem. Eng. 2016, 4, 2376–2386. [Google Scholar] [CrossRef]
- Fedorova, E.A.; Kardash, T.Y.; Kibis, L.S.; Stonkus, O.A.; Slavinskaya, E.M.; Svetlichnyi, V.A.; Pollastri, S.; Boronin, A.I. Unraveling the low-temperature activity of Rh-CeO2 catalysts in CO oxidation: Probing the local structure and Red-Ox transformation of Rh3+ species. Phys. Chem. Chem. Phys. 2022, 25, 2862–2874. [Google Scholar] [CrossRef]
- Landa, L.; Remiro, A.; Valecillos, J.; Valle, B.; Bilbao, J.; Gayubo, A.G. Performance of NiAl2O4 spinel derived catalyst + dolomite in the sorption enhanced steam reforming (SESR) of raw bio-oil in cyclic operation. Int. J. Hydrogen Energy 2024, 58, 1526–1540. [Google Scholar] [CrossRef]
- Lee, J.; Kang, S.; Lee, E.; Kang, M.; Sung, J.; Kim, T.J.; Christopher, P.; Park, J.; Kim, D.H. Aggregation of CeO2 particles with aligned grains drives sintering of Pt single atoms in Pt/CeO2 catalysts. J. Mater. Chem. A 2022, 10, 7029–7035. [Google Scholar] [CrossRef]
- Li, J.; Du, C.; Feng, Q.; Zhao, Y.; Liu, S.; Xu, J.; Hu, M.; Zeng, Z.; Zhang, Z.; Shen, H.; et al. Evolution and performances of Ni single atoms trapped by mesoporous ceria in Dry Reforming of Methane. Appl. Catal. B Environ. 2024, 354, 124069. [Google Scholar] [CrossRef]
- Song, S.; Wang, X.; Zhang, H. CeO2-encapsulated noble metal nanocatalysts: Enhanced activity and stability for catalytic application. NPG Asia Mater. 2015, 7, e179. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Li, M.; Amari, H.; van Veen, A.C. Metal-oxide interaction enhanced CO2 activation in methanation over ceria supported nickel nanocrystallites. Appl. Catal. B Environ. 2018, 239, 27–35. [Google Scholar] [CrossRef]
- Landa, L.; Remiro, A.; Valecillos, J.; Valle, B.; Bilbao, J.; Gayubo, A.G. Unveiling the deactivation by coke of NiAl2O4 spinel derived catalysts in the bio-oil steam reforming: Role of individual oxygenates. Fuel 2022, 321, 124009. [Google Scholar] [CrossRef]
- Remiro, A.; Valle, B.; Aguayo, A.T.; Bilbao, J.; Gayubo, A.G. Steam reforming of raw bio-oil in a fluidized bed reactor with prior separation of pyrolytic lignin. Energy Fuels 2013, 27, 7549–7559. [Google Scholar] [CrossRef]
- Landa, L.; Valecillos, J.; Remiro, A.; Valle, B.; Bilbao, J.; Gayubo, A.G. Comparison of the NiAl2O4 derived catalyst deactivation in the steam reforming and sorption enhanced steam reforming of raw bio-oil in packed and fluidized-bed reactors. Chem. Eng. J. 2023, 458, 141494. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2 g−1) | Smicro (m2 g−1) | Vpore (cm3 g−1) | Vmicro (cm3 g−1) | dpore (nm) |
|---|---|---|---|---|---|
| Fresh | 57.5 | 2.17 | 0.24 | 1.31 × 10−3 | 17.0 |
| CSDR-1 | 38.4 | 0.82 | 0.15 | 0.49 × 10−3 | 15.9 |
| CSDR-2 | 14.8 | 2.16 | 0.04 | 0.85 × 10−3 | 10.9 |
| 1st regeneration (CSDR-1) | 39.6 | 2.16 | 0.20 | 1.00 × 10−3 | 20.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valecillos, J.; Landa, L.; Elordi, G.; Remiro, A.; Bilbao, J.; Gayubo, A.G. Are Rh Catalysts a Suitable Choice for Bio-Oil Reforming? The Case of a Commercial Rh Catalyst in the Combined H2O and CO2 Reforming of Bio-Oil. Catalysts 2024, 14, 571. https://doi.org/10.3390/catal14090571
Valecillos J, Landa L, Elordi G, Remiro A, Bilbao J, Gayubo AG. Are Rh Catalysts a Suitable Choice for Bio-Oil Reforming? The Case of a Commercial Rh Catalyst in the Combined H2O and CO2 Reforming of Bio-Oil. Catalysts. 2024; 14(9):571. https://doi.org/10.3390/catal14090571
Chicago/Turabian StyleValecillos, José, Leire Landa, Gorka Elordi, Aingeru Remiro, Javier Bilbao, and Ana Guadalupe Gayubo. 2024. "Are Rh Catalysts a Suitable Choice for Bio-Oil Reforming? The Case of a Commercial Rh Catalyst in the Combined H2O and CO2 Reforming of Bio-Oil" Catalysts 14, no. 9: 571. https://doi.org/10.3390/catal14090571