Abstract
The quest for efficient green hydrogen production through Alkaline Water Electrolysis (AWE) is a critical aspect of the clean energy transition. The hydrogen evolution reaction (HER) in alkaline media is central to this process, with the performance of electrocatalysts being a determining factor for overall efficiency. Theoretical studies using energy-based descriptors are essential for designing high-performance alkaline HER electrocatalysts. This review summarizes various descriptors, including water adsorption energy, water dissociation barrier, and Gibbs free energy changes of hydrogen and hydroxyl adsorption. Examples of how to apply these descriptors to identify the active site of materials and better design high-performance alkaline HER electrocatalysts are provided, highlighting the previously underappreciated role of hydroxyl adsorption-free energy changes. As research progresses, integrating these descriptors with experimental data will be paramount in advancing AWE technology for sustainable hydrogen production.
1. Introduction
The shift towards hydrogen gas as an alternative energy source is a significant step in combating climate change. Green hydrogen via electrocatalytic water splitting is directly competitive with fossil fuels, which decreases overall greenhouse gas emissions [1]. The intricacies of electrocatalytic water splitting involve the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) [2]. There are several types of water electrolyzers (WE): solid oxide water electrolyzer (SOWE), proton exchange membrane water electrolyzer (PEMWE), and alkaline electrolyzer (AWE) [3]. SOWE can be adapted for continuous operation in industrial areas under high temperatures. However, the technology has seen limited applications due to long start-up times, mechanical compatibility, and chemical instabilities [4,5]. PEMWE in acidic environments is the most efficient for green hydrogen production due to the abundance of the proton reactant in the solution [6]. However, the main challenge associated with PEMWE is the high cost of the electrocatalysts, which are based on platinum and platinum group metals (PGMs) to resist acid corrosion [7]. In contrast, alkaline HER in AWE offers a promising alternative allowing for the exploration of earth-abundant transition metal-based catalysts because their hydro-oxide counterparts are stable in this environment [8,9,10]. Shifting towards an alkaline environment will reduce reliance on expensive noble metals and reduce the hydrogen production cost. Green hydrogen via electrocatalytic water splitting is directly competitive with fossil fuels, which decreases overall greenhouse gas emissions.
The operational principle of AWEs involves the conduction of hydroxide anions through a liquid electrolyte between two electrodes [8]. The main challenges of the current AWE are its low current densities, the inability to operate with intermittent renewable energy sources, and slow kinetics [11]. These limitations are associated with early versions of atmospheric electrolyzers designed for continuous operation at industrial sites with stable and inexpensive electricity sources. Modern pressurized high-temperature AWEs have shown the capability to reach performance and adaptability on par with PEMWEs [12]. The adaptability to various energy sources, coupled with improved design and control systems, positions pressurized AWEs as a promising solution for sustainable and flexible hydrogen generation in the evolving energy technology landscape [12].
Moreover, the anion exchange membrane water electrolyzer (AEMWE) replaces the conventional diaphragm of AWE with an anion exchange membrane [13]. A membrane reduces the gas cross-over and the need for pressure difference operation between the anode and cathode. Additionally, the thickness of the anion exchange membrane is less than the diaphragm, which leads to a lower ohmic over-voltage. The AEMWE uses an alkaline solution with low concentration. Conventional electrolyzers use concentrated potassium hydroxide electrolytes, which increase corrosion. Therefore, AEMWE has several benefits, including no leakage, ease of installation, and control. However, the low energy conversion efficiency of AWEs due to the slow kinetics remains a bottleneck, requiring high-performance electrocatalyst design.
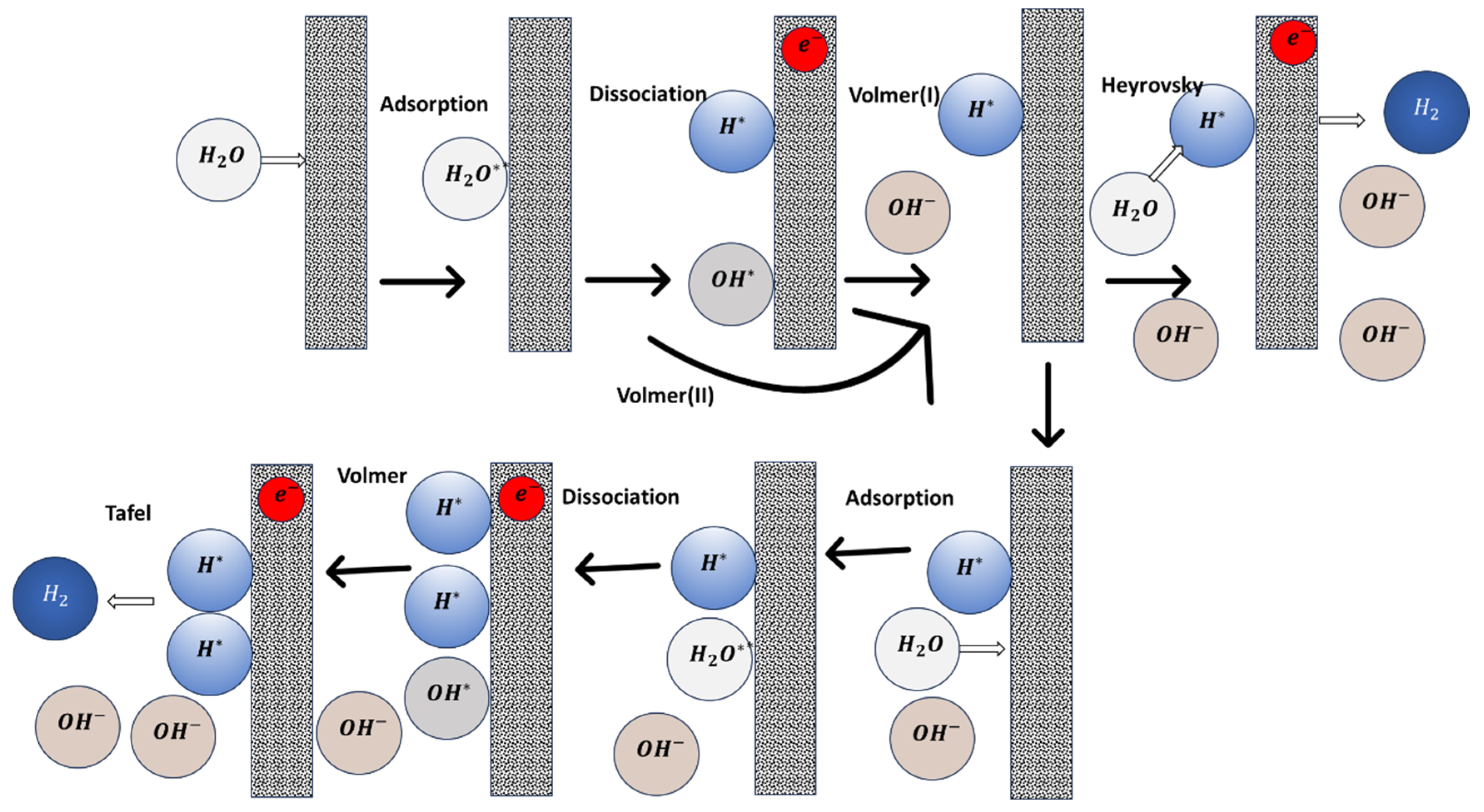
The pursuit of an effective catalyst for AWE is multifaceted, necessitating a blend of experimental and theoretical studies to unlock the full potential of electrocatalysts [8,11,14]. The challenges for the alkaline HER electrocatalyst design are twofold: the inherently lower reaction rates of catalysts and a limited understanding of the complex kinetics involved. The HER process varies significantly depending on the electrolyte medium, which can be either acidic or alkaline. In acidic media, the HER mechanism involves the electrochemical reduction of a proton (H+), leading to the release of hydrogen gas (H2) [15,16,17]. The HER efficiency is associated with the adsorption of hydrogen atom intermediate (H*), which consists of the Volmer/Heyrovsky or Volmer/Tafel steps as delineated below [18]:
Conversely, alkaline HER operates through the electrochemical reduction of water molecules, resulting in the formation of hydrogen gas and hydroxide ions (OH−) [19].
The HER pathway in alkaline media involves complex Volmer-Heyrovsky or Volmer-Tafel steps, with the intermediate hydrogen formed after an initial water adsorption step (Equation (4)). Additionally, the adsorbed hydroxyl intermediate may form during the Volmer step if its binding energy with the active site is strong enough (Equation (5)). Otherwise, the solution-phase hydroxide (OH−) can directly form after the water dissociation (Equation (6)) during the Volmer reaction. The H2 product can be produced through an adsorbed H atom intermediate and water in the Heyrovsky reaction (Equation (7)) or through the combination of two adsorbed H* atom intermediates in the Tafel reaction (Equation (8)). Figure 1 indicates the possible alkaline HER pathway. It shows that the performance of alkaline HER can be influenced by four main factors: (1) water adsorption strength on active sites, (2) water dissociation ability, (3) hydrogen binding energy, and (4) OH adsorption ability. To this end, the properties relevant to these four factors have been used to screen the alkaline HER electrocatalysts.
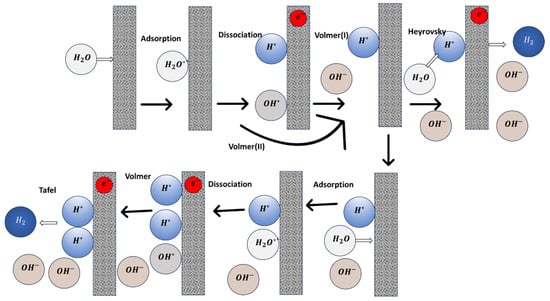
Figure 1.
Possible hydrogen evolution reaction processes in alkaline media.
The step-by-step process that governs HER needs to be theoretically revealed by delving into the intricate details of reaction mechanisms through atomic-scale simulation [8]. Theoretical investigations can offer insights into the energetic aspects of water adsorption, water dissociation, intermediates adsorption, and hydrogen desorption [15,20,21,22]. This granular view allows researchers to identify and optimize the performance of active sites that influence both catalytic efficiency and stability. Such knowledge is instrumental in recognizing the rate-determining steps and potential energy barriers, which are critical for designing catalysts with optimal performance [15]. Furthermore, theoretical models serve as a blueprint for predicting the performance of different materials under various reaction conditions. Therefore, computational studies enable the fine-tuning of working conditions to enhance the overall efficiency of the process. Additionally, these studies contribute significantly to the green hydrogen field by offering a cost-effective strategy for material screening, circumventing the need for laborious and expensive experimental procedures [20,23]. Ultimately, the insights gained from theoretical research facilitate the development of innovative solutions for alkaline hydrogen production [8].
Recent advances have highlighted the importance of atomically precise electrocatalysts, which utilize theoretical descriptors to correlate the properties of catalysts with their alkaline HER performance [8,19,24]. Since different descriptors related to the different reaction steps shown in Figure 1 have been proposed and previously utilized [25,26,27,28,29,30], it is necessary to devise guidelines for selecting descriptors that can accurately predict the performance of HER electrocatalysts in alkaline conditions. Density functional theory (DFT) calculations are essential in computing the proposed energy descriptors related to water adsorption, water dissociation, intermediates adsorption [18,31,32,33], and product desorption. These descriptors are crucial to understanding alkaline HER electrocatalysts and electrocatalytic processes in the alkaline electrolyzer (AEL). Additionally, identifying important descriptors can improve computational accuracy while minimizing costs [23,34]. While several reviews on the recent progress of alkaline HER electrocatalysis have been presented [8,11,35], a summary and discussion of the most appropriate descriptors for guiding the design of efficient AEL is still rare. To fill this gap, this review will provide a framework by which the desired theoretical descriptor can be selected via DFT computations to design electrocatalysts rather than a comprehensive overview of AWEs.
2. Theoretical Descriptors
As indicated by Equations (4)–(8) and Figure 1, the performance of alkaline HER electrocatalysts is a complex interplay of various factors. The water adsorption strength on active sites, the ability of the catalyst to dissociate water molecules, the hydrogen binding energy, and the OH adsorption processes are all critical in determining the efficiency of the HER process. These factors are intricately linked to the intrinsic properties of the electrocatalysts and their interaction with the operational reaction environment, e.g., electrolyte. Therefore, different theoretical energy-related descriptors have been employed as indicators to screen electrocatalysts for alkaline HER [25,26,27,28,29,30,36].
2.1. Water Adsorption and Dissociation
The first step of the electrocatalytic water splitting in an alkaline solution is water adsorption, followed by water dissociation to provide the H+ cation for hydrogen production (Equations (4)–(6)). HER performance in alkaline or neutral media is primarily determined by the equilibrium between water dissociation (Volmer step) and the subsequent chemisorption of water-splitting intermediates (OH and H) on the surface of the electrocatalysts [37]. To this end, it is critical to strengthen water adsorption at the active sites and decrease the water dissociation activation energy during the Volmer step to increase the overall HER activities of these electrocatalysts. Consequently, the adsorption energy of water (*) and its dissociation energy barriers (ΔEa) have been used as descriptors to evaluate the performance of alkaline HER electrocatalysts. The * value can be calculated as shown below:
and are the energies of the surface with and without adsorbed water, respectively. and n represents the energy of an isolated water molecule and the number of adsorbed water molecules in each surface cell, respectively. According to Equation (9), a more negative value of corresponds to stronger water adsorption.
To calculate the water dissociation energy barrier on the active site, the nudged elastic band (NEB) is widely used [38,39]. The NEB method is highly effective for locating the maximum energy point between specified initial and final configurations of a given transition. The NEB approach for climbing images involves finding a transition path where the saddle point corresponds to the image with the highest energy. The image does not depict the spring forces acting on the band. The image attempts to optimize its energy along the band while minimizing it in all other directions. Once this image reaches convergence, it can be precisely located at the saddle point [40]. The climbing image NEB method enhances the possibility of finding the transition state (TS) for the reaction at the highest energy point. The ΔEa value is the energy difference between the TS state and the surface with the adsorbed water.
2.2. Gibbs Free Energy Change of Hydrogen Atom Adsorption
Hydrogen atoms adsorbed at the active site are the HER intermediates. The adsorption and desorption of hydrogen atoms on the catalyst surface are commonly viewed as competing processes. During the HER process, the active catalytic site must have a strong affinity for a hydrogen atom. However, a weak adsorption between the active catalytic site and the hydrogen atom is required [41,42]. The high adsorption strength of hydrogen atoms can promote hydrogen atom intermediate formation but impede the formation of hydrogen molecules. Comparatively, weak adsorption can benefit the formation of H2 but hinder the formation of hydrogen atom intermediates. A balance must be achieved where the hydrogen intermediate must form but not compete with/prevent the generation and release of the hydrogen molecule. As a result, the optimal adsorption strength of the hydrogen atom intermediate is essential for improving the HER. The adsorption strength of hydrogen atom intermediates on active sites in catalysts can be evaluated using the Gibbs free energy change of hydrogen atom adsorption (ΔGH*) [43].
The ΔGH* is obtained by using the computational hydrogen electrode (CHE) method:
where ΔEH* describes the binding energy, which is calculated as follows:
ΔGH* = ΔEH* + ΔZPEH* − TΔSH*
Here, EH* is the total energy of the system with one adsorbed H atom and represents the energy of an isolated H2 molecule. ΔZPEH* is calculated by , where the ZPEH* denotes the vibrational energy of the adsorbed H atom on the active site and the value of is 0.27 eV. Further, ΔSH* is the entropy change of H* adsorption, which is obtained by 1/2 . The TΔSH* value is equal to 1/2 , which is −0.20 eV at 300 K, obtained by Nørskov et al. [32]. The ideal ΔGH* value is 0 eV.
2.3. Gibbs Free Energy Change of Hydroxyl Adsorption
While hydrogen adsorption energy (ΔGH) primarily determines the alkaline HER activity, the Gibbs free energy change of OH adsorption energy (ΔGOH*) is found to be equally important since it is responsible for the water dissociation and the amount of hydrogen available [44]. Therefore, the adsorption and desorption of both reaction intermediates (OH* and H*, see Equation (2)) dictate the overall alkaline HER efficiency [45].
The ΔGOH* is calculated by using the computational hydrogen electrode (CHE) method:
where ΔEOH* describes the binding energy of an OH group at the active site, which is calculated as follows:
where EOH* is the total energy of the system with one adsorbed OH intermediate, the ΔZPE − TΔS value is 0.35 eV at 300 K, obtained by Nørskov et al. The ideal ΔGOH* value is −0.3 eV for the alkaline HER [46].
ΔGOH* = ΔEOH* + ΔZPE − TΔS
3. Applications of Descriptors
3.1. Water Adsorption and Dissociation
Figure 1 shows that water adsorption is the first step in generating hydrogen atom intermediates for alkaline HER. The H2O dissociation becomes a rate-determining step that regulates the catalytic HER activity in alkaline media [47,48]. To this end, the adsorption energy of water and its dissociation energy barriers have been used as descriptors in some research to explain the performance of alkaline HER electrocatalysts.
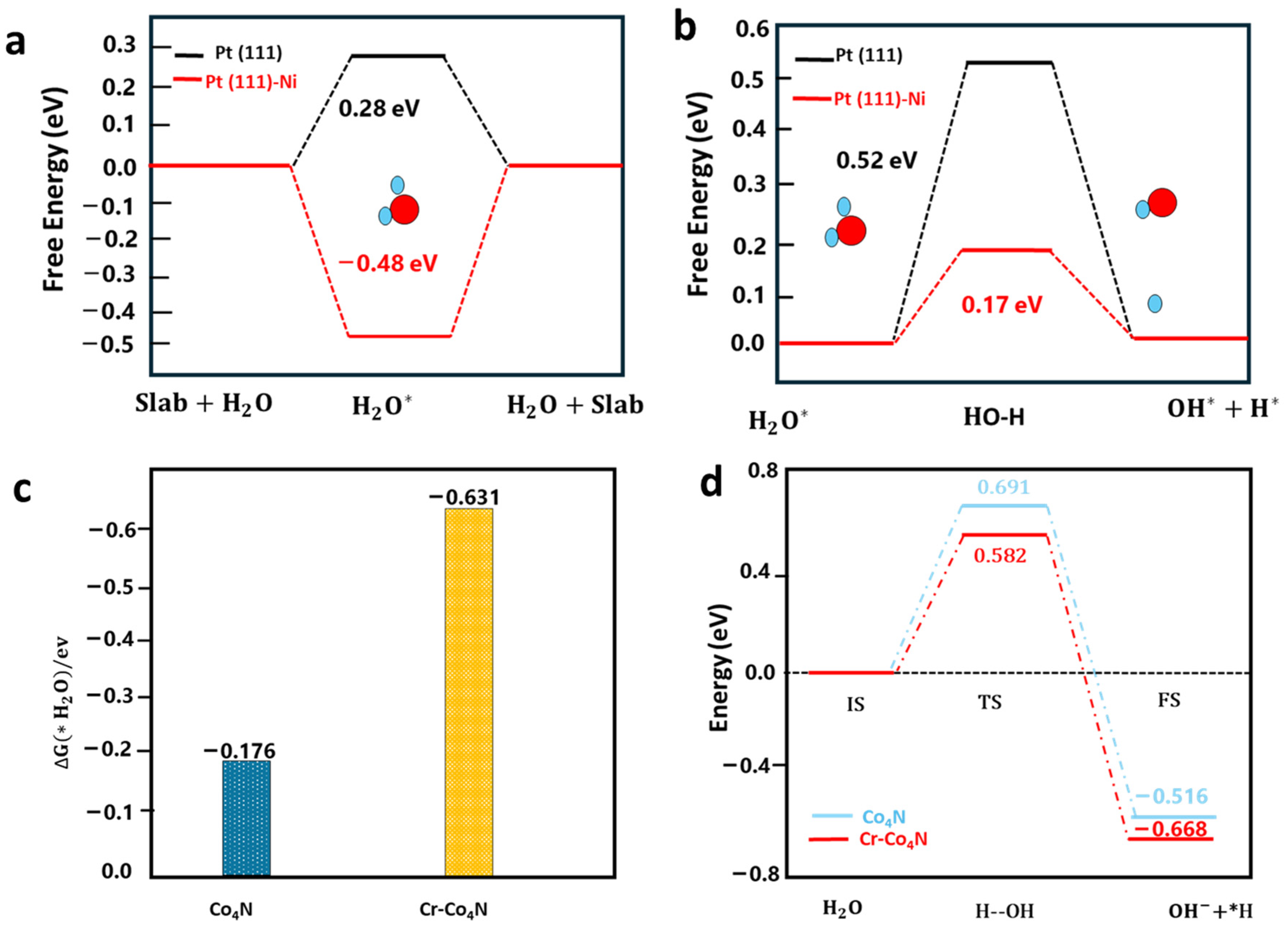
For example, Chen et al. experimentally synthesized a Ni-doped Pt catalyst on carbon support at 500 °C (termed as NiPt-C-500), which has a low overpotential of 14.0 mV at 10 mA/cm2 in the alkaline media [49], using the adsorption energy of water molecules and their dissociation energy barriers as descriptors through DFT computations at the PBE-GGA level to identify the active sites of the NiPt-C-500 catalyst. Figure 2a shows that H2O can be adsorbed on the Ni sites with an adsorption energy of −0.48 eV. As a comparison, the water adsorption energy on Pt is +0.37 eV. The positive value suggests that the adsorption on Pt is energetically unfavorable. Moreover, a lower H2O-dissociation barrier on Ni-doped Pt (111) of 0.17 eV was theoretically revealed compared to that on Pt(111), as illustrated in Figure 2b. This suggests a rapid dissociation of H2O catalyzed by surface Ni atoms on the Pt surface in NiPt-C-500 electrocatalysts, which can increase the rate of H* generation for alkaline HER. Using the same descriptors, Yao et al. resorted to DFT computations to explain the high performance of Cr-doped Co4N nanorods on carbon cloth (Cr-Co4N) for alkaline HER [50]. Figure 2c shows that the calculated H2O adsorption energy on the top of the Cr dopant within Cr-Co4N is −0.63 eV, which is much lower than that on the surface Co of Co4N (−0.18 eV). Therefore, the water adsorption on Cr dopants is substantially stronger than on undoped Co4N. This is because the oxygen atom of water carries a negative Bader charge of −1.13 |e|. As a result, Cr atoms with a higher positive charge (0.72 |e|) than surface Co (0.21 |e|) can more strongly adsorb water molecules via higher electrostatic interactions. The enhanced adsorption strength of water can further facilitate the dissociation of the adsorbed water and lower the dissociation barrier, as illustrated in Figure 2d. It explains the experimental observation that introducing Cr dopants can boost the alkaline HER activity.
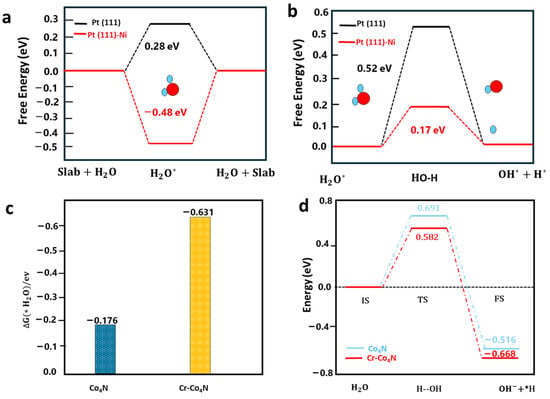
Figure 2.
(a) DFT-calculated water adsorption energies () and (b) water dissociation energy barriers (ΔEa) on Pt (1 1 1) and Pt (1 1 1)-Ni surfaces. Reprinted with permission from Ref. [49]. Copyright 2023 Elsevier. (c) DFT-calculated water adsorption energies () and (d) water dissociation energy barriers (ΔEa) on Co4N Cr-Co4N. Reprinted with permission from Ref. [50]. Copyright 2019 Wiley.
More examples of studies using the * to identify the active sites of the alkaline HER electrocatalysts can be found in Table 1. Similarly, some examples of using the ΔEa as the descriptor to explain the performance of alkaline HER electrocatalysts are listed in Table 2.

Table 1.
Water adsorption energies as descriptors to identify the active site of the alkaline HER electrocatalyst.
Table 2.
The dissociation energy barrier of the adsorption water as descriptors to identify the active site of the alkaline HER electrocatalyst.
3.2. Hydrogen Atom Adsorption
The Gibbs free energy change of the intermediate H atom adsorption (ΔGH*) was proposed as a descriptor for HER by Nørskov and his co-workers. This descriptor has successfully identified the active site of acidic HER electrocatalysts and the relevant reaction mechanism [32]. A volcano plot is a valuable tool for understanding and optimizing catalyst performance in acidic HER. It is usually a plot of ∆GH∗ against activities (such as overpotential, specific current density, and turnover frequency) [64,65]. The highest point on the volcano map is the ideal ∆GH∗ value at which the catalytic activity is maximized. Catalysts located on the left side of the volcano plot exhibit excessive hydrogen binding (very strong), while those on the right side have insufficient hydrogen binding (very weak). According to the Sabatier principle, an optimum catalyst should exhibit adsorption energies that are neither too strong nor too weak. In the alkaline HER, the adsorption of H atoms at the active site is also important since it is related to the water dissociation, Volmer, Heyrovsky, and Tafel steps, as suggested by Equations (5)–(8) and Figure 1. To this end, the ΔGH* was also widely used in the analysis of the performance of the alkaline HER electrocatalysts [66].
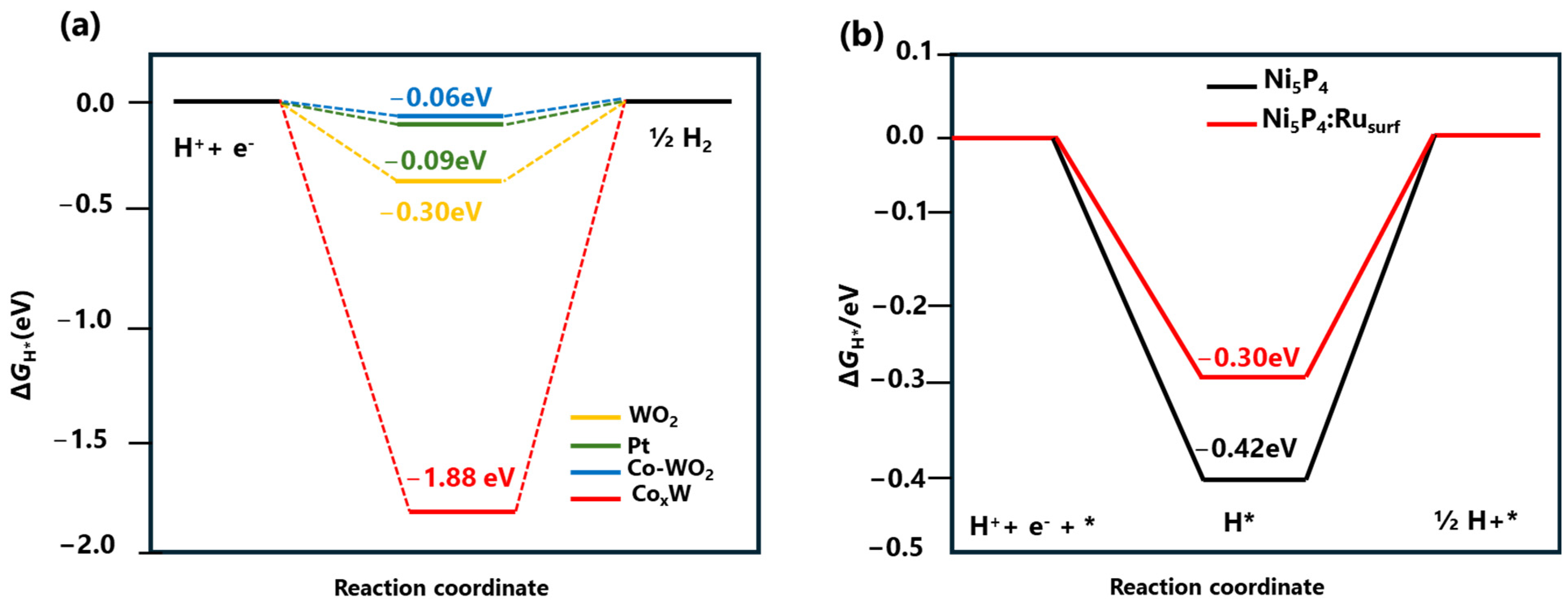
For example, Chen et al. adopted a dual approach to simultaneously promote water dissociation and hydrogen desorption kinetics with Co-doped WO2/amorphous CoxW hybrid catalysts using DFT [51]. As shown in Figure 3a, the ΔGH* on amorphous CoxW is −1.88 eV, indicating a strong hydrogen-binding interaction that prevents H2 production and desorption (Figure 4a). The most catalytically active site on Co−WO2(011) has a ΔGH* of −0.06 eV, closer to thermoneutral than Pt’s −0.09 eV, indicating significant hydrogen adsorption capability. According to DFT calculations, the HER on Co-doped WO2/amorphous COxW hybrid catalyst follows the Volmer-Tafel step, where water molecules are activated and cleaved to form H atoms on the surface of amorphous CoxW. These H atoms then rapidly combine to form H2 on the surface of Co-doped WO2. He et al. incorporated a single Ru atom into Ni5P4 to achieve an effective electrocatalyst for alkaline HER [67]. The DFT calculations were conducted to comprehend the impact of single-atomic Ru incorporation into Ni5P4 on its catalytic and structural properties. The structures of Ni5P4 were determined, both with and without Ru incorporation, and they revealed several potential catalytic sites. Experimental analysis and the established model structural parameters were remarkably congruent. In the case of Ni5P4-Ru, the computed ΔGH* value at the Ru-doped sites was −0.30 eV, which was comparatively higher than the value of −0.42 eV observed at the site of P of pristine Ni5P4 (see Figure 3b). This finding indicated that the Ru dopant exhibited favorable energetics for the desorption of absorbed H atoms to form the H2 gas.
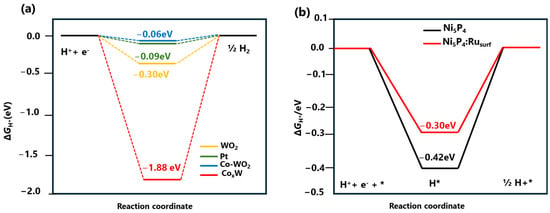
Figure 3.
(a) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*) on WO2, Co-WO2, CoxW, and Pt. Reprinted with permission from Ref. [51]. Copyright 2019 American Chemical Society. (b) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*) on Ni5P4 and Ni5P4: Ru. Reprinted with permission from Ref. [67]. Copyright 2020 Wiley.
Table 3 lists more examples of using ΔGH* as the descriptor to identify the active site of the alkaline HER electrocatalysts and evaluate their performance.
Table 3.
Gibbs free energy change of H intermediate adsorption as descriptors to identify the active site of the alkaline HER electrocatalyst.
3.3. Hydrogen and Hydroxyl Adsorption
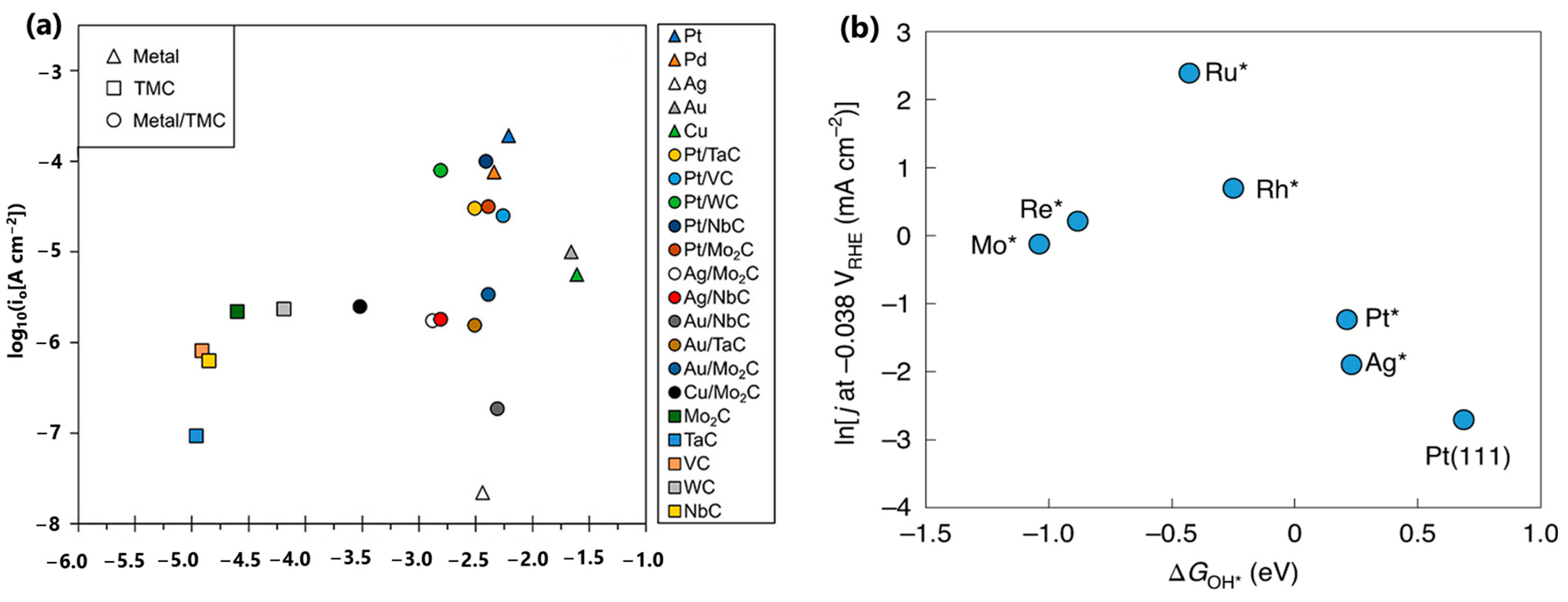
In recent studies, the volcano plot has been adopted to describe HER activities in alkaline media by plotting ΔGOH* against activities. In contrast to the volcano-shaped association observed with ΔGH*, Zhang et al. demonstrated that ΔGOH* exhibits a significantly weaker correlation with alkaline HER activity than the volcano-shaped relationship established with ΔGH* [29]. Au-modified TMCs exhibit OHBEs comparable to those of Pt and Pt-modified TMCs; however, the alkaline exchange current densities of Au/TMCs are two to three orders of magnitude lower than those of their Pt counterparts. Furthermore, carbides modified with Ag and Cu exhibit a broad spectrum of ΔGOH*, but there is no discernible HER trend within this spectrum. According to the research of Zhang et al., ΔGOH* is not an appropriate descriptor for alkaline HER on TMCs and metal-modified TMCs. It also implies that the adsorbed hydroxyl group does not directly participate in the rate-determining step of alkaline HER kinetics on these surfaces [29].
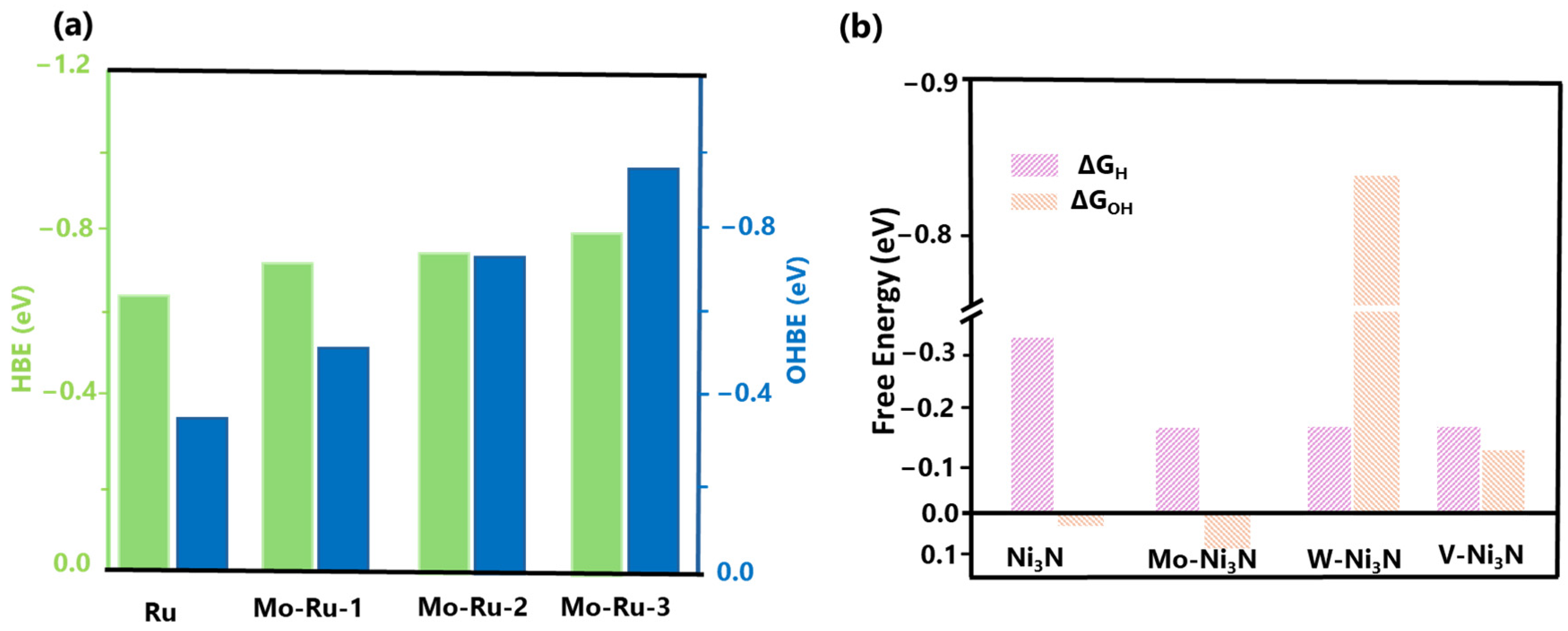
Markovic et al. were the first to study the impact of adsorbed OH on Pt-based electrocatalysts, emphasizing its poisoning effect on surface sites and its influence on the kinetics of the alkaline HER [69]. The hydrogen binding energy (HBE) and hydroxyl binding energy (OHBE) values for pure Ru and various Mo-Ru compounds were computed (Figure 4a) by Zhao et al. Through DFT, the optimal H and OH adsorption sites on distinct catalysts for alkaline HER were identified [70]. In the Mo-Ru-1 structure, one of the Ru atoms is doped with Mo, while Mo-Ru-2 and Mo-Ru-3 are doped with two and four atoms of Mo, respectively. Figure 4a displays the HBE (ΔGH*) values of Ru, Mo-Ru-1, Mo-Ru-2, and Mo-Ru-3, which are −0.64, −0.69, −0.67, and −0.76 eV, respectively. These results indicate that the adsorption energy of H is similar for all catalysts and nearly identical to experimental values [25,71,72]. The OHBE (ΔGOH*) values of Mo-Ru-1 (−0.53 eV), Mo-Ru-2 (−0.68 eV), and Mo-Ru-3 (−0.97 eV) exhibited a substantial increase after Mo-doping, in comparison to native Ru (−0.31 eV). Consequently, the OHBE progressively rose as the Mo atom content increased. Furthermore, the increased OHBE after Mo-doping can be ascribed to the augmented HOR/HER activity of Mo-Ru.
Zhang et al. designed high-efficiency alkaline HER electrocatalysts using a unique dual descriptor of optimal free energies (ΔGH* and ΔGOH*). Ni3N surface reactivity was tailored using theory to balance the adsorption energies of hydrogen and hydroxyl species. Nickel-based materials are highly promising non-noble metal electrocatalysts for the hydrogen evolution reaction (HER) [73,74]. In this study, the metallic Ni3N was considered a case study to illustrate the application of dual-descriptor-driven design. Ni3N has been regarded as a suitable electrocatalyst for cleaving OH-H bonds in the Volmer phase. According to Figure 4b, the DFT calculations demonstrate that the Ni3N (111) surface exhibits a facile hydroxyl adsorption free energy of 0.03 eV. This characteristic is advantageous for the energetics of water dissociation and subsequent hydroxyl desorption [75]. Such findings align with prior studies indicating that Ni3N can function as a promoter of water dissociation [76]. Nevertheless, the DFT calculations indicate that the average GH* value observed on the surface of Ni3N (111) is approximately −0.31 eV, which surpasses the ideal value of ΔGH* = 0 eV by a wide margin. The excessive reactivity of the surface results in unfavorable desorption of hydrogen and subsequent formation of H2. This approach has successfully enhanced the hydrogen evolution reaction (HER) performance by incorporating various transition-metal dopants into Ni3N, including Mo-Ni3N, W-Ni3N, and V-Ni3N [30]. The hydrogen and hydroxyl binding energy results on Mo-Ni3N effectively balance the dual descriptors.
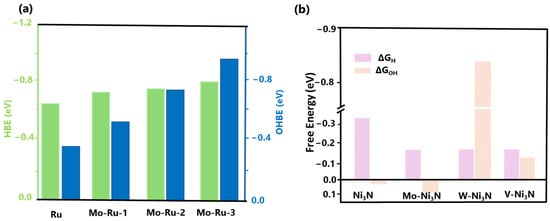
Figure 4.
(a) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*, HBE) and the Gibbs free energy change of OH adsorption energy (ΔGOH*, OHBE) on Ru, Mo-Ru-1, Mo-Ru-2, Mo-Ru-3. Reprinted with permission from Ref. [70]. Copyright 2022 American Chemical Society. (b) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*) and the Gibbs free energy change of OH adsorption energy (ΔGOH*) on Ni3N, Mo-Ni3N, W-Ni3N, and V-Ni3N. Reprinted with permission from Ref. [30]. Copyright 2019 American Chemical Society.
Figure 4.
(a) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*, HBE) and the Gibbs free energy change of OH adsorption energy (ΔGOH*, OHBE) on Ru, Mo-Ru-1, Mo-Ru-2, Mo-Ru-3. Reprinted with permission from Ref. [70]. Copyright 2022 American Chemical Society. (b) DFT-calculated Gibbs free energy change of the intermediate H atom adsorption (ΔGH*) and the Gibbs free energy change of OH adsorption energy (ΔGOH*) on Ni3N, Mo-Ni3N, W-Ni3N, and V-Ni3N. Reprinted with permission from Ref. [30]. Copyright 2019 American Chemical Society.
More examples of using ΔGH* and ΔGOH* as the descriptor to identify the active site of the alkaline HER electrocatalysts and evaluate their performance are listed in Table 4.
Table 4.
Gibbs free energy change of OH intermediate adsorption as descriptors to identify the active site of the alkaline HER electrocatalyst.
4. Discussion
The selection of the right descriptor for screening alkaline HER electrocatalysts is a long-standing debate, especially using the ΔGOH* descriptor. For example, Zhang et al. demonstrated that ΔGOH* exhibits a significantly weaker correlation with alkaline HER activity than the volcano-shaped relationship established with ΔGH* (Figure 5a), which implies that the adsorbed hydroxyl group does not directly participate in the rate-determining step of alkaline HER kinetics on these surfaces [29]. Contrastingly, McCrum et al. found that the performance of alkaline HER electrocatalysts shows a volcano relationship between the natural logarithm of the experimentally measured rate of hydrogen evolution and the DFT-calculated ΔGOH* [19]. The reaction is bifunctional as it involves both ΔGH* and ΔGOH* on the too-strong OH binding side of the volcano. On the too-weak OH binding side of the volcano, ΔGH* is only apparently bifunctional; ΔGOH* is not a useful descriptor for evaluating the performance of catalysts. The different conclusions of these two studies may be ascribed to the scopes of ΔGOH* values. All the ΔGOH* values from Zhang et al. shown in Figure 5a are less than −1.0 eV, which belong to the too-strong OH binding side suggested in Figure 5b. As a result, no volcanic relationship can be identified in Figure 5a. Since all ΔGOH* values are far from the optimal value of −0.3 eV, the performance of these metal-modified transition metal carbides is more affected by the ΔGH* value, as suggested by Zhang et al. [29].
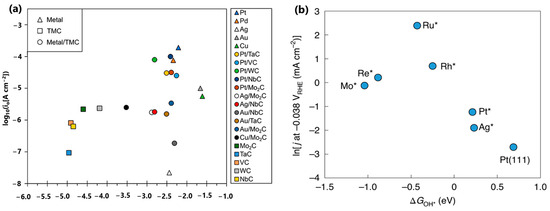
Figure 5.
(a) The logarithm of the experimentally measured rate of hydrogen evolution of metal-modified transition metal carbides in 0.1 M KOH and the corresponding DFT-calculated ΔGOH*. Reprinted with permission from Ref. [29]. Copyright 2019 American Chemical Society. (b) The natural logarithm of the experimentally measured rate of hydrogen evolution of Pt (553), Pt(553) with Mo*, Re*, Ru*, Rh*, and Ag* adsorbed at the step, and Pt(111) in 0.1 M NaOH and the corresponding DFT-calculated ΔGOH*. Reprinted with permission from Ref. [19]. Copyright 2020 Springer Nature.
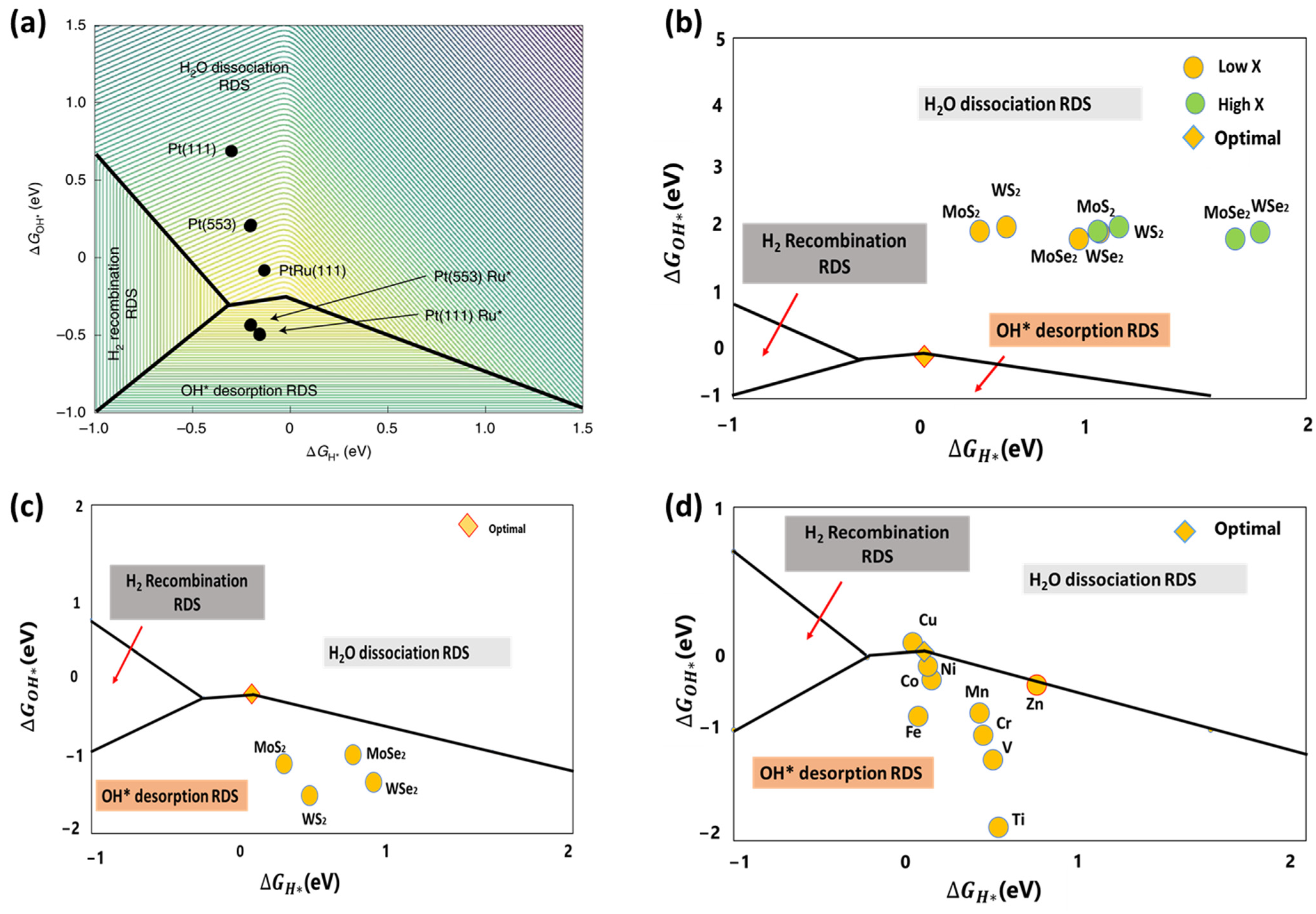
To better screen the alkaline HER electrocatalysts, McCrum et al. adopted a three-dimensional (3D) volcano plot depicting the rate of hydrogen evolution in relation to the ΔGOH* and ΔGH*, as illustrated in Figure 6a. Catalysts exhibiting minimal hydrogen and hydroxide binding energies yield the lowest rates, as indicated by the purple area on the upper right of Figure 6a. The catalysts show the highest performances when they bind hydrogen at an intermediate strength at 0 VRHE (near 0 eV) and hydroxide strongly (near −0.3 eV) (yellow in the middle of Figure 6a). The too-strong H and OH binding leads the H2 or OH* desorption to become the rate-determining step. Consequently, Figure 6a qualitatively represents trends in alkaline HER kinetics and provides design guidelines for efficient catalysts. For example, to improve the hydrogen evolution rate of the well-studied Pt(111) electrocatalysts in an alkaline solution, the ΔGH* must be increased by approximately 0.2 eV. In comparison, the ΔGOH* must be significantly reduced by approximately 0.9 eV through appropriate manipulation strategies such as doping, surface engineering, and defect engineering [19].
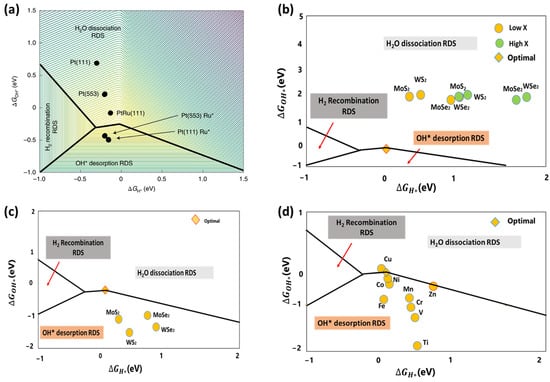
Figure 6.
(a) Logarithm of the rate of hydrogen evolution (contours) as a function of DFT-calculated ΔGH* and ΔGOH* on Pt(111), Pt(553), Ru* adsorbed at the step of Pt(553), PtRu(111) alloy and Ru* clusters on Pt(111). Reprinted with permission from Ref. [19]. Copyright 2020 Springer Nature. (b) Plot of ΔGOH* against ΔGH* of pristine MX2 TMDs (M = Mo, W and X = S, Se), (c) plot of ΔGOH* against ΔGH* of MX2 TMDs with a single X vacancy; and (d) plot of ΔGOH* against ΔGH* of MX2 TMDs with 3d TM dopant next to the single X vacancy. Reprinted with permission from Ref. [77]. Copyright 2024 IOPscience.
Following these guidelines, we employed both ΔGH* and ΔGOH* descriptors to investigate the catalytic HER performance of 1T’ transition metal dichalcogenides (TMDs such as MoSe2, MoS2, WSe2, and WS2) in an alkaline solution using DFT [77]. Our findings indicate that the pristine sulfides exhibited superior alkaline HER performance compared to their selenide counterparts. Nevertheless, the activities of all pristine 1T’ TMDs are insufficient to dissociate water (See Figure 6b). Defect engineering techniques were employed to improve the reactivity of TMD-based electrocatalysts. Our DFT results indicate that the reactivities of TMD materials can be enhanced by introducing single S/Se vacancy defects, as shown in Figure 6c. However, the rate-determining phase is the desorption of OH. The reactivities of active sites were further regulated to achieve optimal OH desorption by doping defective MoS2 with late 3d transition metal (TM) atoms, particularly Cu, Ni, and Co, as illustrated in Figure 6d. Consequently, the TM-doped defective 1T’ MoS2 can substantially improve the alkaline HER performance, which matches the recently reported experimental observations.
It is crucial to remember that each descriptor has limitations and challenges when studying alkaline HER in alkaline media. Descriptors such as water adsorption energy and water dissociation energy barriers serve as indicators of material reactivity. They are closely linked to the Volmer step, which encompasses the adsorption of water molecules and their subsequent dissociation into adsorbed hydrogen (H*) and hydroxide ions (OH−). These steps are fundamental to the overall HER process, yet they do not provide a complete picture of the catalytic site’s capabilities, especially concerning the Heyrovsky and Tafel steps. The Heyrovsky step involves the electrochemical desorption of H* to form hydrogen gas.
The ΔGH* has been the most prevalent descriptor for assessing HER performance at the Heyrovsky and Tafel steps. It offers a measure of the free energy change when H* is adsorbed on the catalyst surface, which is a critical factor in determining the rate of the HER. However, ΔGH* alone is insufficient for evaluating the Volmer step or the desorption efficiency of adsorbed OH, which are also essential for a complete understanding of the HER mechanism.
The Gibbs free energy of hydroxide adsorption (ΔGOH*) can be used to evaluate the desorption efficiency of adsorbed OH, providing insights into the potential for water dissociation, as suggested by the Bronsted-Evans-Polanyi (BEP) relationship [19]. This relationship posits a linear correlation between the activation energy of a reaction and the reaction enthalpy, allowing for the prediction of reaction barriers based on thermodynamic parameters. However, it does not offer information on the catalyst’s performance during the Heyrovsky or Tafel steps. Therefore, relying solely on ΔGOH* would give an incomplete assessment of a catalyst’s overall activity and efficiency.
Given these considerations, it is evident that no single descriptor can fully encapsulate the complexities of the HER process. Researchers must carefully select and combine multiple descriptors to gain a comprehensive understanding of the catalytic activity and to design more efficient catalysts. This approach allows for the evaluation of catalysts across all steps of the HER, ensuring a more accurate prediction of their performance in real-world applications.
It is also worth noting that different descriptors may be required to understand the performance of alkaline HER composite electrocatalysts. For example, Chen et al. designed a complicated composite electrocatalyst including a Co dopant on WO2/amorphous CoxW hybrid materials [51]. Using the water adsorption energy and dissociation energy barrier energy as the descriptor, they found that the amorphous CoxW is the active site. However, the analysis of the ΔGH* descriptor indicates that Co-doped WO2 is the active site for H2 formation. The DFT calculations then suggest that the individual component of this composite electrocatalyst has a different function. The synergy between them enables the high alkaline HER performance of this Co-Doped WO2/amorphous CoxW hybrid electrocatalyst.
5. Conclusions and Outlook
The hydrogen evolution reaction (HER) in alkaline media is a prominent method for large-scale hydrogen production from an electrolyzer. Alkaline Water Electrolysis (AWE) has demonstrated superior cost-effectiveness to acidic proton exchange membrane (PEM). One of the biggest challenges of AWE is ascribed to the relatively low energy conversion efficiency of their electrocatalysts. To address this issue, selecting the right computational tools for theoretical studies is essential in designing high-performance alkaline HER electrocatalysts. The descriptors used to evaluate the energy conversion efficiency of electrocatalysts include water adsorption energy, water dissociation barrier, Gibbs free energy change of hydrogen adsorption, and Gibbs free energy change of hydroxyl adsorption from DFT calculations. In the review, some of the latest examples have been used to illustrate the applications of different descriptors. It reveals that the hydroxyl (ΔGOH*) adsorption process is one of the important parameters often ignored in many previous theoretical studies. When the adsorption strength of hydroxyl is weak, it suggests a high water dissociation energy barrier due to the BEP relationship [19]. The water cannot effectively interact with the active site to provide an H atom intermediate. At the same time, the weak adsorption of OH also indicates that the dissolved hydroxide is via the Volmer (II) mechanism (Equation (6)), as shown in Figure 1. In this case, the ΔGH* becomes a more important descriptor to analyze and predict the efficiency of an active site. It explains the great matches between some experimental measurements and the trend of the ΔGH* descriptor in some combined studies.
The CHE model has evolved to include previously overlooked factors such as solvent and electrolyte effects, reflecting its adaptability [18]. Despite these advancements, a knowledge gap persists, particularly in the dynamic aspects of water splitting. A significant hurdle remains in developing a more accurate model of the electrode–electrolyte interface, which is crucial for pinpointing active sites in HER and connecting microscopic interactions with macroscopic observations. The electrified solid–liquid interface in alkaline HERs largely determines the charge transfer rate of electrochemical redox reactions [78]. Innovative approaches, such as engineering a localized acid-like environment within an alkaline medium, have significantly boosted HER performance significantly [79]. This is achieved by tailoring the local reaction conditions, which are critical for the process. Moreover, understanding the impact of variables like electrolyte concentration on HER allows for further refinement of reaction conditions [16]. The descriptors described in this review can only provide limited dynamic information about the electrochemical processes at the electrode–electrolyte interface [15,21,22]. This is because the atomistic models used in most of these studies make it difficult to comprehend the thermodynamic state and dynamic properties of interfacial processes [21]. The interaction between water molecules and the electrified surface must be investigated by examining scenarios in which water molecules are introduced to the catalyst surface in various configurations [80]. Insights into the hydrogen bonding network and structural characteristics of the water layer can be derived from the average dipole orientation of water molecules relative to the surface normal [80]. Furthermore, the complex relationship among adsorbed hydrogen, water molecules, and the electrochemical environment, which influences the behaviors of adsorption and the overall characteristics of the interface between solid and water, needs to be investigated [81]. To comprehend the activities of HER intermediates in alkaline media, it is critical to know the potential and pH-dependent adsorption energies of these intermediates [82]. To this end, it is clear that theoretical studies on alkaline HER are still an area ripe for further research.
The intersection of machine learning (ML) with density functional theory and multiscale modeling is a burgeoning field that holds great promise for advancing materials science and chemistry [83,84,85,86]. By leveraging ML’s ability to analyze vast datasets and identify patterns, researchers can significantly reduce the computational resources required for DFT calculations. This synergy enables the prediction of complex chemical behaviors and the design of new materials with tailored properties. However, the success of the ML methods hinges on the quality and consistency of the data fed into the ML models [84]. As such, creating comprehensive and reliable databases is crucial for training algorithms that can accurately predict electrochemical behaviors and guide the development of efficient alkaline HER electrocatalysts. This integrated approach is set to transform the landscape of computational chemistry, offering a more streamlined and precise method for exploring the vast potential of chemical space. They can predict the performance of electrocatalysts, thereby informing the design of next-generation materials with enhanced catalytic properties. These concerted efforts in theoretical research are essential for the progression of clean energy technologies.
In sum, the exploration of electrocatalytic mechanisms in alkaline HER is a complex field that necessitates a comprehensive approach. Advancements beyond the DFT-calculated descriptors are critical for a deeper understanding. Operando simulations offer a dynamic perspective by considering actual working conditions, providing insights into the real-time structural and chemical changes during the reaction process. Meanwhile, ML-based force fields for classical molecular dynamics and Monte Carlo simulations represent a significant leap in mesoscale modeling, enabling simulations that capture the nuanced interactions within molecular systems. These ML models can bridge the gap between classical and quantum mechanical accuracy, offering a more detailed view of the catalytic processes. Lastly, ML-driven high-throughput screening is revolutionizing the way electrocatalysts are discovered and optimized. By analyzing vast datasets, ML algorithms can predict performance, stability, and efficiency, thereby accelerating the development of new materials for HER. Together, these methodologies form a multi-faceted approach that could significantly advance the field of electrocatalysis.
Author Contributions
Methodology, Formal analysis, Writing–original draft, S.A.O.; Writing—review & editing, F.M.; Writing—review & editing, Funding Request, A.B.; Writing—review & editing, J.J.H.; Writing—review & editing, L.Z.; Writing–review & editing, Z.W.; Writing—review & editing, S.B.; Writing—review & editing, Y.Z.; Project administration, Funding Request, Supervision, Conceptualization, Resources, Writing—review & editing, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge financial support from the Australian Research Council Discovery Project (Grant No. DP210103266, DP210104010). This research was conducted on the supercomputers in National Computational Infrastructure (NCI) in Canberra, Australia, which is supported by the Australian Commonwealth Government, and Pawsey Supercomputing Centre in Perth with the funding from the Australian government and the Government of Western Australia. Calculations were performed by using resources from Grand Equipement National de Calcul Intensif (GENCI, grant no. A0120913426). Computational resources provided by the computing facilities Mésocentre de Calcul Intensif Aquitain (MCIA) of the Université de Bordeaux and of the Université de Pau et des Pays de l’Adour. We acknowledge funding from CNRS Institute of Chemistry through the “International Emerging Actions 2022” mobility grant (2DH2 project) and from Agence national de recherche under the RECIFE ANR-DFG project (Grant Number ANR-21-CE08-0036-01).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- de Kleijne, K.; Huijbregts, M.A.J.; Knobloch, F.; van Zelm, R.; Hilbers, J.P.; de Coninck, H.; Hanssen, S.V. Worldwide greenhouse gas emissions of green hydrogen production and transport. Nat. Energy 2024, 1–14. [Google Scholar] [CrossRef]
- Wei, C.; Xu, Z.J. The Comprehensive Understanding of as an Evaluation Parameter for Electrochemical Water Splitting; Wiley Online Library: Hoboken, NJ, USA, 2018; Volume 2, p. 1800168. [Google Scholar]
- Horri, B.A.; Ozcan, H. Green hydrogen production by water electrolysis: Current status and challenges. Curr. Opin. Green. Sust. 2024, 47, 100932. [Google Scholar] [CrossRef]
- Ghosh, S.; Basu, S. Solid Oxide Electrolysis Cell for Hydrogen Generation: General Perspective and Mechanism. In Climate Action and Hydrogen Economy: Technologies Shaping the Energy Transition; Springer: Berlin/Heidelberg, Germany, 2024; pp. 231–260. [Google Scholar]
- Laguna-Bercero, M.A.; Wang, Y.; Zhou, X.-D.; Zhu, L. Fundamentals of Solid Oxide Electrolysis Cells (SOEC). In High. Temperature Electrolysis; Springer: Berlin/Heidelberg, Germany, 2023; pp. 5–34. [Google Scholar]
- Tao, H.B.; Liu, H.; Lao, K.J.; Pan, Y.P.; Tao, Y.B.; Wen, L.R.; Zheng, N.F. The gap between academic research on proton exchange membrane water electrolysers and industrial demands. Nat. Nanotechnol. 2024, 19, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhao, C.F.; Li, H.Y.; Shen, S.Y.; Yan, X.H.; Zhang, J.L. Rational electrode design for low-cost proton exchange membrane water electrolyzers. Cell Rep. Phys. Sci. 2024, 5, 101880. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Vasileff, A.; Qiao, S.Z. The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts. Angew. Chem. Int. Edit. 2018, 57, 7568–7579. [Google Scholar] [CrossRef]
- Pitchai, C.; Vedanarayanan, M.; Chen, C.-M.; Gopalakrishnan, S.M. Enhanced electrochemical efficiency of the open porous sandrose structured electrocatalyst for sustainable hydrogen and oxygen evolution reactions. Int. J. Hydrogen Energy 2024, 72, 755–763. [Google Scholar] [CrossRef]
- Pitchai, C.; Gopalakrishnan, S.M.; Chen, C.-M. Ultra-efficient Nitrogen-Doped Carbon Dots-Supported Nickel Sulfide as a Platinum-Free Electrocatalyst for Overall Water Splitting in Basic Medium. Energy Fuels 2024, 38, 2235–2247. [Google Scholar] [CrossRef]
- Hu, C.L.; Zhang, L.; Gong, J.L. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 2019, 12, 2620–2645. [Google Scholar] [CrossRef]
- Ehlers, J.C.; Feidenhans’l, A.A.; Therkildsen, K.T.; Larrazabal, G.O. Affordable Green Hydrogen from Alkaline Water Electrolysis: Key Research Needs from an Industrial Perspective. ACS Energy Lett. 2023, 8, 1502–1509. [Google Scholar] [CrossRef]
- Du, N.Y.; Roy, C.; Peach, R.; Turnbull, M.; Thiele, S.; Bock, C. Anion-Exchange Membrane Water Electrolyzers. Chem. Rev. 2022, 122, 11830–11895. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jing, Z.; Bai, H.; Chen, Z.; Osman, A.I.; Farghali, M.; Rooney, D.W.; Yap, P.-S. Optimizing hydrogen production by alkaline water decomposition with transition metal-based electrocatalysts. Environ. Chem. Lett. 2023, 21, 2583–2617. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y. Theoretical Identification and Understanding of Catalytic Active Sites for Water Splitting Reactions. Catalysis 2022, 34, 1–16. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Liu, J.X.; Al-Mamun, M.; Liew, A.W.C.; Yin, H.J.; Wen, W.; Zhong, Y.L.; Liu, P.R.; Zhao, H.J. Electrolyte Effect on Electrocatalytic Hydrogen Evolution Performance of One-Dimensional Cobalt-Dithiolene Metal-Organic Frameworks: A Theoretical Perspective. ACS Appl. Energy Mater. 2018, 1, 1688–1694. [Google Scholar] [CrossRef]
- Liu, J.X.; Yin, H.J.; Liu, P.R.; Chen, S.; Yin, S.W.; Wang, W.L.; Zhao, H.J.; Wang, Y. Theoretical Understanding of Electrocatalytic Hydrogen Production Performance by Low-Dimensional Metal-Organic Frameworks on the Basis of Resonant Charge-Transfer Mechanisms. J. Phys. Chem. Lett. 2019, 10, 6955–6961. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jónsson, H.; Nørskov, J.K. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt (111) electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3250. [Google Scholar] [CrossRef]
- McCrum, I.T.; Koper, M.T. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat. Energy 2020, 5, 891–899. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, S.; Wang, Y. Mathematical Modeling for Enhanced Electrochemical Properties. In Metal-Air Batteries; CRC Press: Boca Raton, FL, USA, 2023; pp. 45–63. [Google Scholar]
- Wang, Y. Multiscale Modeling of Electrochemical Reactions and Processes; AIP Publishing LLC: Melville, NY, USA, 2021. [Google Scholar]
- Hinsch, J.J.; Wang, Y. DFT and Simulation of Solid-Liquid Interface Properties and Processes; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Norskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Zheng, J.; Nash, J.; Xu, B.; Yan, Y. Perspective—Towards establishing apparent hydrogen binding energy as the descriptor for hydrogen oxidation/evolution reactions. J. Electrochem. Soc. 2018, 165, H27. [Google Scholar] [CrossRef]
- Fung, V.; Hu, G.; Wu, Z.; Jiang, D.-E. Descriptors for hydrogen evolution on single atom catalysts in nitrogen-doped graphene. J. Phys. Chem. C 2020, 124, 19571–19578. [Google Scholar] [CrossRef]
- Mao, B.; Sun, P.; Jiang, Y.; Meng, T.; Guo, D.; Qin, J.; Cao, M. Identifying the transfer kinetics of adsorbed hydroxyl as a descriptor of alkaline hydrogen evolution reaction. Angew. Chem. 2020, 132, 15344–15349. [Google Scholar] [CrossRef]
- Jia, Q.; Liang, W.; Bates, M.K.; Mani, P.; Lee, W.; Mukerjee, S. Activity descriptor identification for oxygen reduction on platinum-based bimetallic nanoparticles: In situ observation of the linear composition–strain–activity relationship. ACS Nano 2015, 9, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, Z.; Tackett, B.M.; Denny, S.R.; Tian, B.; Chen, X.; Wang, B.; Chen, J.G. Trends and descriptors of metal-modified transition metal carbides for hydrogen evolution in alkaline electrolyte. ACS Catal. 2019, 9, 2415–2422. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Liu, J.; Zhang, L.; Wan, H.; Miao, L.; Jiang, J. Dual-descriptor tailoring: The hydroxyl adsorption energy-dependent hydrogen evolution kinetics of high-valance state doped Ni3N in alkaline media. ACS Catal. 2019, 9, 9332–9338. [Google Scholar] [CrossRef]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Ostergaard, F.C.; Bagger, A.; Rossmeisl, J. Predicting catalytic activity in hydrogen evolution reaction. Curr. Opin. Electrochem. 2022, 35, 101037. [Google Scholar] [CrossRef]
- Norskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Ďurovič, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the hydrogen evolution reaction in alkaline and neutral media. A comparative review. J. Power Sources 2021, 493, 229708. [Google Scholar] [CrossRef]
- Ledezma-Yanez, I.; Wallace, W.D.Z.; Sebastian-Pascual, P.; Climent, V.; Feliu, J.M.; Koper, M.T.M. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat. Energy 2017, 2, 1–7. [Google Scholar] [CrossRef]
- Zhang, L.; Gao, X.; Zhu, Y.; Liu, A.; Dong, H.; Wu, D.; Han, Z.; Wang, W.; Fang, Y.; Zhang, J. Electrocatalytically inactive copper improves the water adsorption/dissociation on Ni 3 S 2 for accelerated alkaline and neutral hydrogen evolution. Nanoscale 2021, 13, 2456–2464. [Google Scholar] [CrossRef]
- Guha, A.; Sahoo, M.; Alam, K.; Rao, D.K.; Sen, P.; Narayanan, T.N. Role of water structure in alkaline water electrolysis. Iscience 2022, 25, 104835. [Google Scholar] [CrossRef]
- Zheng, X.; Yao, Y.; Ye, W.; Gao, P.; Liu, Y. Building up bimetallic active sites for electrocatalyzing hydrogen evolution reaction under acidic and alkaline conditions. Chem. Eng. J. 2021, 413, 128027. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- He, R.; Hua, J.; Zhang, A.; Wang, C.; Peng, J.; Chen, W.; Zeng, J. Molybdenum disulfide–black phosphorus hybrid nanosheets as a superior catalyst for electrochemical hydrogen evolution. Nano Lett. 2017, 17, 4311–4316. [Google Scholar] [CrossRef]
- Lin, L.; Zhou, W.; Gao, R.; Yao, S.; Zhang, X.; Xu, W.; Zheng, S.; Jiang, Z.; Yu, Q.; Li, Y.-W. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 2017, 544, 80–83. [Google Scholar] [CrossRef]
- Cuenya, B.R.; Behafarid, F. Nanocatalysis: Size-and shape-dependent chemisorption and catalytic reactivity. Surf. Sci. Rep. 2015, 70, 135–187. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, P.; Sheng, W. Hydrogen evolution and oxidation: Mechanistic studies and material advances. Adv. Mater. 2019, 31, 1808066. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Zhao, Z.; Zhang, Q.-H.; Huang, L.-B.; Gu, L.; Lu, G.; Hu, J.-S.; Wan, L.-J. Steering elementary steps towards efficient alkaline hydrogen evolution via size-dependent Ni/NiO nanoscale heterosurfaces. Natl. Sci. Rev. 2020, 7, 27–36. [Google Scholar] [CrossRef]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Chang, K.C.; Paulikas, A.; Stamenkovic, V.; Markovic, N.M. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew. Chem. 2012, 124, 12663–12666. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Kang, L.; Cai, D.; Geng, S.; Liu, Y.; Chen, J.; Song, S.; Wang, Y. Ultrafine Pt-based catalyst decorated with oxygenophilic Ni-sites accelerating alkaline H2O dissociation for efficient hydrogen evolution. J. Colloid Interface Sci. 2023, 650, 1715–1724. [Google Scholar] [CrossRef]
- Yao, N.; Li, P.; Zhou, Z.; Zhao, Y.; Cheng, G.; Chen, S.; Luo, W. Synergistically tuning water and hydrogen binding abilities over Co4N by Cr doping for exceptional alkaline hydrogen evolution electrocatalysis. Adv. Energy Mater. 2019, 9, 1902449. [Google Scholar] [CrossRef]
- Chen, J.; Jin, Q.; Li, Y.; Li, Y.; Cui, H.; Wang, C. Design superior alkaline hydrogen evolution electrocatalyst by engineering dual active sites for water dissociation and hydrogen desorption. ACS Appl. Mater. Interfaces 2019, 11, 38771–38778. [Google Scholar] [CrossRef]
- Kim, J.; Jung, H.; Jung, S.-M.; Hwang, J.; Kim, D.Y.; Lee, N.; Kim, K.-S.; Kwon, H.; Kim, Y.-T.; Han, J.W. Tailoring binding abilities by incorporating oxophilic transition metals on 3D nanostructured Ni arrays for accelerated alkaline hydrogen evolution reaction. J. Am. Chem. Soc. 2020, 143, 1399–1408. [Google Scholar] [CrossRef]
- Maslovara, S.; Anićijević, D.V.; Brković, S.; Georgijević, J.; Tasić, G.; Kaninski, M.M. Experimental and DFT study of CoCuMo ternary ionic activator for alkaline HER on Ni cathode. J. Electroanal. Chem. 2019, 839, 224–230. [Google Scholar] [CrossRef]
- You, B.; Liu, X.; Hu, G.; Gul, S.; Yano, J.; Jiang, D.-E.; Sun, Y. Universal surface engineering of transition metals for superior electrocatalytic hydrogen evolution in neutral water. J. Am. Chem. Soc. 2017, 139, 12283–12290. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, J.; Sun, M.; Lin, F.; Huang, B.; Lv, F.; Zeng, L.; Zhang, Q.; Gu, L.; Luo, M. Cu-Doped Heterointerfaced Ru/RuSe2 Nanosheets with Optimized H and H2O Adsorption Boost Hydrogen Evolution Catalysis. Adv. Mater. 2023, 35, 2300980. [Google Scholar] [CrossRef]
- Zou, Y.; Kazemi, S.A.; Shi, G.; Liu, J.; Yang, Y.; Bedford, N.M.; Fan, K.; Xu, Y.; Fu, H.; Dong, M. Ruthenium single-atom modulated Ti3C2Tx MXene for efficient alkaline electrocatalytic hydrogen production. EcoMat 2023, 5, e12274. [Google Scholar] [CrossRef]
- Chen, Z.; Gong, W.; Wang, J.; Hou, S.; Yang, G.; Zhu, C.; Fan, X.; Li, Y.; Gao, R.; Cui, Y. Metallic W/WO2 solid-acid catalyst boosts hydrogen evolution reaction in alkaline electrolyte. Nat. Commun. 2023, 14, 5363. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Song, H.; Zhang, B.; Liu, J.; Shai, X.; Miao, L. Water dissociation kinetic-oriented design of nickel sulfides via tailored dual sites for efficient alkaline hydrogen evolution. Adv. Funct. Mater. 2021, 31, 2008578. [Google Scholar] [CrossRef]
- Xu, K.; Ding, H.; Zhang, M.; Chen, M.; Hao, Z.; Zhang, L.; Wu, C.; Xie, Y. Regulating water-reduction kinetics in cobalt phosphide for enhancing HER catalytic activity in alkaline solution. Adv. Mater. 2017, 29, 1606980. [Google Scholar] [CrossRef]
- Wang, J.; Xin, S.; Xiao, Y.; Zhang, Z.; Li, Z.; Zhang, W.; Li, C.; Bao, R.; Peng, J.; Yi, J. Manipulating the Water Dissociation Electrocatalytic Sites of Bimetallic Nickel-Based Alloys for Highly Efficient Alkaline Hydrogen Evolution. Angew. Chem. Int. Ed. 2022, 61, e202202518. [Google Scholar] [CrossRef]
- Kumar, A.; Bui, V.Q.; Lee, J.; Jadhav, A.R.; Hwang, Y.; Kim, M.G.; Kawazoe, Y.; Lee, H. Modulating interfacial charge density of NiP2–FeP2 via coupling with metallic Cu for accelerating alkaline hydrogen evolution. ACS Energy Lett. 2021, 6, 354–363. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Zhao, H.; Qi, W.; Zhang, D.; Yu, Y.; Cai, W.; Li, S.; Lai, J.; Huang, B. Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis. Nat. Commun. 2020, 11, 5437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, M.T.; Cao, A.; Chan, K.; Nørskov, J.K. Insights into the hydrogen evolution reaction on 2D transition-metal dichalcogenides. J. Phys. Chem. C 2022, 126, 5151–5158. [Google Scholar] [CrossRef]
- Laursen, A.B.; Varela, A.S.; Dionigi, F.; Fanchiu, H.; Miller, C.; Trinhammer, O.L.; Rossmeisl, J.; Dahl, S. Electrochemical hydrogen evolution: Sabatier’s principle and the volcano plot. J. Chem. Educ. 2012, 89, 1595–1599. [Google Scholar] [CrossRef]
- Laursen, A.B.; Wexler, R.B.; Whitaker, M.J.; Izett, E.J.; Calvinho, K.U.; Hwang, S.; Rucker, R.; Wang, H.; Li, J.; Garfunkel, E. Climbing the volcano of electrocatalytic activity while avoiding catalyst corrosion: Ni3P, a hydrogen evolution electrocatalyst stable in both acid and alkali. ACS Catal. 2018, 8, 4408–4419. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasché, F.; Herranz, J.; Gasteiger, H. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- He, Q.; Tian, D.; Jiang, H.; Cao, D.; Wei, S.; Liu, D.; Song, P.; Lin, Y.; Song, L. Achieving efficient alkaline hydrogen evolution reaction over a Ni5P4 catalyst incorporating single-atomic Ru sites. Adv. Mater. 2020, 32, 1906972. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhang, Y.; Shen, B.; Wan, L.; Hou, G.; Cao, H.; Zheng, G.; Zhao, Y.; Zhang, H. Multi-site synergistic hydrogen evolution reactions on porous homogeneous FeCoNiCu high-entropy alloys fabricated by solution combustion synthesis and hydrogen reduction. J. Alloys Compd. 2024, 1002, 175356. [Google Scholar] [CrossRef]
- Markovic, N. The Hydrogen Electrode Reaction and the Electrooxidation of CO and H2/CO Mixtures on Well-Characterized Pt and Pt-Bimetallic Surfaces; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2001. [Google Scholar]
- Zhao, Y.; Wu, D.; Luo, W. Correlating alkaline hydrogen electrocatalysis and hydroxide binding energies on Mo-modified Ru catalysts. ACS Sustain. Chem. Eng. 2022, 10, 1616–1623. [Google Scholar] [CrossRef]
- Sheng, W.; Bivens, A.P.; Myint, M.; Zhuang, Z.; Forest, R.V.; Fang, Q.; Chen, J.G.; Yan, Y. Non-precious metal electrocatalysts with high activity for hydrogen oxidation reaction in alkaline electrolytes. Energy Environ. Sci. 2014, 7, 1719–1724. [Google Scholar] [CrossRef]
- Sheng, W.; Myint, M.; Chen, J.G.; Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 2013, 6, 1509–1512. [Google Scholar] [CrossRef]
- Gong, M.; Wang, D.-Y.; Chen, C.-C.; Hwang, B.-J.; Dai, H. A mini review on nickel-based electrocatalysts for alkaline hydrogen evolution reaction. Nano Res. 2016, 9, 28–46. [Google Scholar] [CrossRef]
- De, S.; Zhang, J.; Luque, R.; Yan, N. Ni-based bimetallic heterogeneous catalysts for energy and environmental applications. Energy Environ. Sci. 2016, 9, 3314–3347. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Liu, P.; Liu, S.; Dong, R.; Zhuang, X.; Chen, M.; Feng, X. Engineering water dissociation sites in MoS2 nanosheets for accelerated electrocatalytic hydrogen production. Energy Environ. Sci. 2016, 9, 2789–2793. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Yu, X.; Wang, Y.; Zheng, G. Superb alkaline hydrogen evolution and simultaneous electricity generation by pt-decorated Ni3N nanosheets. Adv. Energy Mater. 2017, 7, 1601390. [Google Scholar] [CrossRef]
- Ogunkunle, S.A.; Bouzid, A.; Hinsch, J.J.; Allen, O.J.; White, J.J.; Bernard, S.; Wu, Z.; Zhu, Y.; Wang, Y. Defect engineering of 1T’MX 2 (M = Mo, W and X = S, Se) transition metal dichalcogenide-based electrocatalyst for alkaline hydrogen evolution reaction. J. Phys. Condens. Matter 2024, 36, 145002. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Numerical Simulation of Electrified Solid–Liquid Interfaces; AIP Publishing LLC: Melville, NY, USA, 2021; pp. 3-1–3-18. [Google Scholar]
- Tan, H.; Tang, B.; Lu, Y.; Ji, Q.Q.; Lv, L.Y.; Duan, H.L.; Li, N.; Wang, Y.; Feng, S.H.; Li, Z.; et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 2022, 13, 2024. [Google Scholar] [CrossRef]
- Hinsch, J.J.; Bouzid, A.; Barker, J.C.; White, J.J.; Mortier, F.; Zhao, H.; Wang, Y. Revisiting the Electrified Pt (111)/Water Interfaces through an Affordable Double-Reference Ab Initio Approach. J. Phys. Chem. C 2023, 127, 19857–19866. [Google Scholar] [CrossRef]
- Eslamibidgoli, M.J.; Eikerling, M.H. Approaching the self-consistency challenge of electrocatalysis with theory and computation. Curr. Opin. Electrochem. 2018, 9, 189–197. [Google Scholar] [CrossRef]
- Duan, Z.; Henkelman, G. Theoretical resolution of the exceptional oxygen reduction activity of Au (100) in alkaline media. ACS Catal. 2019, 9, 5567–5573. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, X.; Liu, X.; Xiao, W.; Luo, Z.; Wang, W.; Zhang, Y.; Liu, J.-C. Understanding the hydrogen evolution reaction activity of doped single-atom catalysts on two-dimensional GaPS4 by DFT and machine learning. J. Energy Chem. 2023, 81, 93–100. [Google Scholar] [CrossRef]
- Botu, V.; Batra, R.; Chapman, J.; Ramprasad, R. Machine Learning Force Fields: Construction, Validation, and Outlook. J. Phys. Chem. C 2016, 121, 511–522. [Google Scholar] [CrossRef]
- Raccuglia, P.; Elbert, K.C.; Adler, P.D.F.; Falk, C.; Wenny, M.B.; Mollo, A.; Zeller, M.; Friedler, S.A.; Schrier, J.; Norquist, A.J. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533, 73–76. [Google Scholar] [CrossRef]
- Ooka, H.; Wintzer, M.E.; Nakamura, R. Non-Zero Binding Enhances Kinetics of Catalysis: Machine Learning Analysis on the Experimental Hydrogen Binding Energy of Platinum. ACS Catal. 2021, 11, 6298–6303. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).