Abstract
Antibiotic pollution, particularly via tetracycline (TC), poses significant environmental risks due to its recalcitrance and potential to induce antibiotic resistance. This study employed density functional theory (DFT) and transition state theory (TST) to investigate TC degradation by hydroxyl radicals (·OH), focusing on hydrogen atom transfer (HAT) and radical adduct formation (RAF) pathways. Geometry optimizations and vibrational analysis validated stationary points, while intrinsic reaction coordinate (IRC) calculations confirmed transition states. Key findings reveal that RAF pathways exhibit lower activation barriers (1.23–30.33 kJ/mol) and greater exothermicity (−164.42 kJ/mol) compared to HAT pathways (3.51–42.04 kJ/mol, −109.58 kJ/mol), making them kinetically and thermodynamically dominant. Frontier molecular orbital (FMO) analysis links HAT to TC’s HOMO (π-orbital character on aromatic rings) and RAF to its LUMO (electrophilic sites). Rate constants calculated at 298 K (TST with Wigner correction) confirm RAF’s kinetic superiority (up to 7.0 × 1011 s−1), surpassing HAT’s fastest pathway (6.2 × 1011 s−1). These insights advance the understanding of TC degradation mechanisms and help with the design of efficient photocatalytic oxidation processes for antibiotic removal.
1. Introduction
The widespread use of antibiotics, particularly tetracycline (TC), has led to their persistent accumulation in aquatic environments, posing significant ecological and public health risks due to the emergence of antibiotic-resistant genes and pathogens [1,2,3,4,5]. Conventional wastewater treatment methods often fail to effectively eliminate these recalcitrant compounds, necessitating advanced oxidation processes (AOPs, such as photocatalytic oxidation) that leverage highly reactive and oxidizing radicals, such as hydroxyl radicals (·OH), to achieve efficient degradation [6,7,8]. ·OH exhibits the highest standard oxidation potential (+2.8 V vs. SHE) among reactive oxygen species (ROS), enabling non-selective oxidative attacks on the complex molecular structure of TC. This superior reactivity is experimentally validated: ·OH-driven advanced oxidation processes (AOPs), such as Zn-doped FeOCl photo-Fenton systems, achieve >90% TC removal within 60 min [9]. The degradation kinetics further highlight ·OH dominance, with second-order rate constants ranging from 109 to 1010 L·mol−1·s−1—orders of magnitude higher than those of sulfate radicals (SO4−·: 107–108 L·mol−1·s−1) and superoxide radicals (O2−·), which exhibit negligible direct oxidation capacity. Crucially, ·OH-mediated pathways mineralize TC into environmentally benign end products (e.g., CO2, H2O, and low-molecular-weight carboxylic acids), whereas SO4−·-based systems risk generating persistent chlorinated byproducts. These combined attributes—unmatched oxidative power, rapid kinetics, and minimal ecological risk—establish ·OH as the most critical species for TC destruction. Consequently, this study focuses on elucidating the mechanistic and kinetic roles of ·OH in TC degradation [10].
However, the intricate reaction mechanisms between ·OH and complex antibiotic molecules like TC remain poorly understood, hindering the rational design of efficient photocatalytic systems [11,12]. Current experimental studies on photocatalytic TC degradation primarily focus on macroscopic kinetics and product identification, offering limited insight into the atomistic details of reaction pathways, transition states, and thermodynamic/kinetic preferences [13]. These gaps impede the optimization of photocatalytic conditions and the development of targeted photocatalysts. Computational chemistry, particularly density functional theory (DFT), provides a powerful tool to unravel reaction mechanisms at the molecular level. Yet, systematic investigations into the interplay of hydrogen atom transfer (HAT), radical adduct formation (RAF) [14], and other pathways for TC and ·OH reactions are scarce, leaving critical questions unanswered: Which pathways dominate under varying conditions? How do the structural and electronic properties of TC influence reactivity? What roles do tunneling effects and temperature play in reaction kinetics? Although there have been attempts to use computational methods to study the reactions related to TC degradation, most of them have only considered a limited number of reaction pathways or have lacked any comprehensive analysis.
In this study, we uniquely combine density functional theory (DFT) calculations and transition state theory (TST) in a comprehensive manner [15,16]. By doing so, we map out as many as 19 distinct reaction channels, precisely 9 HAT and 10 RAF paths. This detailed identification of reaction channels far exceeds the scope of previous studies, enabling us to have a more complete understanding of the reaction network between TC and ·OH. Second, we conduct a thorough analysis of these reaction channels. We not only calculate their energy barriers but also evaluate their thermodynamic feasibility and temperature-dependent kinetics. This multi-faceted analysis provides a more in-depth understanding of the reaction mechanisms at the molecular level. Finally, we focus on evaluating the photocatalytic implications of our findings. By bridging the molecular-level insights obtained from DFT and TST with macroscopic kinetics, we provide a practical framework for optimizing photocatalytic oxidation processes. This framework can be directly applied to combat antibiotic pollution, which is a crucial aspect that previous studies have failed to address comprehensively. In summary, our study offers new perspectives and methods for understanding and optimizing TC degradation in photocatalytic systems.
2. Results and Discussion
2.1. Optimized Structures of TC
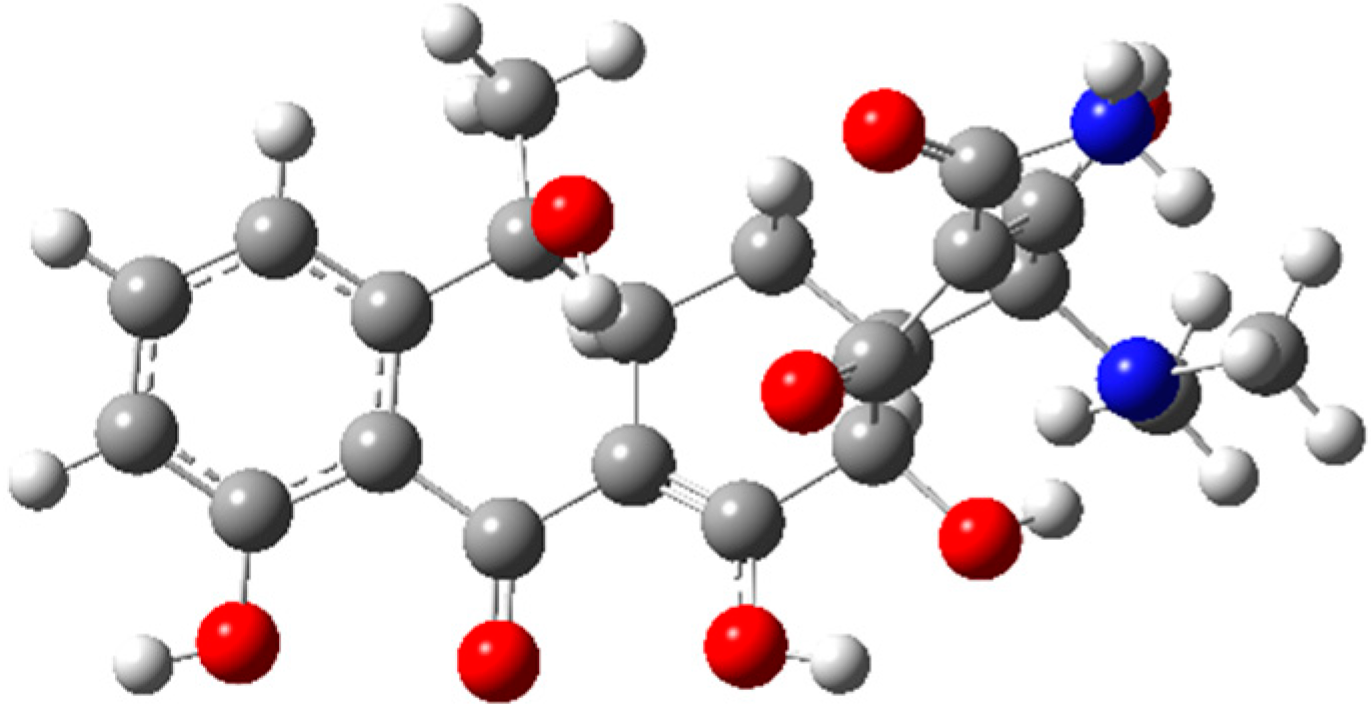
Based on the Monte Carlo conformational sampling analysis, ten possible structures of TC were selected, which would be further optimized to determine the energy minimum by the Gaussian 16 A.03 program. The most stable conformation with the lowest energy is presented in Figure 1. Reasonable bond angles and atomic arrangements reflect the stability of the molecular structure, laying a geometric foundation for subsequent reaction mechanism research.
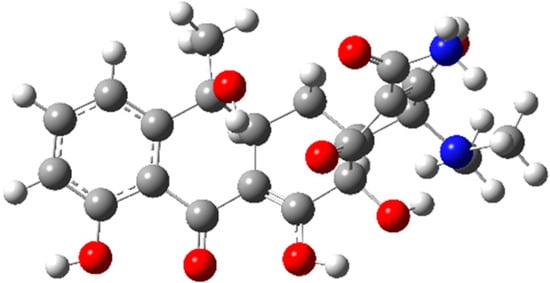
Figure 1.
The optimized geometry of tetracycline at the B3LYP-D3/6-31G (d, p) level. Bond angles are in degrees. Atom representation: red for O, gray for C, white for H, and blue for N.
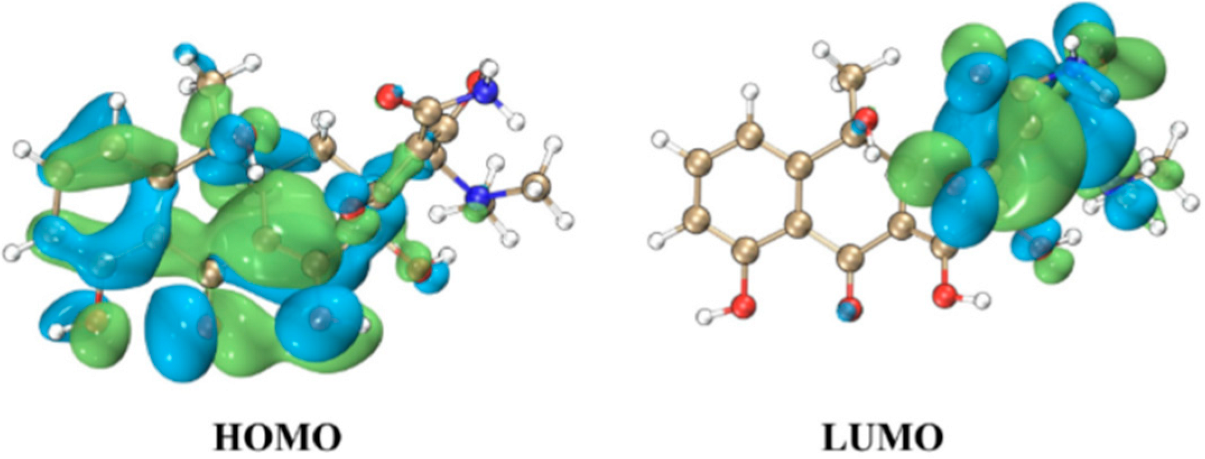
To investigate the structural characteristics of the TC molecule, the distributions of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of TC are illustrated in Figure 2, with an isosurface value set to 0.015 atomic units (a. u.). The HOMO-LUMO energy gap (Δε) is calculated to be 3.81 eV, reflecting the electronic structure and reactivity potential of TC in redox processes involving hydroxyl radicals, which provides theoretical support at the molecular orbital level for the analysis of reaction pathways. The HOMO of TC is predominantly localized over the left-ring moiety of the molecule, with the aromatic ring region exhibiting π-orbital characteristics, and the hydrogen atoms on the aromatic ring are easily extracted by ·OH, leading to HAT reactions. While the LUMO is primarily distributed over the right side of the molecule, carbon atoms near unsaturated bonds are prone to undergo addition reactions with ·OH, resulting in RAF reactions.
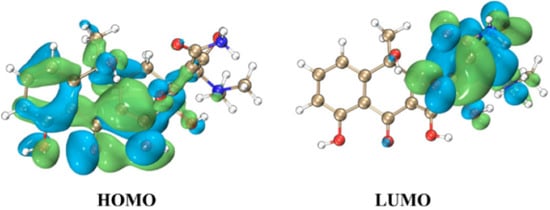
Figure 2.
Structures of frontier molecular orbitals for TC. Light green positive value contour surface, light blue negative value contour surface.
2.2. Reaction Mechanisms of TC with ·OH Radicals
To unravel detailed reaction mechanisms, the initial reaction pathways of TC with ·OH radicals were systematically explored. Three reaction patterns were identified [17,18,19,20]:
- (1)
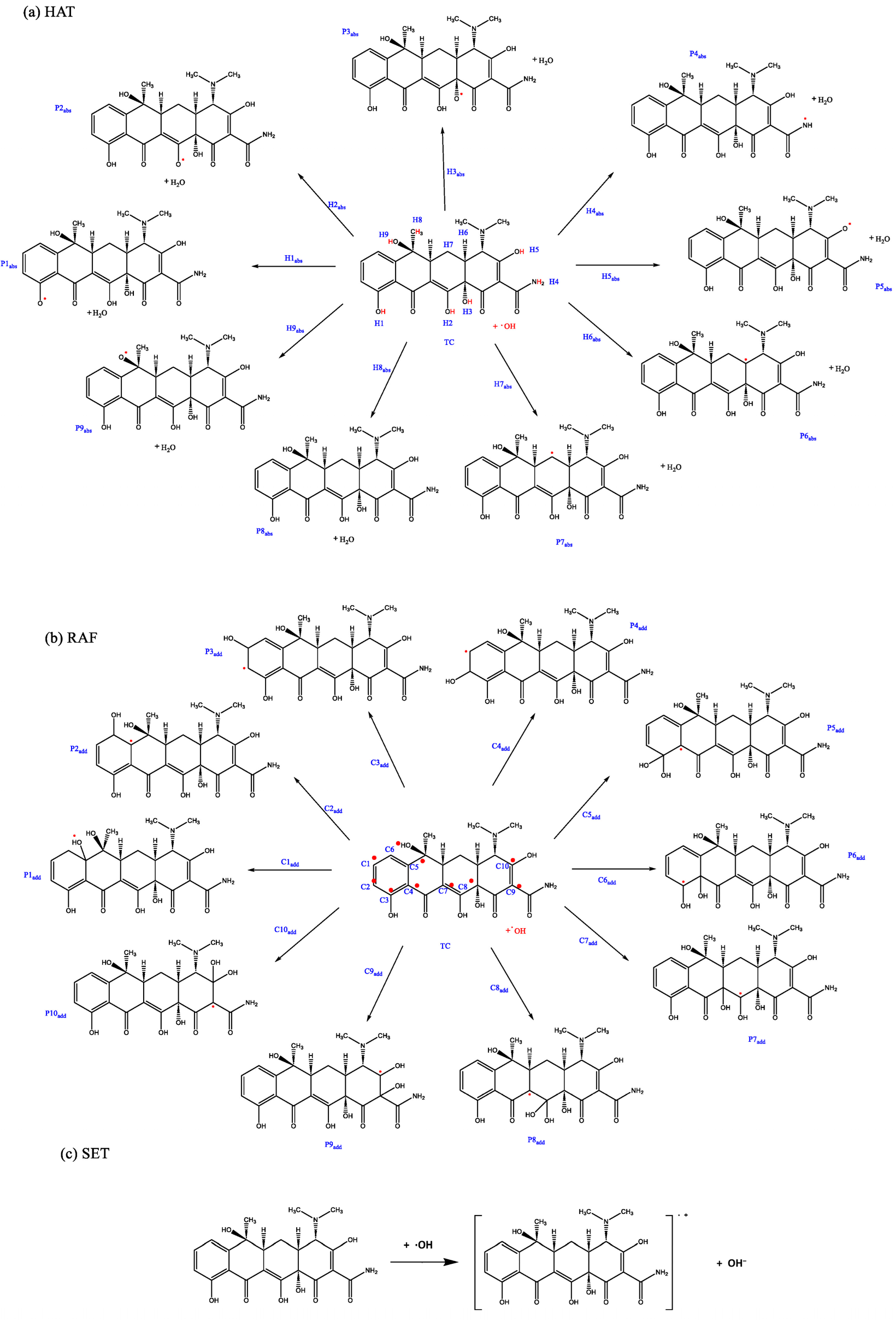
- HAT: ·OH abstracts labile hydrogen atoms from electron-rich functional groups of TC, including phenolic hydroxyls (on benzene rings), enolic hydroxyls (on the central tricarbonyl moiety), dimethylamino protons (-N(CH3)2), and amide protons (-NH2) in the side chain (Figure 3a).
- (2)
- RAF: ·OH adds to electrophilic carbon sites, specifically aromatic carbons (e.g., C10, C11 of the phenol ring) and carbonyl carbons (e.g., C11 keto group and C22 amide group), forming hydroxylated intermediates (Figure 3b).
- (3)
- Single electron transfer (SET): A direct electron transfer from TC to ·OH generates cationic radicals (Figure 3c).
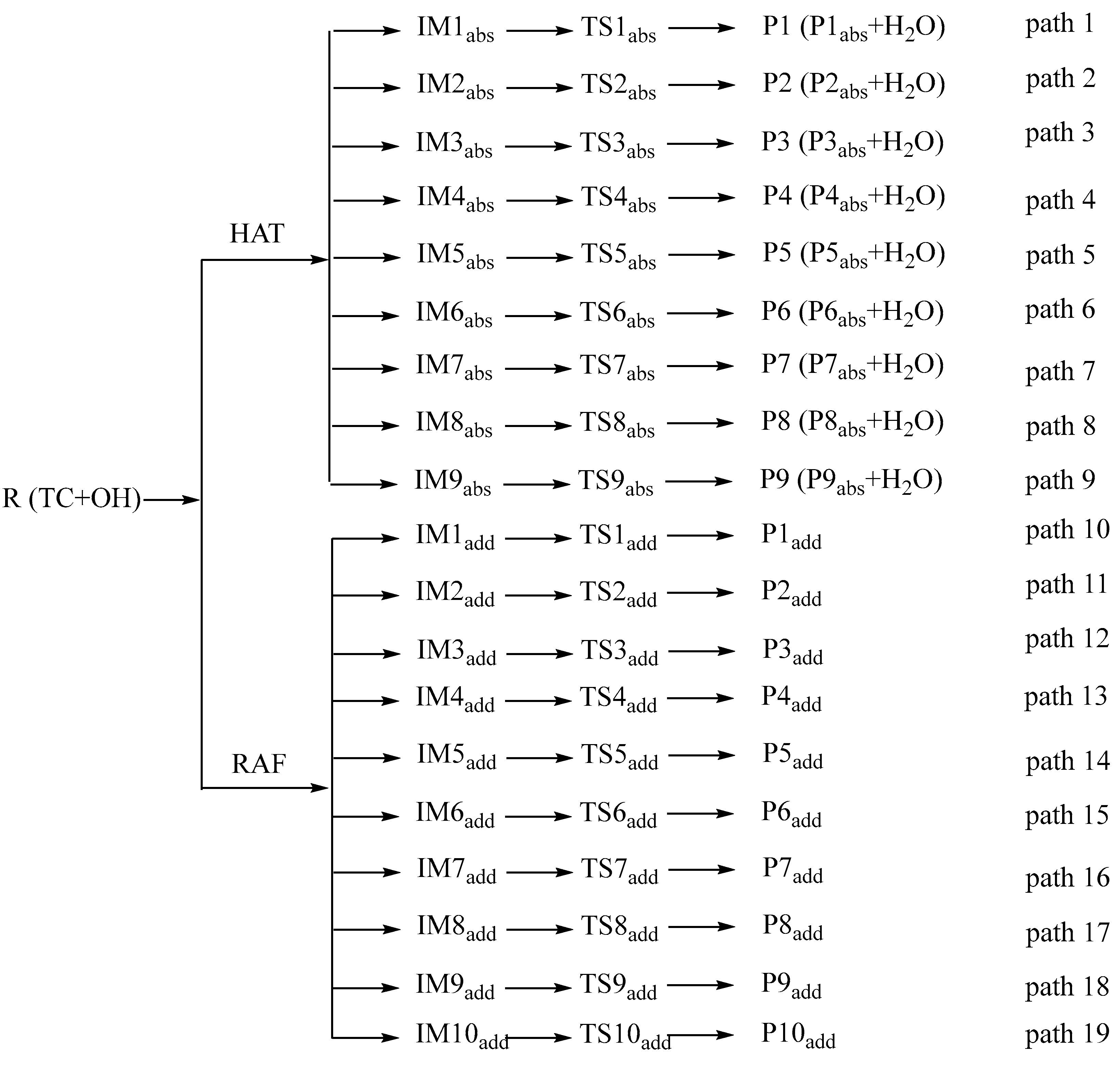
This study focuses on HAT and RAF pathways. For detailed channel diagrams, refer to Figure 4, where “R” represents Reactant, “IM” represents Intermediate, “TS” represents Transition State, “P” represents Product, “abs” represents abstract, and “add” represents adduct.
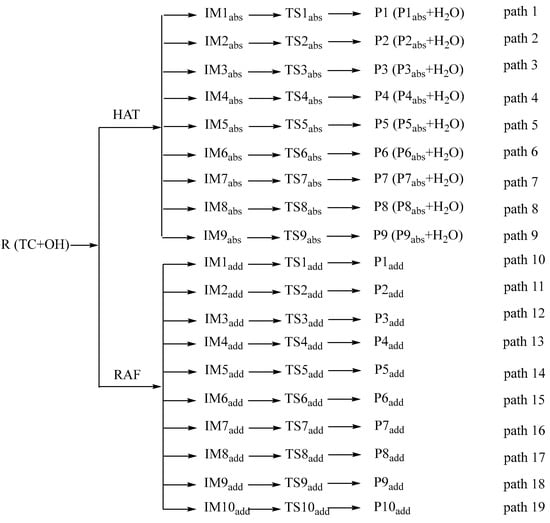
Figure 4.
Detailed reaction pathways of TC and ·OH.
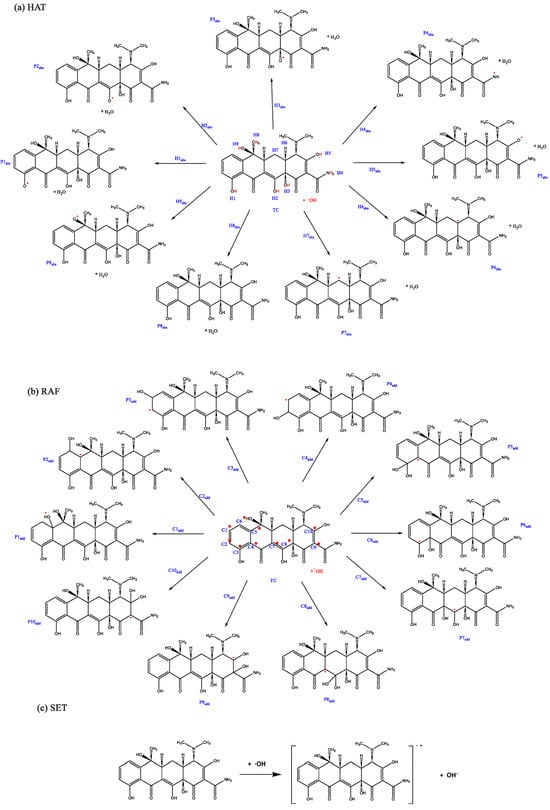
Figure 3.
Three degradation pathways for the reactions of TC with ·OH based on (a) HAT, (b) RAF, and (c) SET pathways. The red dots indicate the key sites of the reaction, showing where in the molecule the reaction takes place.
Figure 3.
Three degradation pathways for the reactions of TC with ·OH based on (a) HAT, (b) RAF, and (c) SET pathways. The red dots indicate the key sites of the reaction, showing where in the molecule the reaction takes place.
2.3. Optimized Structures of Reactants, Transition States, and Products
The reactants, intermediates, transition states, and products were optimized at the B3LYP-D3/6-31G (d, p) level; the optimized geometries, the structural parameters of the transition states, the energies of the stationary points, and the first frequencies of the transition states are shown in the Supplementary Materials.
Based on Figure S1, Table S1, and the vibrational modes of the imaginary frequencies of transition states (TSs), for HAT pathways, the transition state structures involve the transfer of a H atom from N/C/O in TC to the O of the ·OH radical, leading to the formation of hydrogen transfer products and H2O. The imaginary frequency vibrational modes of the TSs consistently show the H atom oscillating between the N/C/O of TC and the O of the ·OH radical. In terms of bond lengths, bond angles, and dihedral angles, the structural parameters of atoms involved in the TSs exhibit similarity. Specifically, the bond length of O1/C/N-H1 ranges from 0.1029 nm to 0.1325 nm. The bond length of H1-O ranges from 0.1099 nm to 0.1520 nm. The bond angle ∠O1-H-O spans from 148.2° to 169.6°. The dihedral angle O1/C/N-H-O-H1 varies from −174.4° to 123.2°. These results indicate a conserved structural motif in the HAT transition states, characterized by partial bond formation/cleavage between the transferring H atom and its original N/C/O atom in TC, as well as the incoming ·OH radical.
Based on Figure S2, Table S2, and the vibrational modes of the imaginary frequencies of transition states (TSs), for RAF pathways, the transition state structure involves the approach of the O atom (bearing its H atom) in the ·OH radical toward N/C/O atoms in TC, followed by additions to these atoms to generate RAF products. The imaginary frequency vibrational modes of the TSs consistently show the mutual approach and retreat between the N/C/O atoms in TC and the O atom of the ·OH radical.
In terms of structural parameters, the atoms involved in the TSs exhibit consistent geometric characteristics: The C-O bond length (between TC’s carbon and the ·OH oxygen) ranges from 0.1975 nm to 0.2265 nm, reflecting the partial bond formation during the RAF process. The O-H bond length in the ·OH radical remains relatively stable, spanning 0.0972 nm to 0.0990 nm, indicating minimal perturbation to the hydroxyl group’s intrinsic structure. The bond angle ∠C-O-H varies between 77.5° and 102.2°, reflecting the geometric rearrangement required for the radical addition. These findings highlight a conserved structural motif in RAF transition states, characterized by the dynamic interaction between the electrophilic ·OH radical and nucleophilic N/C/O sites in TC, providing mechanistic insights into the regioselectivity of the radical addition process.
Vibrational analysis results of each stationary point in the reaction reveal that the eigenvalues of the force constant matrix for reactants, products, and intermediates are all positive, confirming their status as stable points on the potential energy surface. As presented in Tables S3 and S4, all transition states exhibit only one imaginary frequency, which validates their authenticity as transition states according to transition state criterion theory. For HAT pathways, the imaginary frequencies of TS1abs to TS9abs are −148.24 cm−1, −1160.90 cm−1, −445.56 cm−1, −1497.52 cm−1, −823.17 cm−1, −449.50 cm−1, −95.32 cm−1, −1008.53 cm−1, and −629.01 cm−1, respectively. In contrast, RAF pathways (TS1add to TS10add) display imaginary frequencies of −315.07 cm−1, −188.82 cm−1, −362.06 cm−1, −99.64 cm−1, −245.49 cm−1, −253.78 cm−1, −97.72 cm−1, −85.47 cm−1, −236.60 cm−1, and −245.90 cm−1. Notably, HAT TSs exhibit significantly greater imaginary frequencies compared to RAF TSs. A smaller imaginary frequency in RAF TSs suggests a smoother potential energy surface along the reaction coordinate, indicating a less structurally constrained transition state.
Further validation comes from IRC calculations, where the molecular configurations at both ends of the IRC curves correspond directly to the respective reactants and products. This confirms that each TS is a genuine saddle point connecting the reactant and product along the reaction pathway.
2.4. Reaction Mechanism and Potential Energy Surface Analysis
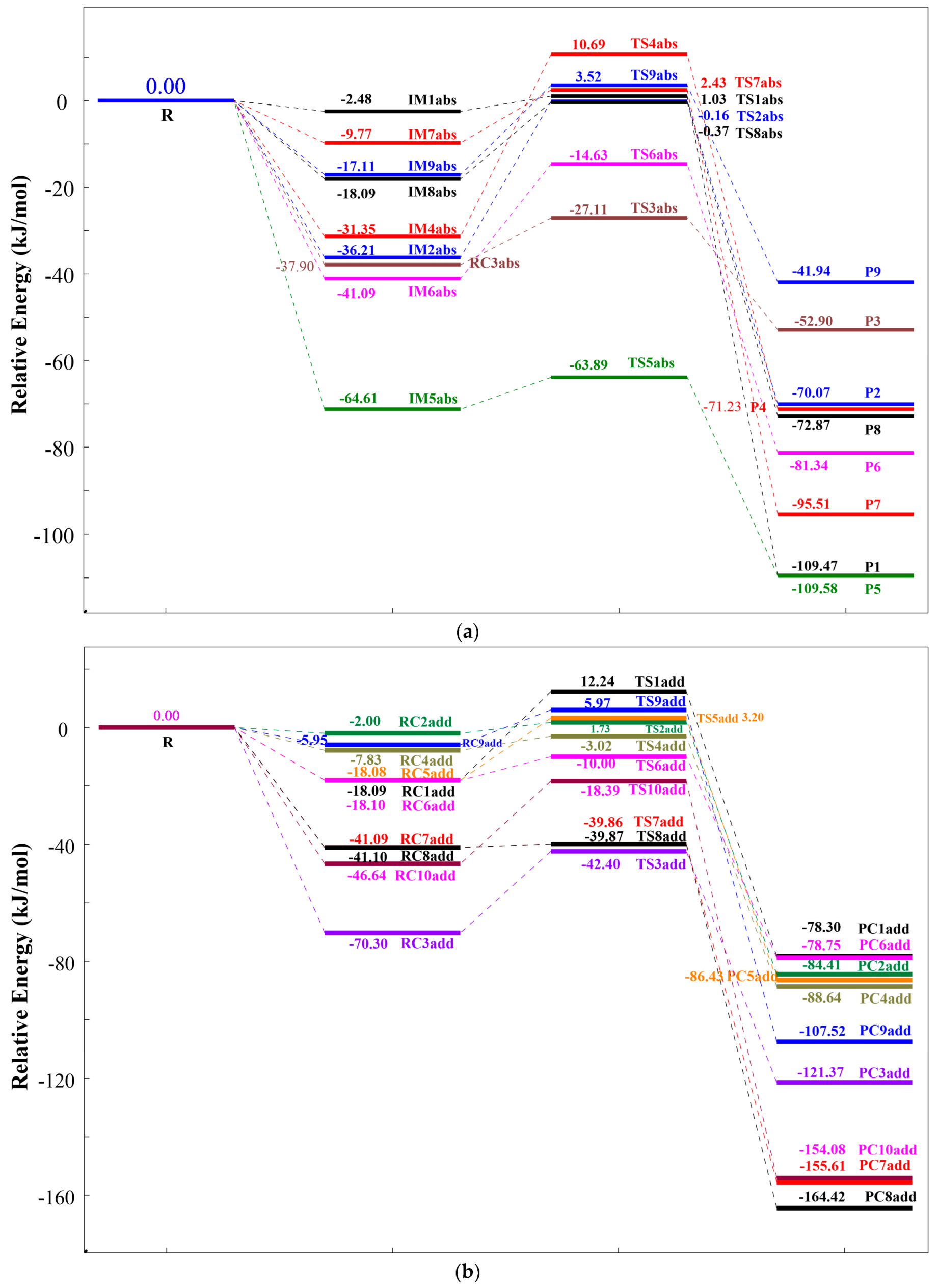
To facilitate the analysis of energy barrier variations, the total energy of the reactants (R(TC+OH)) was set as the zero reference. Tables S5 and S6 present the relative energies of all stationary points, which were used to construct the reaction potential energy surface diagrams illustrated in Figure 5a (HAT pathways) and Figure 5b (RAF pathways).
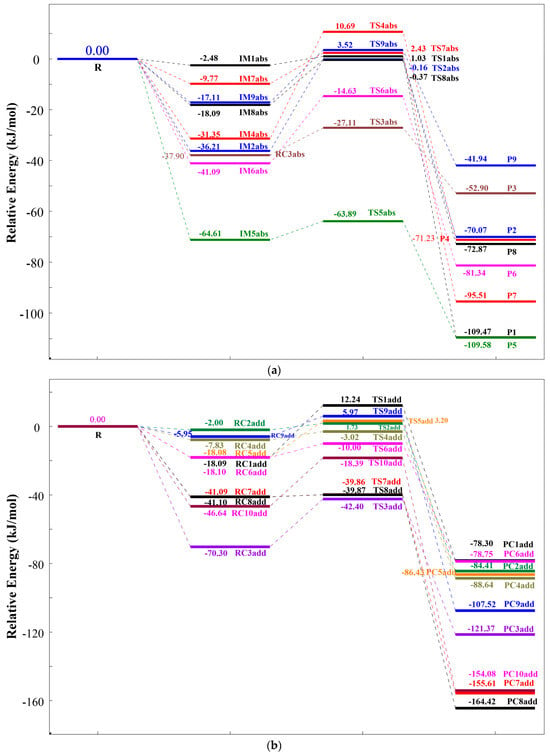
Figure 5.
The potential energy surfaces for the reaction of TC and ·OH via (a) HAT and (b) RAF pathways at the m06-2x/def2TZVP+ZPE level.
For HAT pathways, all reaction routes involve the approach of TC and ·OH radicals. Depending on the reaction site, distinct intermediates (IMabs) form, which resemble reactant complexes with energies lower than the reactants. Among all IMabs (IM1abs to IM9abs), IM5abs exhibits the largest energy reduction (71.23 kJ/mol), while IM1abs shows the smallest decrease (2.48 kJ/mol). These intermediates then proceed through transition states to yield products. The energy barriers for Paths 1 to 9 are 3.51 kJ/mol, 36.05 kJ/mol, 10.79 kJ/mol, 42.04 kJ/mol, 7.34 kJ/mol, 26.46 kJ/mol, 12.2 kJ/mol, 17.72 kJ/mol, and 20.63 kJ/mol, respectively. Path 1, with the lowest barrier (3.51 kJ/mol), is the most facile and dominant HAT pathway. This is attributed to the chemical environment of the reacting hydroxyl group on the aromatic ring, which lacks steric hindrance, facilitating hydrogen abstraction. Conversely, Path 4 (42.04 kJ/mol) is the most challenging, likely due to the abstraction of a hydrogen atom from an amino group (-NH2) adjacent to a carbonyl group, which hinders hydrogen transfer. All HAT products have lower energy than the reactants, confirming the exothermic nature of these pathways and their thermodynamic favorability. P9 has the highest product energy (−41.94 kJ/mol), while P1 is the most stable (−109.58 kJ/mol).
For RAF pathways, TC and ·OH radicals also approach each other, forming intermediates (IMadd) at different carbon sites. These intermediates, analogous to reactant complexes, are lower in energy than the reactants. Among IMadd species (IM1add to IM10add), IM10add shows the largest energy drop (46.64 kJ/mol) and IM2add the smallest (2.00 kJ/mol). Subsequently, the transition state generates new products. The energy barriers for Paths 10 to 19 are in turn 30.33 kJ/mol, 3.73 kJ/mol, 27.9 kJ/mol, 4.81 kJ/mol, 21.28 kJ/mol, 8.1 kJ/mol, 1.23 kJ/mol, 1.23 kJ/mol, 11.92 kJ/mol, and 28.25 kJ/mol. Paths 16 and 17 (1.23 kJ/mol) are the most kinetically favorable and dominant RAF pathways, whereas Path 10 (30.33 kJ/mol) is the most hindered, influenced by the chemical environment of the target carbon atom in TC. Like HAT, RAF products are thermodynamically favorable due to their lower energy relative to the reactants. P1add has the highest product energy (−78.30 kJ/mol), and P8add has the lowest (−164.42 kJ/mol).
A comparison of the two pathways reveals that RAF exhibits lower overall energy barriers (kinetically more favorable) and more exothermic products (thermodynamically more favorable) than HAT. These findings highlight the inherent advantages of RAF in the degradation of TC by ·OH radicals.
2.5. Kinetic Calculations
For a better understanding of the transformation of TC in aquatic environments and quantitatively evaluating the contribution of HAT and RAF pathways during TC degradation, the rate constants (ks) for HAT and RAF reaction pathways were calculated using the KiSThelP 2021 program. Considering the experimental temperature of TC degradation in photocatalytic oxidation and the actual temperature of sewage treatment plants, the temperature was set at 298 K in this study. The rate constants of key elementary reactions are displayed in Table 1 and Table 2, indicating that the HAT and RAF reactions between TC and ·OH can easily occur at room temperature. In HAT pathways, Path 1 exhibits significantly higher rate constants, while Path 9 shows notably lower values, consistent with the previous energy analysis (Path 1 has the lowest energy barrier and the easiest reaction to proceed). For RAF pathways, Path 16 and 17 display relatively larger rate constants, whereas Path 10 exhibits comparably smaller rate constants, which are also consistent with the energy analysis of the RAF pathway (Path16 and 17 have the lowest energy barriers and the reaction is easiest to proceed, while Path10 has a higher energy barrier and the reaction is not easy to proceed). Rate constants computed at 298 K (TST with Wigner tunneling correction) confirm RAF’s kinetic superiority, with Paths 16/17 achieving the highest rate constants (≈7.0 × 1011 s−1), surpassing HAT’s most facile pathway (Path 1, ≈6.2 × 1011 s−1). In summary, compared to HAT, RAF has an advantage in reaction rate.

Table 1.
Predicted rate constants at 298 K of the reaction for HAT paths (the unit of k is in s−1).
Table 2.
Predicted rate constants at 298 K of the reaction for RAF paths (the unit of k is in s−1).
3. Theoretical Calculations
3.1. Electronic Structure Calculations
DFT was employed to investigate the reaction pathways of ·OH with tetracycline hydrochloride. First, the B3LYP-D3 method [21] was used to fully optimize the reactants, intermediates, transition states, and products in the reaction system at the 6-31G (d, p) basis set level, and the correctness of the transition states was verified by vibrational analysis. To obtain more accurate energies, single-point energies were calculated at the m06-2x/def2TZVP level [22], based on which reaction barriers and relative energies (Erel) were calculated with zero-point energy (ZPE) corrections. The connection relationship between reactants, intermediates, transition states, and products in the reaction channel was verified through the calculation of intrinsic reaction coordinates (IRCs) [23]. All the above calculations were completed using the Gaussian 16 A.03 program.
The distribution of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) characterizes the chemical reactivity and charge transfer interaction in the molecule. The energy difference between HOMO and LUMO is called the energy gap (Δε), which can be used as an excellent indicator to study the reactivity of reactants. Therefore, this chapter uses Multiwfn 3.8 software and VMD 1.9.3 software to calculate the FMO diagram of TC.
3.2. Reaction Kinetics Calculations
The traditional transition state theory (TST) has shown remarkable advantages in calculating reaction rate constants. In combination with the Gibbs free energy barrier, the primary reaction rate constants of TC with ·OH radicals were calculated at 101,325 Pa and 298 K using the KiSThelP 2021 program [24], which was based on transition state theory (TST) with Wigner tunneling correction. The reaction rate constants follow the following formula:
where is the ideal gas constant (8.314 J·mol−1·K−1), is the Gibbs free energy, σ is the reaction path degeneracy, and and are the Boltzmann and Planck constants, respectively. The unit of is exactly the inverse of the concentration, and = 0 and 1 denote gas-phase unimolecular and bimolecular processes, respectively.
4. Conclusions
This study systematically elucidated the degradation mechanisms of TC by ·OH using DFT and TST, yielding critical insights into reaction pathways and environmental implications.
Reaction Pathways and Energetics: HAT and RAF dominate TC degradation by ·OH. RAF pathways exhibit distinct advantages, characterized by lower activation barriers (e.g., 1.23 kJ/mol for Paths 16/17) and greater exothermicity (product energy as low as −164.42 kJ/mol), rendering them both kinetically and thermodynamically advantageous compared to HAT. HAT reactivity is governed by the chemical environment of hydrogen abstraction sites, with hydroxyl groups on aromatic rings (Path 1, 3.51 kJ/mol) demonstrating the lowest barriers due to minimal steric hindrance. In contrast, RAF pathways are dictated by electrophilic attacks on unsaturated carbon sites (e.g., C10/C11 of the phenol ring), with preferential additions to electron-deficient moieties.
Molecular Orbital Analysis: Frontier molecular orbital (FMO) distributions provide a mechanistic rationale for observed reactivity patterns. The HOMO of TC, localized on the left aromatic ring with π-orbital characteristics, promotes HAT reactivity by facilitating hydrogen abstraction. Conversely, the LUMO, distributed over electrophilic sites on the molecule’s right side, enables RAF through nucleophilic attack. The calculated HOMO-LUMO energy gap (3.81 eV) underscores TC’s redox potential, aligning with its susceptibility to ·OH-induced degradation.
Kinetic Profiling: Rate constants computed at 298 K (TST with Wigner tunneling correction) confirm RAF’s kinetic superiority, with Paths 16/17 achieving the highest rate constants (≈7.0 × 1011 s−1), surpassing HAT’s most facile pathway (Path 1, ≈6.2 × 1011 s−1). Tunneling effects marginally enhance reaction rates, particularly for HAT, highlighting the importance of quantum mechanical corrections in kinetic modeling.
Environmental and Catalytic Implications: The dominance of RAF pathways validates the efficacy of ·OH-based photocatalytic oxidation for TC removal in wastewater treatment. The low activation barriers and rapid kinetics at ambient temperatures emphasize the practical viability of photocatalytic systems toward treating antibiotic pollutants. These findings provide a theoretical framework for optimizing photocatalytic oxidation, with potential applications in designing photocatalysts that leverage RAF pathways to enhance degradation efficiency.
Contributions and Outlook: This study advances the mechanistic understanding of TC degradation by establishing RAF as the primary pathway, linking reactivity to molecular electronic structure, and providing quantitative kinetic data. However, it is important to note that real-life wastewater is a complex matrix containing various other substances, such as dissolved organic matter, inorganic salts, and competing pollutants. These components may interact with TC, ·OH radicals, or the photocatalyst surface, potentially affecting the reaction rates and pathways. For instance, some substances might scavenge ·OH radicals, reducing the overall degradation efficiency of TC. But overall, considering the high reactivity of ·OH radicals and the favorable thermodynamics and kinetics of the RAF pathways, we believe that the degradation of TC via these pathways is feasible in real-life wastewater treatment scenarios. Future research may explore pH-dependent reactivity, solvent effects, and photocatalyst design to further optimize degradation performance. By bridging computational insights with environmental relevance, this work contributes to the development of sustainable strategies for combating antibiotic contamination.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal15050420/s1, Figure S1. Geometric configurations of the reactants, transition states and products in HAT paths. Figure S2. Geometric configurations of the reactants, transition states and products in RAF paths. Table S1. Geometric parameters of species in HAT paths. Table S2. Geometric parameters of species in RAF paths. Table S3. Stationary point energies and imaginary frequency of transition states in HAT paths. Table S4. Stationary point energies and imaginary frequency of transition states in RAF paths. Table S5. Relative energies (kJ•mol−1) in HAT paths. Table S6. Relative energies (kJ•mol−1) in RAF paths.
Author Contributions
Conceptualization, J.L. and T.S.; methodology, J.L.; software, Y.L.; validation, H.B.; formal analysis, T.S.; investigation, Y.L.; resources, Y.Z.; data curation, J.L.; writing—original draft preparation, J.L.; writing—review and editing, Y.Z.; visualization, C.Z.; supervision, C.Z.; project administration, J.L.; funding acquisition, J.L., C.Z., and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Scientific Research Fund Project of Shandong University of Aeronautics (BZXYLG1924), the Scientific Research Start-up Fund Project for Doctoral Degree Personnel of Shandong University of Aeronautics (2023Y33), the Shandong University of Aeronautics Research Funds (2023Y19), the Youth Backbone Teacher Training Project of Henan Province (2024GGJS123), and the Undergraduate Innovation and Entrepreneurship Training Project of Henan Province (202410478025).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials; further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fu, Y.; Xu, Y.; Mao, Y.; Tan, M.; He, Q.; Mao, H.; Du, D.; Hao, D.; Wang, Q. Multi-functional Ag/Ag3PO4/AgPMo with S-scheme heterojunction for boosted photocatalytic performance. Sep. Purif. Technol. 2023, 317, 123922. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, Q.; Deng, D.; Li, Z.; Xue, B.; Liu, A.; Shen, B.; Hao, D.; Zhu, H.; Wang, Q. Schottky heterojunction-based photocatalysis-in-situ-self-Fenton system: Removal of tetracycline hydrochloride and biotoxicity evaluation of intermediates. Appl. Catal. B 2025, 360, 124533. [Google Scholar] [CrossRef]
- Hu, C.; Chang, L.; Chen, W.; Hsu, W.; Chien, S.; Chen, C.; Lin, Y.; Hsu, T.; Tung, K. 3D-printed Al2O3 framework supported carbon-bridged tri-s-triazine of g-C3N4 for photocatalytic tetracycline oxidation. Chem. Eng. J. 2024, 487, 150504. [Google Scholar] [CrossRef]
- He, S.; Zhai, C.; Fujitsuka, M.; Kim, S.; Zhu, M.; Yin, R.; Zeng, L.; Majima, T. Femtosecond time-resolved diffuse reflectance study on facet engineered charge-carrier dynamics in Ag3PO4 for antibiotics photodegradation. Appl. Catal. B 2021, 281, 119479. [Google Scholar] [CrossRef]
- Lu, P.; Peng, Y.; Bai, J. Polyimide/Ag2WO4 Z-scheme heterojunction for efficient photocatalytic degradation of tetracycline. Langmuir 2024, 40, 12191–12199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zheng, S.Z.; Ma, W.G.; Qian, J.Y.; Huang, L.Y.; Deng, H.; Zhou, Q.; Zheng, S.R.; Li, S.J.; Du, H.; et al. Facile synthesis of direct Z-scheme PPy/NH2-UiO-66 heterojunction for enhanced photocatalytic Cr(VI) reduction, industrial electroplating wastewater treatment, and tetracycline degradation. Appl. Catal. B 2024, 344, 123669. [Google Scholar] [CrossRef]
- Xie, Q.H.; Qin, J.N.; Gao, T.; Li, F.; Zhong, N.B.; Pan, B. Engineering ZnIn2S4 nanosheets with zinc vacancies: Unleashing enhanced photocatalytic degradation of tetracycline. Langmuir 2024, 40, 25327–25333. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.N.; Yang, H.; Xie, J.B.; Teng, G.X.; He, J.; Zhao, Z.L.; Zhang, C. A photocatalyst combined of copper doped ZnO and graphdiyne (Cu/ZnO@GDY) for photocatalytic degradation of tetracycline: Mechanism and application. Water Res. 2025, 278, 123345. [Google Scholar] [CrossRef]
- Lu, C.G.; Jiao, C.J.; Wei, Z.Q. Zn-doped FeOCl nanosheets enable accelerated tetracycline degradation via simulated sunlight-responsive photo-Fenton catalysis. Mater. Sci. Semicond. Process. 2025, 194, 109542. [Google Scholar] [CrossRef]
- Choi, N.; Tang, C.; Park, Y.; Du, A.; Ayoko, G.A.; Hwang, Y.; Chae, S. Visible-light-driven photocatalytic degradation of tetracycline using citric acid and lemon juice-derived carbon quantum dots incorporated TiO2 nanocomposites. Sep. Purif. Technol. 2024, 350, 127836. [Google Scholar] [CrossRef]
- Zhang, Q.; Zheng, D.; Bai, B.; Ma, Z.; Zong, S. Insight into antibiotic removal by advanced oxidation processes (AOPs): Performance, mechanism, degradation pathways, and ecotoxicity assessment. Chem. Eng. J. 2024, 500, 157134. [Google Scholar] [CrossRef]
- Chen, Y.; Yin, R.; Zeng, L.; Guo, W.; Zhu, M. Insight into the effects of hydroxyl groups on the rates and pathways of tetracycline antibiotics degradation in the carbon black activated peroxydisulfate oxidation process. J. Hazard. Mater. 2021, 412, 125256. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, S.; Kang, J.; Tang, Y.; Wang, J.; Xu, Z.; Liu, J. Enhanced tetracycline degradation by NC codoped Fe2O3 with rich oxygen vacancies in peroxymonosulfate assisting photoelectrochemical oxidation system: Performance, mechanism and degradation pathway. Chem. Eng. J. 2023, 451, 138611. [Google Scholar] [CrossRef]
- Biswas, S.; Shukla, P.K. A DFT study on the scavenging activity of curcumin toward methyl and ethyl radicals. Mol. Simulat. 2023, 49, 589–598. [Google Scholar] [CrossRef]
- Yue, T.-C.; Wang, L.-L.; Li, W.; Zhang, J.; Wang, D.-Z.; Bu, X.-H. Efficient degradation of tetracycline hydrochloride via Fe-MOF-Initiated Photo-Self-Fenton Catalysis. Sep. Purif. Technol. 2025, 363, 132127. [Google Scholar] [CrossRef]
- Wang, S.J.; Zhang, J.Q.; Li, B.; Sun, H.Q.; Wang, S.B. Engineered Graphitic Carbon Nitride-Based Photocatalysts for Visible-Light-Driven Water Splitting: A Review. Energy Fuels 2021, 35, 6504–6526. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Hadizadeh, M.H.; Liu, L.; Xu, F. Theoretical insights into the degradation mechanisms, kinetics and eco-toxicity of oxcarbazepine initiated by OH radicals in aqueous environments. J. Environ. Sci. 2023, 129, 189–201. [Google Scholar] [CrossRef]
- Luo, S.; Gao, L.; Wei, Z.; Spinney, R.; Dionysiou, D.D.; Hu, W.-P.; Chai, L.; Xiao, R. Kinetic and mechanistic aspects of hydroxyl radical-mediated degradation of naproxen and reaction intermediates. Water Res. 2018, 137, 233–241. [Google Scholar] [CrossRef]
- Gao, L.; Mao, Q.; Luo, S.; Cao, L.; Xie, X.; Yang, Y.; Deng, Y.; Wei, Z. Experimental and theoretical insights into kinetics and mechanisms of hydroxyl and sulfate radicals-mediated degradation of sulfamethoxazole: Similarities and differences. Environ. Pollut. 2020, 259, 113795. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, X.; Tan, W.; Peng, H. Atmospheric oxidation of Folpet initiated by OH radicals, NO3 radicals, and O3. RSC Adv. 2021, 11, 2346–2352. [Google Scholar] [CrossRef]
- Jena, N.R.; Shukla, P.K. Hydroxyl radical-induced C1’-H abstraction reaction of different artificial nucleotides. J. Mol. Model. 2024, 30, 330. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lv, G.; He, L.; Sun, X. New insight into photodegradation mechanisms, kinetics and health effects of p-nitrophenol by ozonation in polluted water. J. Hazard. Mater. 2021, 403, 123805. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yin, L.; Qu, R.; Pan, X.; Wang, Z. Experimental and quantum chemical study on the transformation behavior of bisphenol S by radical-driven persulfate oxidation. Environ. Sci. Water Res. Technol. 2022, 8, 116–126. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Zhang, Y.; Xu, F.; Zhang, Q.; Wang, W. Theoretical investigation on the mechanisms, kinetics, and product nucleation of OH/O3-initiated atmospheric oxidation of methyl salicylate. J. Environ. Chem. Eng. 2025, 13, 115297. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).