
Sustainable Synthesis of α-Glucosidase Inhibitors by Gas-Free Pd-Carbonylation of Nature-Based Hydroxytyrosol

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
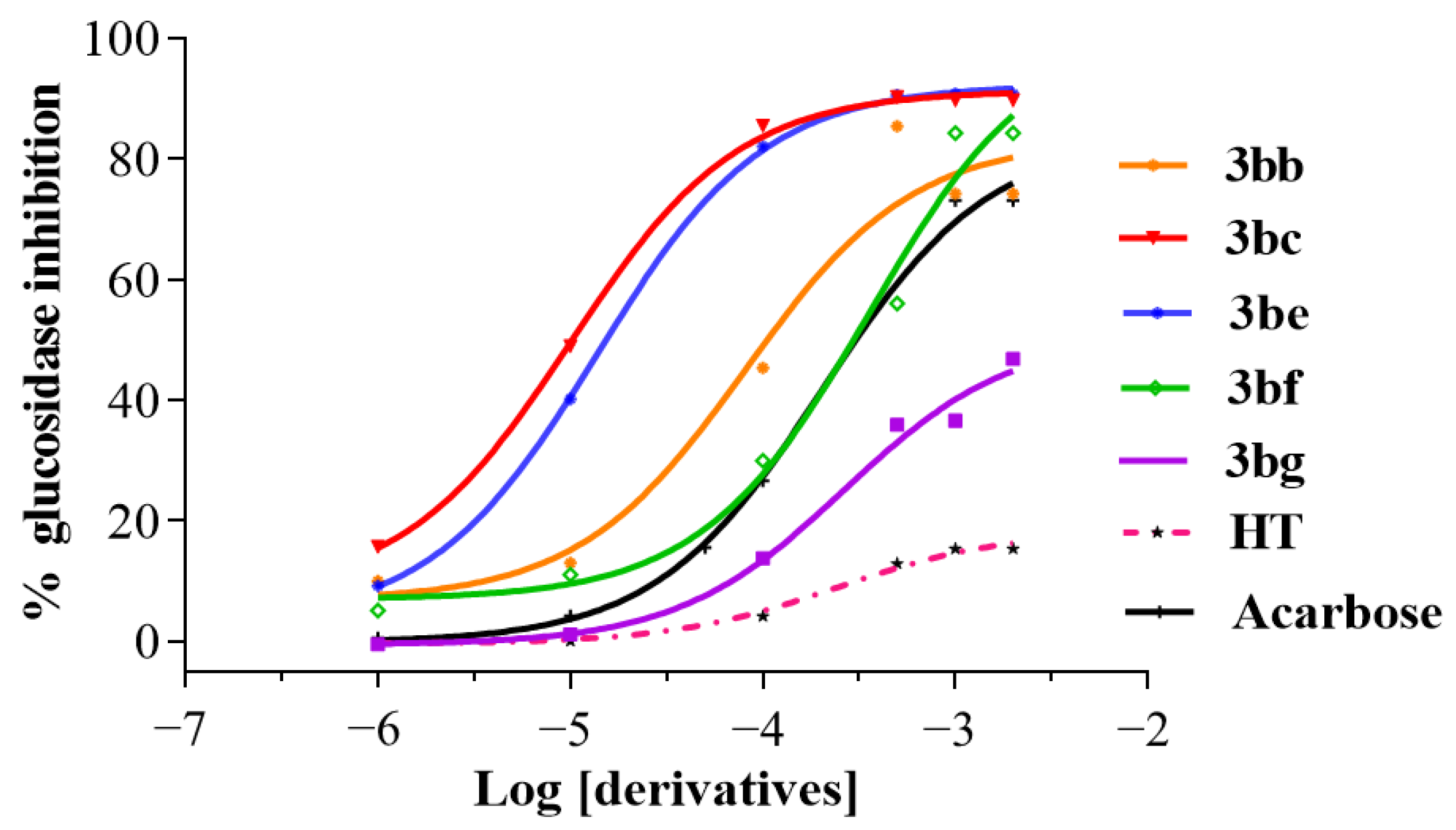
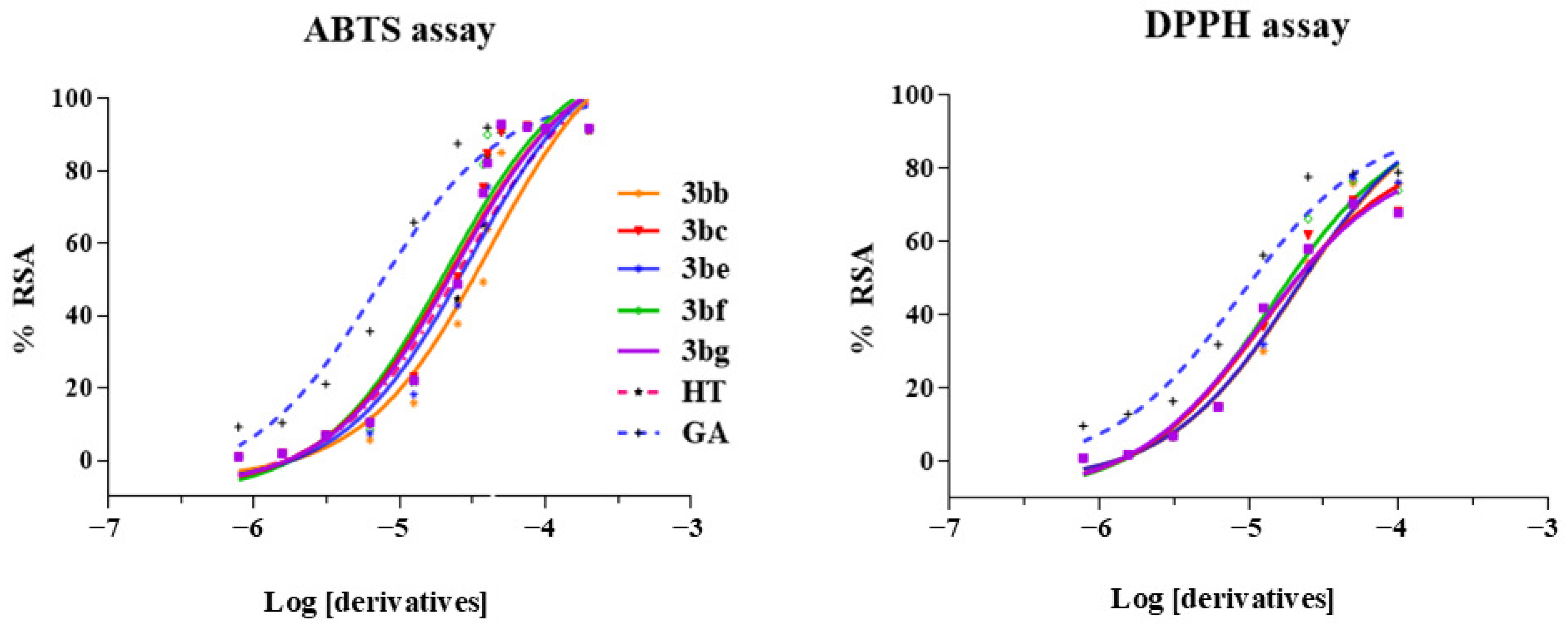
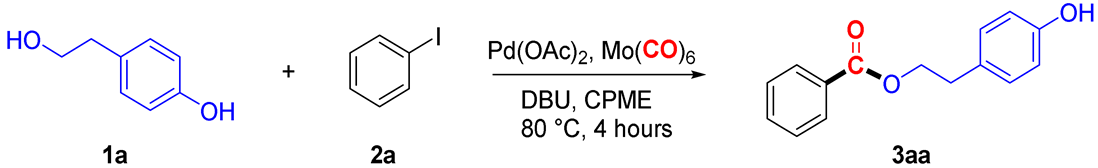
2. Results and Discussion
3. Materials and Methods
3.1. General Aspects
3.2. Experimental Procedure for the Synthesis of Tyrosol (T) and Hydroxytyrosol (HT) by Reduction of the Corresponding Carboxylic Acids
3.3. Experimental Procedure for the Carbonylative Coupling Between Alcohols 1a–b and Aryl Iodides 2a–h in CPME
4. Biological Assays
4.1. α-Glucosidase Inhibition Assay
4.2. Antioxidant Activity with ABTS Protocol
4.3. Antioxidant Activity with DPPH Protocol
4.4. Cell Culture and Cell Viability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Hannah Ritchie and Max Roser CO2 Emissions. Available online: https://ourworldindata.org/co2-emissions (accessed on 16 December 2024).
- Waste Generation in the Chemical Industry. Available online: https://www.eea.europa.eu/en/european-zero-pollution-dashboards/indicators/waste-generation-in-the-chemical-industry?activeTab=658e2886-cfbf-4c2f-a603-061e1627a515 (accessed on 13 December 2024).
- Mori, R. Replacing All Petroleum-Based Chemical Products with Natural Biomass-Based Chemical Products: A Tutorial Review. RSC Sustain. 2023, 1, 179–212. [Google Scholar] [CrossRef]
- Maneffa, A.; Priecel, P.; Lopez-Sanchez, J.A. Biomass-Derived Renewable Aromatics: Selective Routes and Outlook for p-Xylene Commercialisation. ChemSusChem 2016, 9, 2736–2748. [Google Scholar] [CrossRef] [PubMed]
- Al Ghatta, A.; Perry, J.M.; Maeng, H.; Lemus, J.; Hallett, J.P. Sustainable and Efficient Production of Furoic Acid from Furfural through Amine Assisted Oxidation with Hydrogen Peroxide and Its Implementation for the Synthesis of Alkyl Furoate. RSC Sustain. 2023, 1, 303–309. [Google Scholar] [CrossRef]
- Lipshutz, B.H. On the Role of Surfactants: Rethinking “Aqueous” Chemistry. Green Chem. 2024, 26, 739–752. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef]
- Ghandi, K. A Review of Ionic Liquids, Their Limits and Applications. Green Sustain. Chem. 2014, 4, 44–53. [Google Scholar] [CrossRef]
- Lomba, L.; Zuriaga, E.; Giner, B. Solvents Derived from Biomass and Their Potential as Green Solvents. Curr. Opin. Green. Sustain. Chem. 2019, 18, 51–56. [Google Scholar] [CrossRef]
- Beckman, E.J. Supercritical and Near-Critical CO2 in Green Chemical Synthesis and Processing. J. Supercrit. Fluids 2004, 28, 121–191. [Google Scholar] [CrossRef]
- Perrone, S.; Salomone, A.; Caroli, A.; Falcicchio, A.; Citti, C.; Cannazza, G.; Troisi, L. Stereoselective Synthesis of α-Alkylidene β-Oxo Amides by Palladium-Catalyzed Carbonylation. Eur. J. Org. Chem. 2014, 2014, 5932–5938. [Google Scholar] [CrossRef]
- Salomone, A.; Perna, F.M.; Sassone, F.C.; Falcicchio, A.; Bezenšek, J.; Svete, J.; Stanovnik, B.; Florio, S.; Capriati, V. Preparation of Polysubstituted Isochromanes by Addition of Ortho-Lithiated Aryloxiranes to Enaminones. J. Org. Chem. 2013, 78, 11059–11065. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Capua, M.; Salomone, A.; Troisi, L. Multicomponent Synthesis of Uracil Analogues Promoted by Pd-Catalyzed Carbonylation of α-Chloroketones in the Presence of Isocyanates and Amines. J. Org. Chem. 2015, 80, 8189–8197. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Cicco, L.; Messa, F.; Perna, F.M.; Salomone, A.; Capriati, V. Streamlined Routes to Phenacyl Azides and 2,5-Diarylpyrazines Enabled by Deep Eutectic Solvents. Eur. J. Org. Chem. 2019, 2019, 5557–5562. [Google Scholar] [CrossRef]
- Paparella, A.N.; Messa, F.; Dilauro, G.; Troisi, L.; Perrone, S.; Salomone, A. A Glycerol-Based Deep Eutectic Solvent as Natural Medium and Organic Reductant for Homocoupling of (Hetero)Aryl Chlorides: A Green Route to 2,2′-Bipyridine and Biaryl Scaffolds. ChemistrySelect 2022, 7, e202203438. [Google Scholar] [CrossRef]
- Messa, F.; Dilauro, G.; Paparella, A.N.; Silvestri, L.; Furlotti, G.; Iacoangeli, T.; Perrone, S.; Salomone, A. Deep Eutectic Solvents Meet Safe, Scalable and Sustainable Hydrogenations Enabled by Aluminum Powder and Pd/C. Green Chem. 2022, 24, 4388–4394. [Google Scholar] [CrossRef]
- Messa, F.; Giotta, L.; Troisi, L.; Perrone, S.; Salomone, A. Sustainable Extraction of Hydroxytyrosol from Olive Leaves Based on a Nature-Inspired Deep Eutectic Solvent (NADES). ChemistrySelect 2024, 9, e202403476. [Google Scholar] [CrossRef]
- Messa, F.; Paparella, A.N.; Perrone, S.; Salomone, A. Gas-Free Alkoxycarbonylation of Aryl Iodides in a Phosphonium-Based Deep Eutectic Solvent with Mo(CO)6 as a Solid CO Source. Org. Biomol. Chem. 2023, 21, 5164–5170. [Google Scholar] [CrossRef]
- Messa, F.; Paparella, A.N.; Veselý, D.; Krajčovič, J.; Papadia, P.; Perrone, S.; Salomone, A. Gas-Free Amino- and Alkoxycarbonylation of Aryl Iodides in a Bioinspired Deep Eutectic Solvent with Mo(CO)6 as a Safe CO Source. Eur. J. Org. Chem. 2023, 26, e202300309. [Google Scholar] [CrossRef]
- Alder, C.M.; Hayler, J.D.; Henderson, R.K.; Redman, A.M.; Shukla, L.; Shuster, L.E.; Sneddon, H.F. Updating and Further Expanding GSK’s Solvent Sustainability Guide. Green Chem. 2016, 18, 3879–3890. [Google Scholar] [CrossRef]
- Borsari, C.; Trader, D.J.; Tait, A.; Costi, M.P. Designing Chimeric Molecules for Drug Discovery by Leveraging Chemical Biology. J. Med. Chem. 2020, 63, 1908–1928. [Google Scholar] [CrossRef]
- Wannberg, J.; Larhed, M. Increasing Rates and Scope of Reactions: Sluggish Amines in Microwave-Heated Aminocarbonylation Reactions under Air. J. Org. Chem. 2003, 68, 5750–5753. [Google Scholar] [CrossRef] [PubMed]
- Moser, W.R.; Wang, A.W.; Kildahl, N.K. Mechanistic Studies of the Palladium-Catalyzed Reaction of Methanol with Bromobenzene and Carbon Monoxide to Produce Methyl Benzoate. Stoichiometric Study. J. Am. Chem. Soc. 1988, 110, 2816–2820. [Google Scholar] [CrossRef]
- Diabetes. Available online: https://www.who.int/health-topics/diabetes#tab=tab (accessed on 30 January 2025).
- Alssema, M.; Ruijgrok, C.; Blaak, E.E.; Egli, L.; Dussort, P.; Vinoy, S.; Dekker, J.M.; Denise Robertson, M. Effects of Alpha-Glucosidase-Inhibiting Drugs on Acute Postprandial Glucose and Insulin Responses: A Systematic Review and Meta-Analysis. Nutr. Diabetes 2021, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A Review of Alpha-Glucosidase Inhibitors from Plants as Potential Candidates for the Treatment of Type-2 Diabetes. Phytochem. Rev. 2022, 21, 1049–1079. [Google Scholar] [CrossRef]
- Floegel, A.; Kim, D.O.; Chung, S.J.; Koo, S.I.; Chun, O.K. Comparison of ABTS/DPPH Assays to Measure Antioxidant Capacity in Popular Antioxidant-Rich US Foods. J. Food Compos. Anal. 2011, 24, 1043–1048. [Google Scholar] [CrossRef]
- Foti, P.; Randazzo, C.L.; Russo, M.; Di Sanzo, R.; Romeo, F.V.; Scilimati, A.; Miciaccia, M.; Grazia Perrone, M.; Caggia, C. Effect of Microbial Fermentation on Functional Traits and Volatiloma Profile of Pâté Olive Cake. Food Res. Int. 2023, 174, 113510. [Google Scholar] [CrossRef]
- Daglia, M.; Lorenzo, A.; Nabavi, S.; Talas, Z.; Nabavi, S. Polyphenols: Well Beyond The Antioxidant Capacity: Gallic Acid and Related Compounds as Neuroprotective Agents: You Are What You Eat! Curr. Pharm. Biotechnol. 2014, 15, 362–372. [Google Scholar] [CrossRef]
- Schirizzi, A.; Contino, M.; Carrieri, L.; Riganti, C.; De Leonardis, G.; Scavo, M.P.; Perrone, M.G.; Miciaccia, M.; Kopecka, J.; Refolo, M.G.; et al. The Multiple Combination of Paclitaxel, Ramucirumab and Elacridar Reverses the Paclitaxel-Mediated Resistance in Gastric Cancer Cell Lines. Front. Oncol. 2023, 13, 1129832. [Google Scholar] [CrossRef]
- Jiang, L.; Kumar, S.; Nuechterlein, M.; Reyes, M.; Tran, D.; Cabebe, C.; Chiang, P.; Reynolds, J.; Carrier, S.; Sun, Y.; et al. Application of a High-Resolution in Vitro Human MDR1-MDCK Assay and in Vivo Studies in Preclinical Species to Improve Prediction of CNS Drug Penetration. Pharmacol. Res. Perspect. 2022, 10, e00932. [Google Scholar] [CrossRef]
- Snopov, S.A.; Teryukova, N.P.; Sakhenberg, E.I.; Teplyashina, V.V.; Nasyrova, R.F. Use of HepG2 Cell Line for Evaluation of Toxic and Metabolic Antipsychotic Action. Cell Tissue Biol. 2017, 11, 405–415. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, J.; Li, A. Cytotoxicity and Intestinal Permeability of Phycotoxins Assessed by the Human Caco-2 Cell Model. Ecotoxicol. Environ. Saf. 2023, 249, 114447. [Google Scholar] [CrossRef] [PubMed]
- Miciaccia, M.; Rizzo, F.; Centonze, A.; Cavallaro, G.; Contino, M.; Armenise, D.; Baldelli, O.M.; Solidoro, R.; Ferorelli, S.; Scarcia, P.; et al. Harmaline to Human Mitochondrial Caseinolytic Serine Protease Activation for Pediatric Diffuse Intrinsic Pontine Glioma Treatment. Pharmaceuticals 2024, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Xu, Q.; Zhang, L.; Xia, G.; Sun, J.; Zhu, J. Synthesis and Biological Activities of Hydroxytyrosol Ester Derivatives. J. Chem. Soc. Pak. 2020, 42, 109. [Google Scholar] [CrossRef]
- Foti, P.; Occhipinti, P.S.; Russo, N.; Scilimati, A.; Miciaccia, M.; Caggia, C.; Perrone, M.G.; Randazzo, C.L.; Romeo, F.V. Olive Mill Wastewater Fermented with Microbial Pools as a New Potential Functional Beverage. Molecules 2023, 28, 646. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | 1a (mmol) | 2a (mmol) | Solvent | 3aa (Yield%) b |
| 1 | 0.5 | 0.6 | ChCl/Urea | 0 |
| 2 | 0.5 | 0.6 | 2-MeTHF | 36 |
| 3 | 0.5 | 0.6 | DMC | traces |
| 4 | 0.5 | 0.6 | CPME | 50 |
| 5 | 0.75 | 0.5 | CPME | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messa, F.; Armenise, D.; Liturri, A.; Perrone, M.G.; Perrone, S.; Salomone, A. Sustainable Synthesis of α-Glucosidase Inhibitors by Gas-Free Pd-Carbonylation of Nature-Based Hydroxytyrosol. Catalysts 2025, 15, 202. https://doi.org/10.3390/catal15030202
Messa F, Armenise D, Liturri A, Perrone MG, Perrone S, Salomone A. Sustainable Synthesis of α-Glucosidase Inhibitors by Gas-Free Pd-Carbonylation of Nature-Based Hydroxytyrosol. Catalysts. 2025; 15(3):202. https://doi.org/10.3390/catal15030202
Chicago/Turabian StyleMessa, Francesco, Domenico Armenise, Anselma Liturri, Maria Grazia Perrone, Serena Perrone, and Antonio Salomone. 2025. "Sustainable Synthesis of α-Glucosidase Inhibitors by Gas-Free Pd-Carbonylation of Nature-Based Hydroxytyrosol" Catalysts 15, no. 3: 202. https://doi.org/10.3390/catal15030202
APA StyleMessa, F., Armenise, D., Liturri, A., Perrone, M. G., Perrone, S., & Salomone, A. (2025). Sustainable Synthesis of α-Glucosidase Inhibitors by Gas-Free Pd-Carbonylation of Nature-Based Hydroxytyrosol. Catalysts, 15(3), 202. https://doi.org/10.3390/catal15030202