
Engineering of Cyclodextrin Glucosyltransferase from Paenibacillus macerans for Improved Regioselectivity and Product Specificity Toward Hydroxyflavone Glycosylation

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Identification of Key Residues for Regioselective Glycosylation of PmCGTase
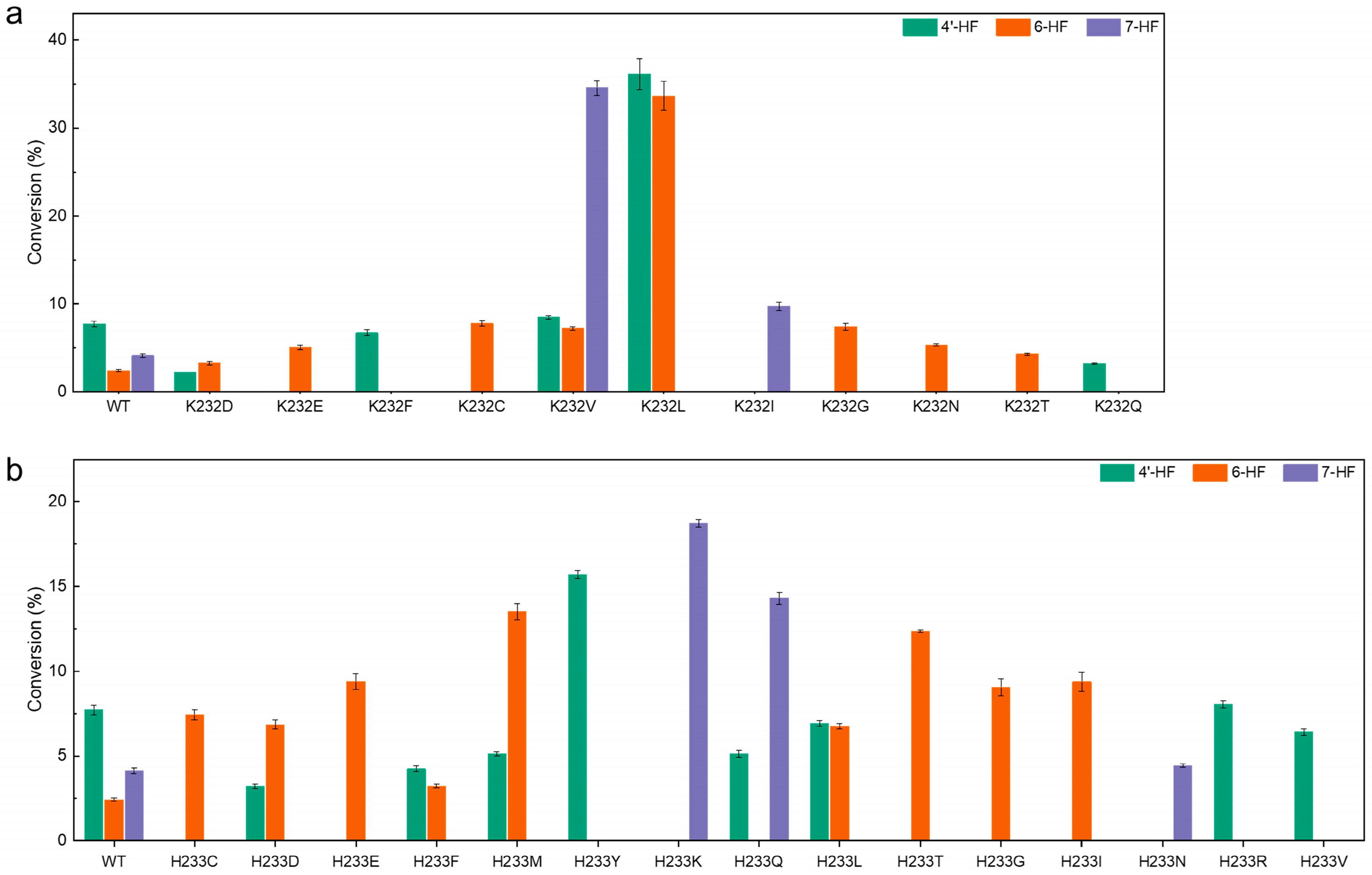
2.2. Regioselectivity Modulation via Saturation Mutagenesis of K232 and H233
2.3. Enhanced Specificity for Short-Chain Glycoside Products by K232 and H233 Variants
2.4. Enhanced Hydrolytic Activity and Altered Functional Profiles of K232 and H233 Variants
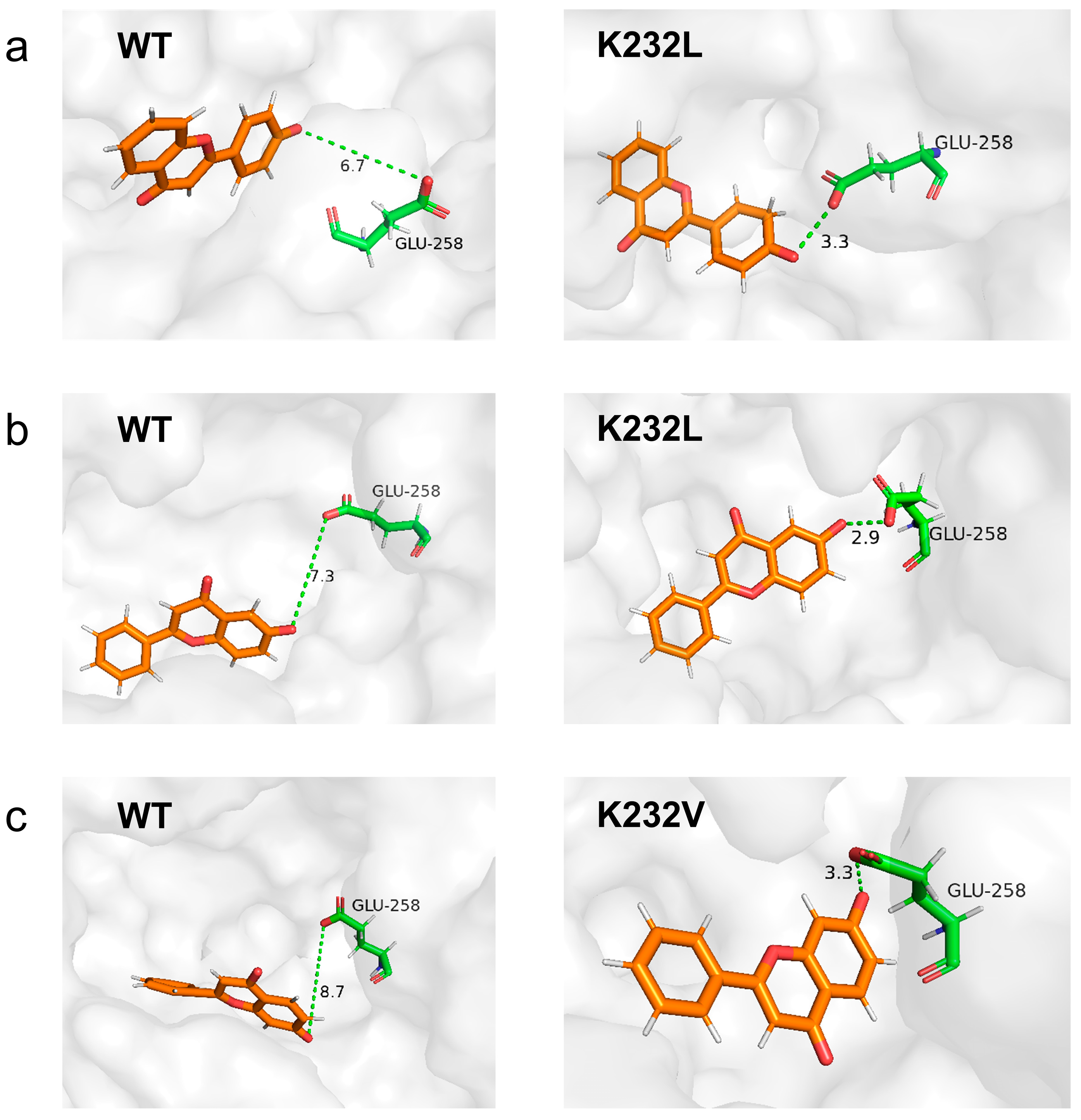
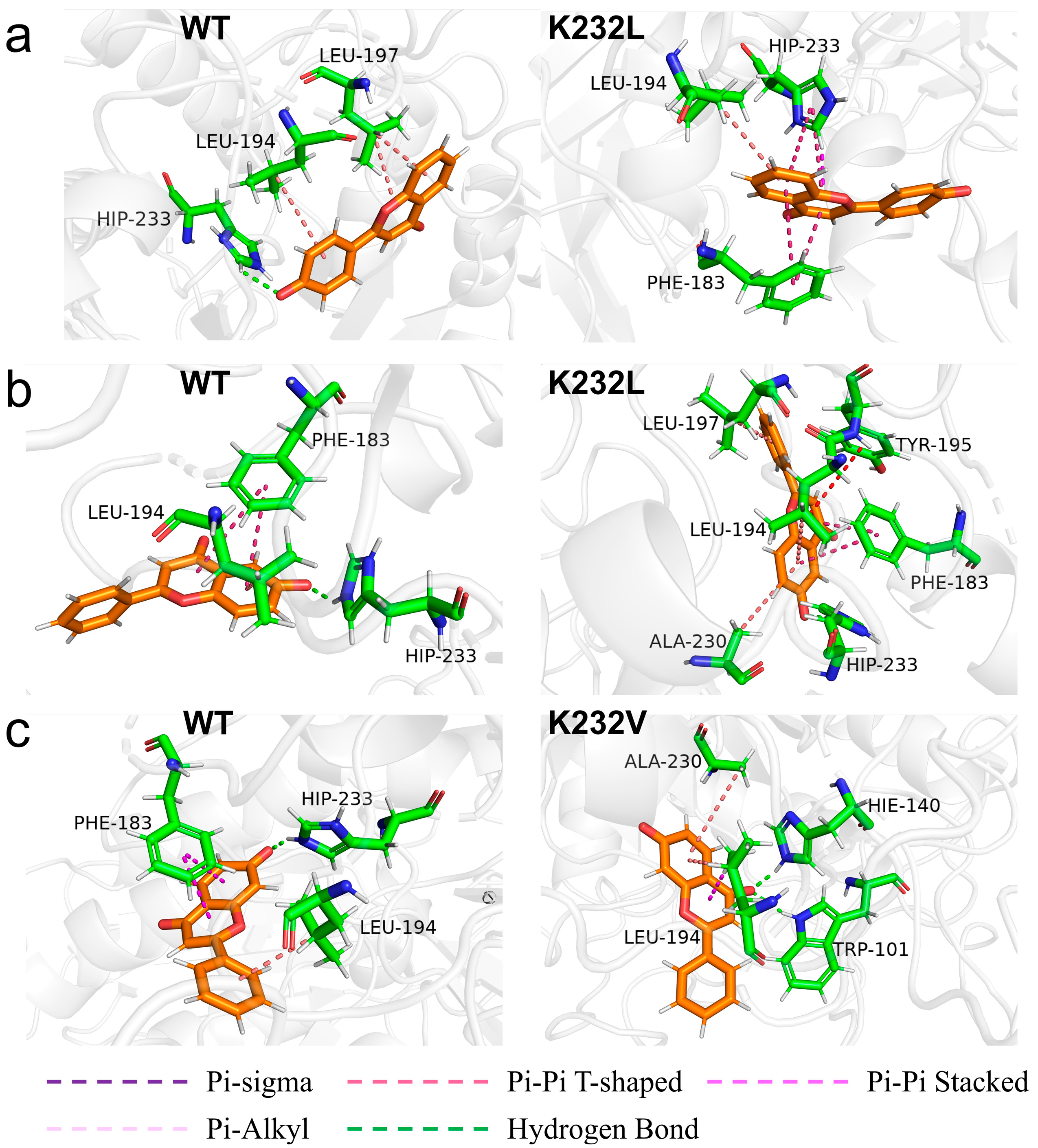
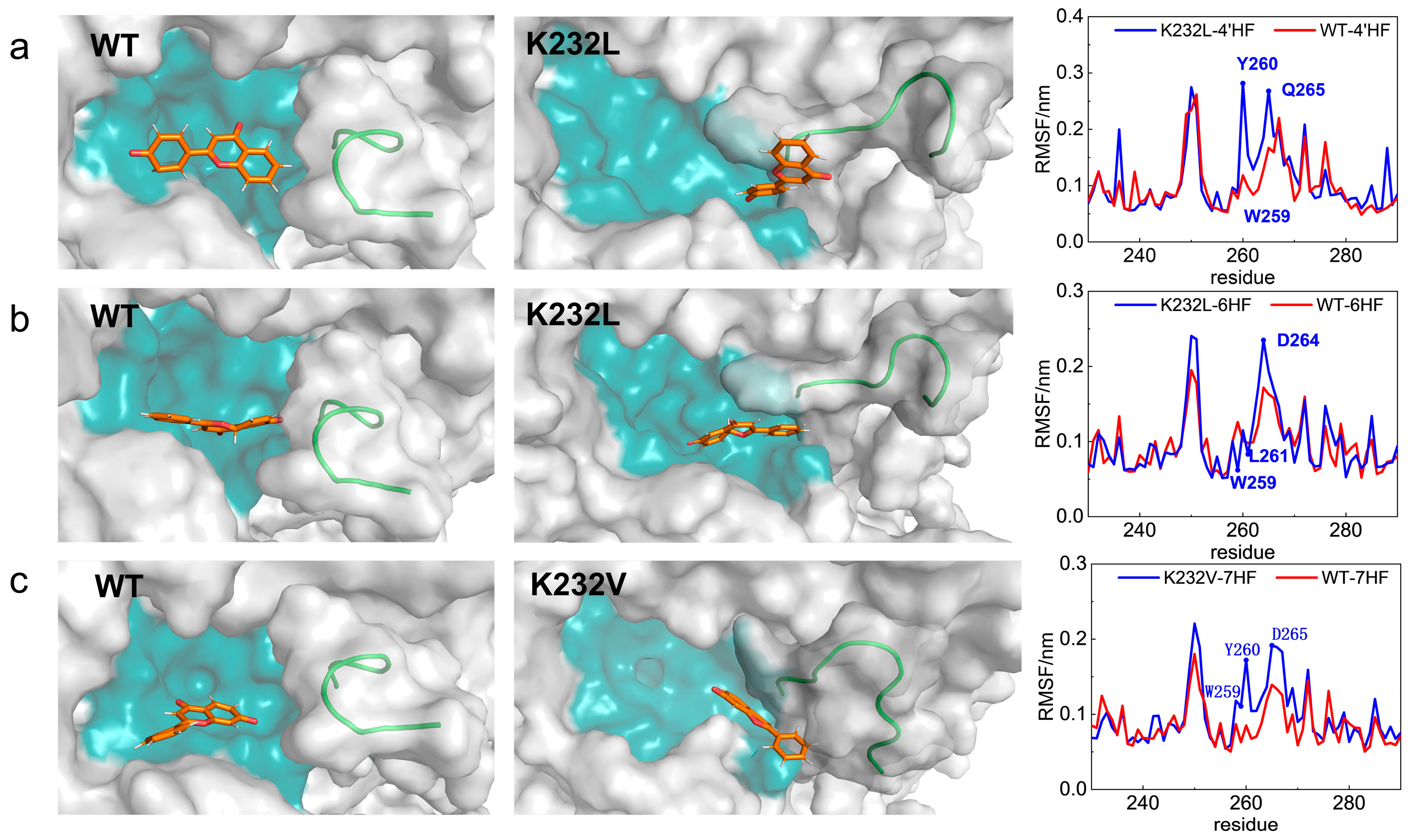
2.5. MD Simulations Analysis Revealing the Improved Properties of Variants
3. Materials and Methods
3.1. Strains and Chemicals
3.2. Saturation Mutagenesis
3.3. Expression and Purification of PmCGTase and Its Mutants
3.4. Enzyme Activity Assays
3.5. Analysis and Identification of Hydroxyflavone Glycosylated Products
3.6. Molecular Docking and MD Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gangopadhyay, A.; Chakraborty, S.; Jash, S.K.; Gorai, D. Cytotoxicity of natural flavones and flavonols against different cancer cells. J. Iran. Chem. Soc. 2022, 19, 1547–1573. [Google Scholar] [CrossRef]
- Chen, X.-H.; Zhao, Y.-Y.; Wang, Q.; Li, Z.-L.; Li, F.-X.; Tan, H.-Y.; Huang, Y.-Y. Chemical constituents from the stems of Acanthopanax senticosus with their cytotoxic activities. J. Asian Nat. Prod. Res. 2022, 24, 673–678. [Google Scholar] [CrossRef]
- Lee, M.; Upadhyay, S.; Mariyam, F.; Martin, G.; Hailemariam, A.; Lee, K.; Jayaraman, A.; Chapkin, R.S.; Lee, S.-O.; Safe, S. Flavone and hydroxyflavones are ligands that bind the orphan nuclear receptor 4A1 (NR4A1). Int. J. Mol. Sci. 2023, 24, 8152. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Roh, S.-A.; Shim, J.-H.; Mikami, B.; Baik, M.-Y.; Park, C.-S.; Park, K.-H. Glycosylation of genistin into soluble inclusion complex form of cyclic glucans by enzymatic modification. J. Agric. Food Chem. 2005, 53, 6516–6524. [Google Scholar] [CrossRef]
- Abdulai, I.L.; Kwofie, S.K.; Gbewonyo, W.S.; Boison, D.; Puplampu, J.B.; Adinortey, M.B. Multitargeted effects of vitexin and isovitexin on diabetes mellitus and its complications. Sci. World J. 2021, 2021, 6641128. [Google Scholar] [CrossRef]
- Chua, L.S.; Abdullah, F.I.; Awang, M.A. Potential of natural bioactive C-glycosyl flavones for antidiabetic properties. Stud. Nat. Prod. Chem. 2020, 64, 241–261. [Google Scholar]
- Zhao, X.-W. Research progress in structure-activity relationship of flavoniods. Chin. Tradit. Herb. Drugs 2015, 46, 3264–3271. [Google Scholar]
- López-Posadas, R.; Ballester, I.; Abadía-Molina, A.C.; Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; de Medina, F.S. Effect of flavonoids on rat splenocytes, a structure–activity relationship study. Biochem. Pharmacol. 2008, 76, 495–506. [Google Scholar] [CrossRef]
- Koirala, N.; Thuan, N.H.; Ghimire, G.P.; Van Thang, D.; Sohng, J.K. Methylation of flavonoids: Chemical structures, bioactivities, progress and perspectives for biotechnological production. Enzym. Microb. Technol. 2016, 86, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Z.; Wu, Y.; Luo, D.; Qiu, L.; Xie, J.; Li, X. Advances on synthesis of flavonoid glycosides. Chin. J. Org. Chem. 2019, 39, 1875. [Google Scholar] [CrossRef]
- Li, Y.-F.; Yu, B.; Sun, J.-S.; Wang, R.-X. Efficient synthesis of baicalin and its analogs. Tetrahedron Lett. 2015, 56, 3816–3819. [Google Scholar] [CrossRef]
- Cao, Z.; Qu, Y.; Zhou, J.; Liu, W.; Yao, G. Stereoselective synthesis of quercetin 3-O-glycosides of 2-amino-2-deoxy-D-glucose under phase transfer catalytic conditions. J. Carbohydr. Chem. 2015, 34, 28–40. [Google Scholar] [CrossRef]
- de Araújo, M.E.M.; Franco, Y.E.; Messias, M.C.; Longato, G.B.; Pamphile, J.A.; Carvalho, P.d.O. Biocatalytic synthesis of flavonoid esters by lipases and their biological benefits. Planta Medica 2017, 83, 7–22. [Google Scholar] [CrossRef]
- Rha, C.-S.; Jung, Y.S.; Seo, D.-H.; Kim, D.-O.; Park, C.-S. Site-specific α-glycosylation of hydroxyflavones and hydroxyflavanones by amylosucrase from Deinococcus geothermalis. Enzym. Microb. Technol. 2019, 129, 109361. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; van der Veen, B.A.; Dijkhuizen, L.; Dijkstra, B.W. Catalytic mechanism and product specificity of cyclodextrin glycosyltransferase, a prototypical transglycosylase from the α-amylase family. Enzym. Microb. Technol. 2002, 30, 295–304. [Google Scholar] [CrossRef]
- van der Veen, B.A.; van Alebeek, G.J.W.; Uitdehaag, J.C.; Dijkstra, B.W.; Dijkhuizen, L. The three transglycosylation reactions catalyzed by cyclodextrin glycosyltransferase from Bacillus circulans (strain 251) proceed via different kinetic mechanisms. Eur. J. Biochem. 2000, 267, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Esteso, M.A.; Romero, C.M. Cyclodextrins: Properties and Applications. Int. J. Mol. Sci. 2024, 25, 4547. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Kubota, N.; Uesugi, D.; Fujitaka, Y.; Okada, S.; Tanigawa, M.; Hamada, H. Regioselective glycosylation of 3-, 5-, 6-, and 7-hydroxyflavones by cultured plant cells. Nat. Prod. Commun. 2015, 10, 1934578X1501000632. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Woo, J.-B.; Ryu, S.-I.; Moon, S.-K.; Han, N.S.; Lee, S.-B. Glucosylation of flavonol and flavanones by Bacillus cyclodextrin glucosyltransferase to enhance their solubility and stability. Food Chem. 2017, 229, 75–83. [Google Scholar] [CrossRef]
- Rha, C.-S.; Kim, E.-R.; Kim, Y.-J.; Jung, Y.S.; Kim, D.-O.; Park, C.-S. Simple and efficient production of highly soluble daidzin glycosides by amylosucrase from Deinococcus geothermalis. J. Agric. Food Chem. 2019, 67, 12824–12832. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Trinkl, J.; Haugeneder, A.; Härtl, K.; Franz-Oberdorf, K.; Giri, A.; Hoffmann, T.; Schwab, W. Semirational design and engineering of grapevine glucosyltransferases for enhanced activity and modified product selectivity. Glycobiology 2019, 29, 765–775. [Google Scholar] [CrossRef]
- Strokopytov, B.; Knegtel, R.M.; Penninga, D.; Rozeboom, H.J.; Kalk, K.H.; Dijkhuizen, L.; Dijkstra, B.W. Structure of cyclodextrin glycosyltransferase complexed with a maltononaose inhibitor at 2.6 Å resolution. Implications for product specificity. Biochemistry 1996, 35, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Uitdehaag, J.C.; van der Veen, B.A.; Dijkhuizen, L.; Elber, R.; Dijkstra, B.W. Enzymatic circularization of a malto-octaose linear chain studied by stochastic reaction path calculations on cyclodextrin glycosyltransferase. Proteins Struct. Funct. Bioinform. 2001, 43, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Rabadiya, K.; Pardhi, D.; Thaker, K.; Patoliya, J.; Rajput, K.; Joshi, R. A review on recent upgradation and strategies to enhance cyclodextrin glucanotransferase properties for its applications. Int. J. Biol. Macromol. 2024, 129315. [Google Scholar] [CrossRef]
- Muniz, I.d.C.B.; Santos, J.B.; de Oliveira, R.M.; Santos, F.G.; de Souza Junior, E.C.; Oyama, L.; Fontan, R.d.C.I.; Bonomo, R.C.F. Current advances in obtaining novel cyclodextrin glycosyltransferases for optimizing the synthesis of cyclodextrins. Process Biochem. 2024. [Google Scholar] [CrossRef]
- Gao, H.-Y.; Liu, Y.; Tan, F.-F.; Zhu, L.-W.; Jia, K.-Z.; Tang, Y.-J. The advances and challenges in enzymatic C-glycosylation of flavonoids in plants. Curr. Pharm. Des. 2022, 28, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Li, J.; Shin, H.-d.; Chen, R.R.; Du, G.; Liu, L.; Chen, J. Recent advances in discovery, heterologous expression, and molecular engineering of cyclodextrin glycosyltransferase for versatile applications. Biotechnol. Adv. 2014, 32, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Chai, B.; Jiang, Y.; Ni, J.; Ni, Y. Engineering of cyclodextrin glycosyltransferase from Paenibacillus macerans for enhanced product specificity of long-chain glycosylated sophoricosides. Mol. Catal. 2022, 519, 112147. [Google Scholar] [CrossRef]
- Han, R.; Ni, J.; Zhou, J.; Dong, J.; Xu, G.; Ni, Y. Engineering of cyclodextrin glycosyltransferase reveals pH-regulated mechanism of enhanced long-chain glycosylated sophoricoside specificity. Appl. Environ. Microbiol. 2020, 86, e00004-20. [Google Scholar] [CrossRef]
- Xie, T.; Song, B.; Yue, Y.; Chao, Y.; Qian, S. Site-saturation mutagenesis of central tyrosine 195 leading to diverse product specificities of an α-cyclodextrin glycosyltransferase from Paenibacillus sp. 602-1. J. Biotechnol. 2014, 170, 10–16. [Google Scholar] [CrossRef]
- Xu, Q.; Han, R.; Li, J.; Du, G.; Liu, L.; Chen, J. Improving maltodextrin specificity by site-saturation engineering of subsite+ 1 in cyclodextrin glycosyltransferase from Paenibacillus macerans. Chin. J. Biotechnol. 2014, 30, 98–108. [Google Scholar]
- Han, R.; Liu, L.; Shin, H.-d.; Chen, R.R.; Du, G.; Chen, J. Site-saturation engineering of lysine 47 in cyclodextrin glycosyltransferase from Paenibacillus macerans to enhance substrate specificity towards maltodextrin for enzymatic synthesis of 2-O-D-glucopyranosyl-L-ascorbic acid (AA-2G). Appl. Microbiol. Biotechnol. 2013, 97, 5851–5860. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Relative Activity (%) | ||
|---|---|---|---|
| Cyclization | Hydrolytic | Disproportionation | |
| WT | 100 | 100 | 100 |
| K232L | 79 ± 2.1 | 147 ± 3.2 | 105 ± 1.8 |
| K232V | 75 ± 1.2 | 172 ± 2.3 | 96 ± 2.4 |
| H233K | 83 ± 2.3 | 144 ± 1.5 | 98 ± 2.9 |
| H233T | 89 ± 3.4 | 130 ± 2.3 | 164 ± 2.4 |
| H233Y | 94 ± 2.3 | 151 ± 2.0 | 153 ± 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, B.; Zhou, J.; Dong, J.; Ni, Y.; Han, R. Engineering of Cyclodextrin Glucosyltransferase from Paenibacillus macerans for Improved Regioselectivity and Product Specificity Toward Hydroxyflavone Glycosylation. Catalysts 2025, 15, 120. https://doi.org/10.3390/catal15020120
Wang J, Wang B, Zhou J, Dong J, Ni Y, Han R. Engineering of Cyclodextrin Glucosyltransferase from Paenibacillus macerans for Improved Regioselectivity and Product Specificity Toward Hydroxyflavone Glycosylation. Catalysts. 2025; 15(2):120. https://doi.org/10.3390/catal15020120
Chicago/Turabian StyleWang, Jin, Binhao Wang, Jieyu Zhou, Jinjun Dong, Ye Ni, and Ruizhi Han. 2025. "Engineering of Cyclodextrin Glucosyltransferase from Paenibacillus macerans for Improved Regioselectivity and Product Specificity Toward Hydroxyflavone Glycosylation" Catalysts 15, no. 2: 120. https://doi.org/10.3390/catal15020120
APA StyleWang, J., Wang, B., Zhou, J., Dong, J., Ni, Y., & Han, R. (2025). Engineering of Cyclodextrin Glucosyltransferase from Paenibacillus macerans for Improved Regioselectivity and Product Specificity Toward Hydroxyflavone Glycosylation. Catalysts, 15(2), 120. https://doi.org/10.3390/catal15020120