Abstract
This article reviews work done at authors’ laboratories about catalysts based on combinations between copper and ceria for preferential oxidation of CO in H2-rich streams (CO-PROX). The main focus of this review is the characterization of active sites for the process on the basis of spectroscopic analysis of the systems under reaction conditions (operando techniques). On such a basis, it is exposed the state of the art in this field in connection with results obtained in other laboratories.
1. Introduction
Production of H2 for polymer fuel cells (PEMFC) is usually accomplished by a multi-step process that includes catalytic reforming of hydrocarbons or oxygenated hydrocarbons followed by water-gas shift (WGS) [1,2]. The gas stream obtained after these processes presents, in most cases, a relatively high CO concentration that disallows efficient handling of the fuel by the Pt alloy anode usually employed in the PEMFC. Preferential (or selective) oxidation of CO in the H2-rich stream resulting from such processes (CO-PROX) has been recognized as one of the most straightforward and cost-effective methods to achieve acceptable CO concentrations (below ca. 100 ppm) [3,4,5,6,7].
Different types of catalysts have shown their efficiency for the CO-PROX process. These can be classified into three general groups as a function of their nature and/or respective catalytic properties. The first group involves supported noble metal catalysts (mainly Pt ones) and follows from the first developments done by Engelhard researchers in the context of processes related to production of clean hydrogen for ammonia synthesis [8]. These are mostly employed and commercially available catalysts, although they exhibit a relatively low selectivity for the reaction of interest (CO oxidation) at practical operating temperature (between 373 and 473 K), which can make necessary interstage cooling operations to avoid extensive heating as a consequence of the exothermicity of the (H2 and CO) oxidation reactions involved [9]. A second group of active catalysts involves supported gold catalysts, well known for their outstanding performance for CO oxidation [10,11,12,13,14]. These show a high CO-PROX activity with a good match between their activity window and the PEMFC anode operating temperature (353–403 K). However, they can have the drawback of their poor resistance to the presence of CO2 in the reactant mixture [3,11,12,15]. The third group is constituted by catalysts based on closely interacting copper oxide and ceria, which have shown promising properties in terms of activity, selectivity and resistance to CO2 and H2O, while their lower cost (particularly in comparison to catalysts based on supported platinum, taking also into account the relatively high platinum loading required to optimize CO-PROX performance) could make them strongly competitive [3,4,6,7,16,17,18,19,20,21,22,23,24].
The particular ability of copper-ceria catalysts for the CO-PROX or related processes has been essentially attributed to the synergistic redox properties exhibited upon formation of copper oxide-ceria interfacial sites [4,6,17,18,25,26,27,28,29,30,31,32,33,34,35]. In this sense, generally speaking, the properties of copper oxide entities for CO oxidation promotion depend strongly on their dispersion degree and/or related degree of interaction with ceria [18,25,26,36,37]. Nevertheless, although reaction models for CO oxidation (competing or not with H2 oxidation, as it occurs under CO-PROX conditions) have been proposed on the basis of indirect analysis of redox or catalytic properties for this type of catalyst [38,39]; direct evidence on redox changes taking place in the catalyst under the reactant atmosphere, which could provide details on the nature of active sites for the processes taking place under CO-PROX conditions in this type of catalyst, has, to the best of our knowledge, only recently been reported in a series of contributions from our laboratory [18,40,41]. In turn, such redox characteristics have been shown recently to depend strongly on the type of face exposed by the ceria support, which has important consequences for the CO-PROX performance of this type of catalyst [42].
The present contribution reviews such studies and also analyzes issues related to catalytic/redox correlations on catalysts of this type in order to attempt to establish the most relevant aspects of the state of the art in this field.
2. Results and Discussion
2.1. Redox and Catalytic Properties as a Function of Changes in the Ceria-Based Support Nature
The ability of catalysts based on combinations between copper oxide and ceria for CO oxidation or the CO-PROX process has been related to the promotion of redox properties, which is achieved upon combination of both oxides, taking into account that they appear to operate under a redox-type catalytic mechanism [6]. The existence of a redox promotion has been mainly based on intensive investigation done with different spectroscopic techniques, as well as temperature-programmed reduction studies in which it has generally been shown that the CO oxidation rate can be correlated with the degree of ceria-promoted reducibility attained on the dispersed copper oxide entities [6,26,29,39,43,44]. In turn, the level of promotion of such reduction can be also affected by modifying the nature of the support within structurally related doped ceria materials [18,27]. In this sense, the first approach of the authors’ laboratory to the catalytic properties of this type of system was done in a work in which a set of catalysts prepared by impregnation with the same copper loading (1 wt.%)—but differing in the nature of the ceria-related support that was employed—were tested for the CO-PROX process [18]. Supports were chosen on the basis of previous experience in the field of three-way catalysts (TWC), as well as considering differences in oxygen transport properties [45]. Thus, in addition to the simple pure ceria support, Ce-Zr and Ce-Tb mixed oxide supports were employed, taking into account that they could provide enhanced physicochemical and catalytic characteristics based on previous experience in the TWC field, particularly in terms of enhanced redox (oxygen transport) properties [46]. Preparation and characterization details for these systems can be found elsewhere [18]; it must be noted that SBET values around 100 m2/g were exhibited by all these systems. As displayed in Figure 1, all catalysts are essentially constituted by fluorite nanocrystals (stable structure for the corresponding supports; also in agreement with HREM investigation displaying the presence of more or less rounded ca. 5–8 nm nanocrystals [46,47]) onto which copper oxide entities have been dispersed. The chemical state of copper has been shown to be similar for all samples and, in turn, similar to that observed for a CuO reference, with small differences being attributed to interactions with the ceria-related support [18,37], as evidenced by XANES (Figure 1). The existence of a fully oxidized state, Cu(II), of copper in the initial calcined catalysts is also in agreement with XPS investigation, as shown in 2. In turn values of XPS atomic ratios are in agreement with achievement of a relatively high dispersion degree in the copper oxide entities in all cases ). However, in spite of the fact that mixed oxide supports could provide enhanced redox properties to the catalysts [46], overall CO-PROX performance is shown to be optimized (both in terms of CO conversion and CO2 selectivity—i.e., O2 selectivity towards oxidizing CO in competition with H2) for the system supported on pure ceria, as displayed in 3. In turn such behavior cannot be attributed to an enhanced copper oxide dispersion in the case of CuC according to analysis of Ar +-sputtering XPS experiments (Figure 2), which point to an increase in the dispersion as CuC ≈ CuCT4 < CuCT1 < CuCZ [18]. In order to attempt to rationalize this behavior, the catalysts of the Ce-Tb series have been examined by means of CO-TPR tests [48]. In this sense, recent work by Luo et al. [32] provides an interesting rationalization of redox/catalytic correlations in catalysts combining copper and cerium oxides; on the basis of classical CO-TPR results, they could differentiate between the reducibilities of the three types of oxidized copper entities generically proposed to be present in this type of catalyst (in decreasing order of reducibility or CO oxidation activity): finely dispersed CuO; bulk-like CuO; and, finally, Cu2+ in the ceria lattice, likely substitutionally. Nevertheless, such an investigation has been undertaken on a series of catalysts combining in all cases copper oxide and ceria while, as previously mentioned, changes in the support nature can modify such redox or catalytic properties [46]. In this sense, as shown in Figure 4, the reduction profile (under CO) of CuC is characterized by a low intensity reduction peak and a somewhat stronger peak at about 170 °C and 225 °C, respectively, which are referred to as α and β peaks, according to the usual nomenclature employed in the literature [32,33,34]. In addition, there is a high intensity peak at 298 °C (γ peak). These peaks below 300 °C can be assigned to the reduction of Cu2+ ions belonging to different types of oxidized copper entities with differing degrees of interaction with the underlying ceria support. The presence of more than one reduction peak in the CuC catalyst, as opposed to one peak in the case of pure CuO (Figure 4), is consistent with the existence of more than one type of copper oxide species in CuC, which is in agreement with the irregular shape detected in the Cu/Ce atomic ratio evolution during Ar+-sputtering experiments (Figure 2) [18]. The reduction of copper species responsible for α and β peaks occurs at lower temperatures compared to pure CuO, while the γ peak maximum appears fairly close to that of bulk CuO. This justifies attribution of the former two peaks to well-dispersed copper oxide species whose reduction would be appreciably promoted by ceria, while the latter accounts for the presence of less dispersed (even though still diffraction silent; see Figure 1) copper oxide entities. Such an interpretation is also in agreement with results achieved by EPR in previous studies [29]. Apparent downward shifts of the reduction of copper species are observed in the presence of Tb in the support (Figure 4). Thus, the reduction profile of CuCT4 contains a low temperature peak at 120 °C, whereas two peaks appear at higher temperatures (190 °C and 250 °C). The presence of multiple peaks must reflect, as in the case of CuC, the heterogeneity of copper oxide entities in these samples, which is in agreement also with Ar+-sputtering experiments (Figure 2). Concerning CuCT1, the reduction starts around 50 °C and an intense peak at 155 °C is observed. There is also a hump at 250 °C in this case, which could in part be related to the existence of residual WGS activity (as a consequence of interaction of CO with surface hydroxyls) [49]. Similar results can be found in reports by Wang et al. [38,50], in which the formation of a low temperature α peak in case of CuO on samarium-doped ceria has been attributed to the effect of the presence of surface oxygen vacancies within the oxygen ion conducting support. They have inferred that an interfacial metal oxide-support interaction mechanism is involved at the inception of the reduction, followed by the induced successive reduction of bulk copper oxides, somewhat in accordance with a proposed model for the reduction of components of this type of system upon interaction with CO [29]. Similarly, our CO-TPR results (Figure 4) suggest that the low temperature reducibility of finely dispersed CuO on CuCT4 and CuCT1 is enhanced in comparison to CuC.
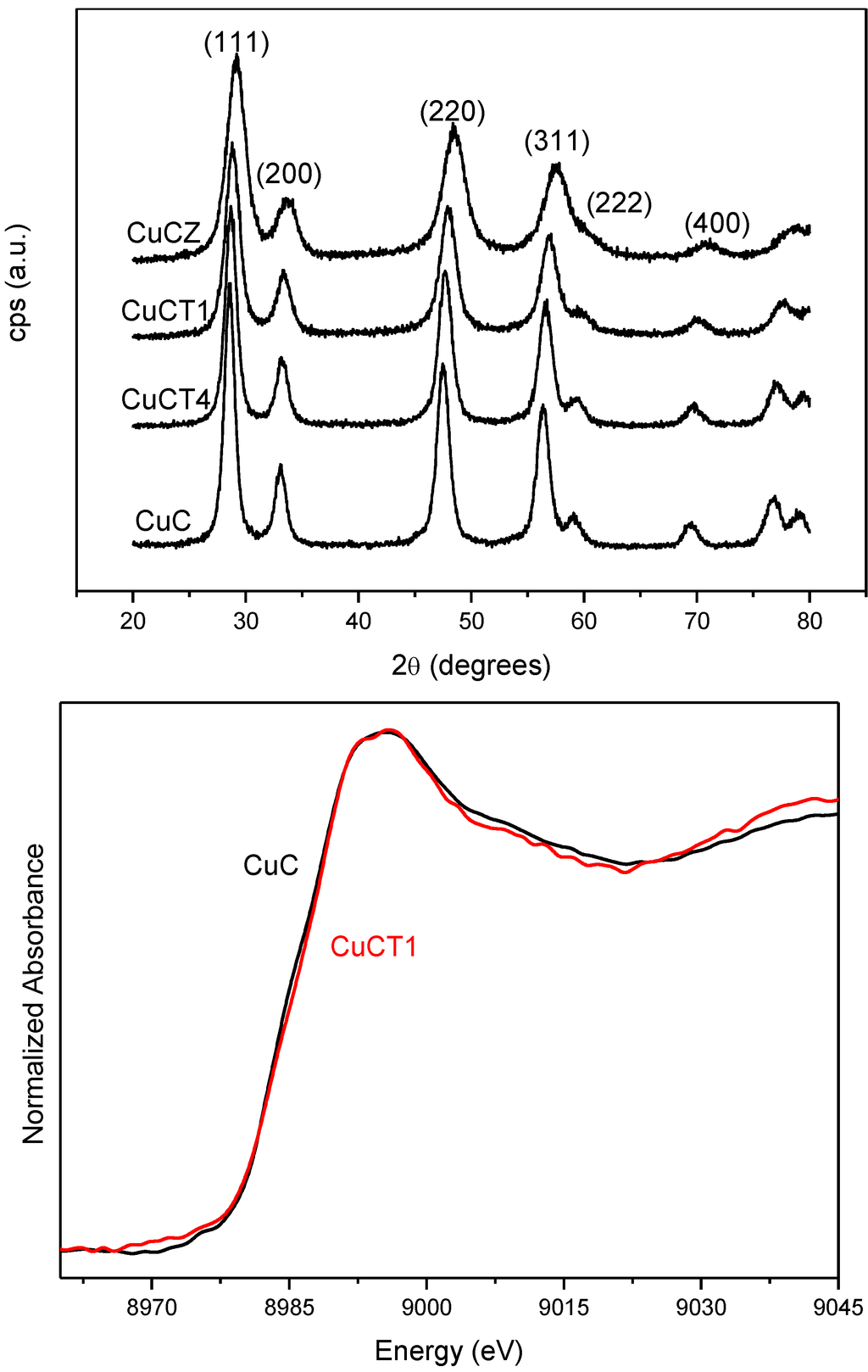
Figure 1.
Top: X-ray diffractograms of catalysts with 1 wt.% copper oxide dispersed on ceria (CuC), Ce-Zr mixed oxide (CuCZ) and Ce-Tb mixed oxides with 4:1 (CuCT4) and 1:1 (CuCT1) Ce:Tb atomic ratios. Bottom: XANES spectra for the indicated catalysts. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
Figure 1.
Top: X-ray diffractograms of catalysts with 1 wt.% copper oxide dispersed on ceria (CuC), Ce-Zr mixed oxide (CuCZ) and Ce-Tb mixed oxides with 4:1 (CuCT4) and 1:1 (CuCT1) Ce:Tb atomic ratios. Bottom: XANES spectra for the indicated catalysts. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
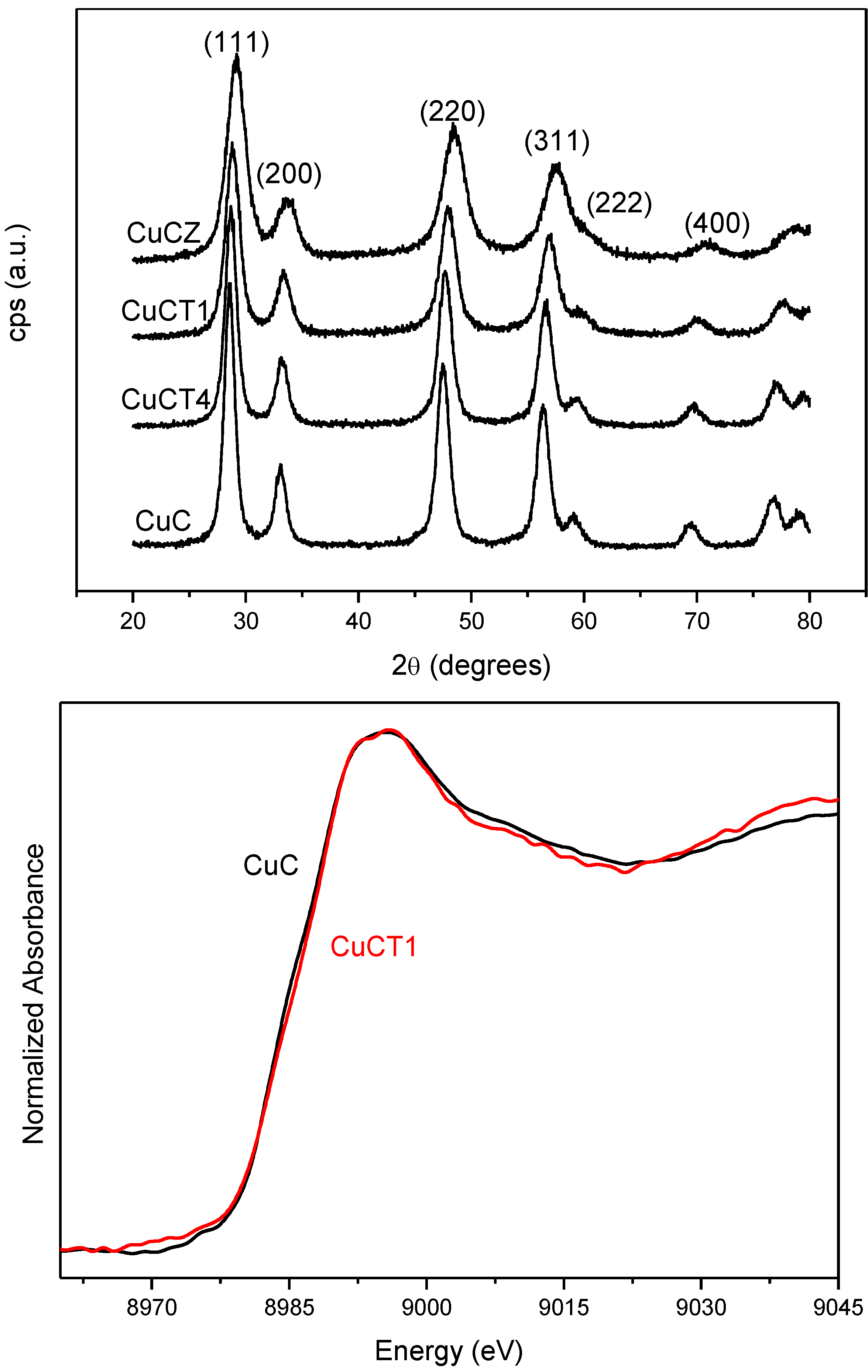
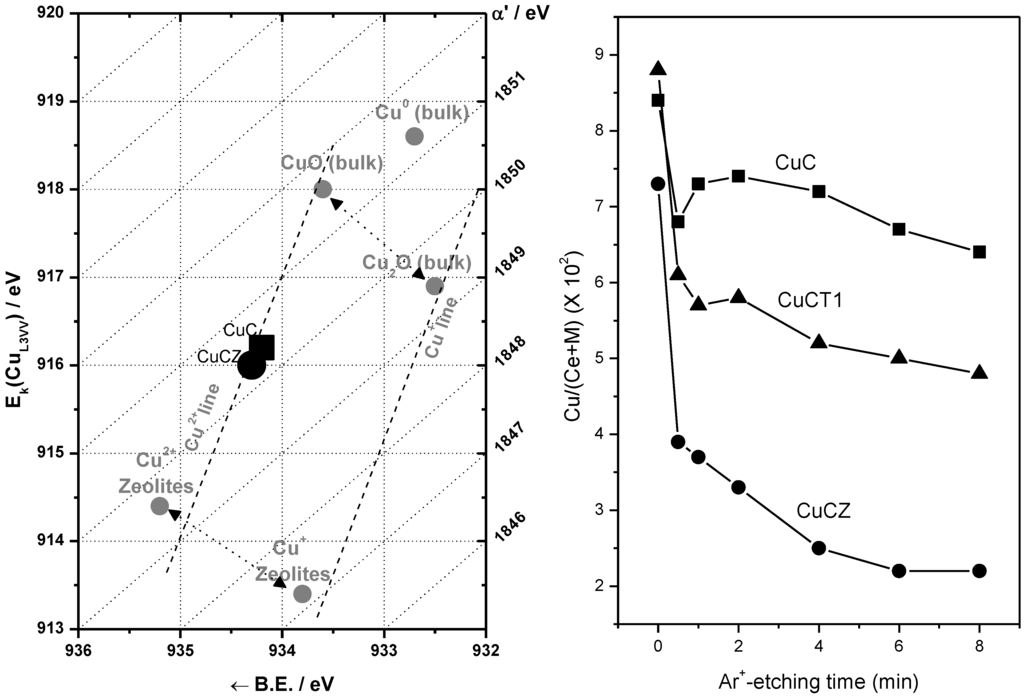
Figure 2.
Left: Wagner diagram showing Cu (2p and AES) XP parameters observed for CuC and CuCZ samples. Right: Profile of the Cu/(Ce + M), M = Zr or Tb, atomic ratio as a function of the Ar+-sputtering time for the indicated samples. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
Figure 2.
Left: Wagner diagram showing Cu (2p and AES) XP parameters observed for CuC and CuCZ samples. Right: Profile of the Cu/(Ce + M), M = Zr or Tb, atomic ratio as a function of the Ar+-sputtering time for the indicated samples. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
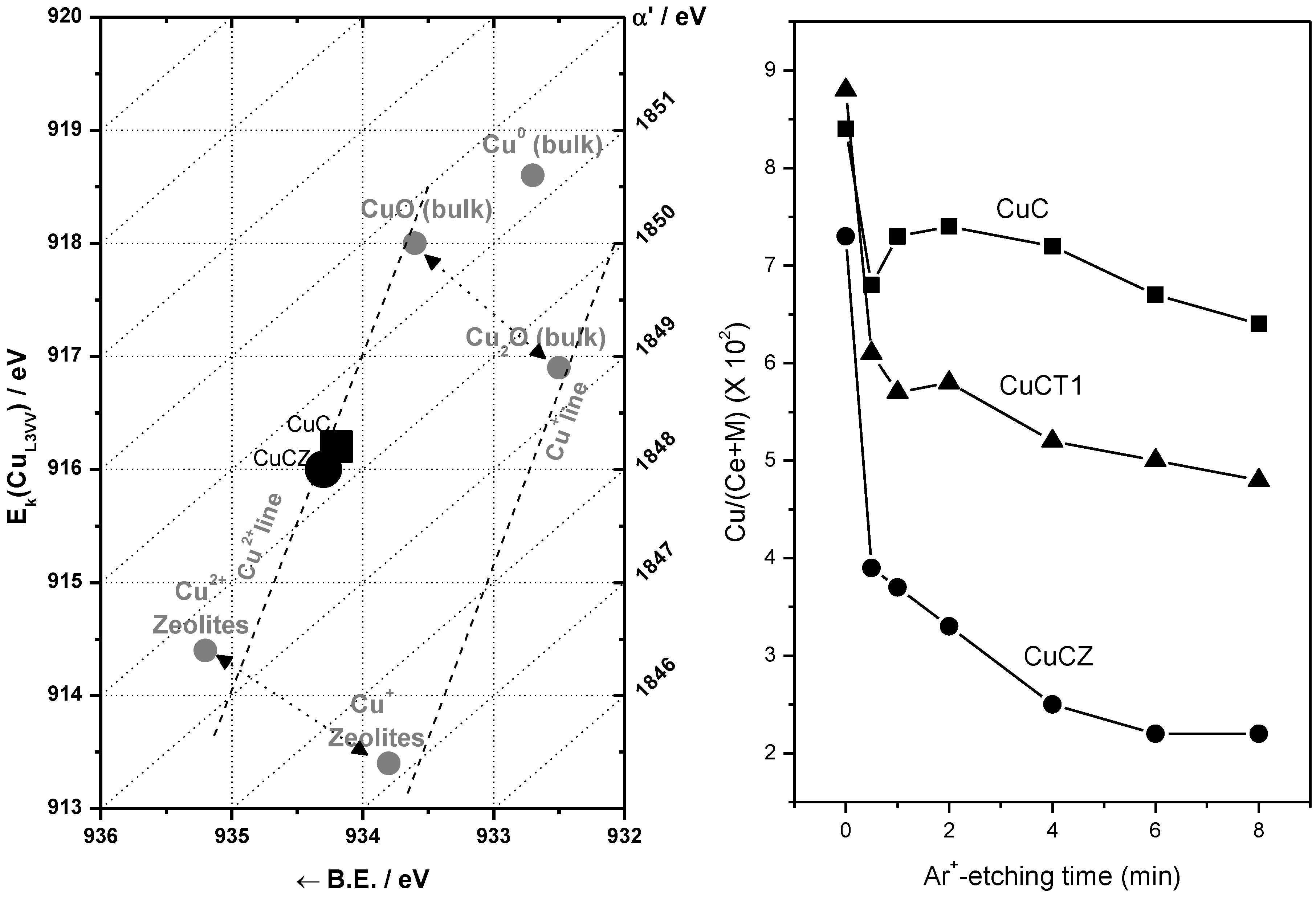
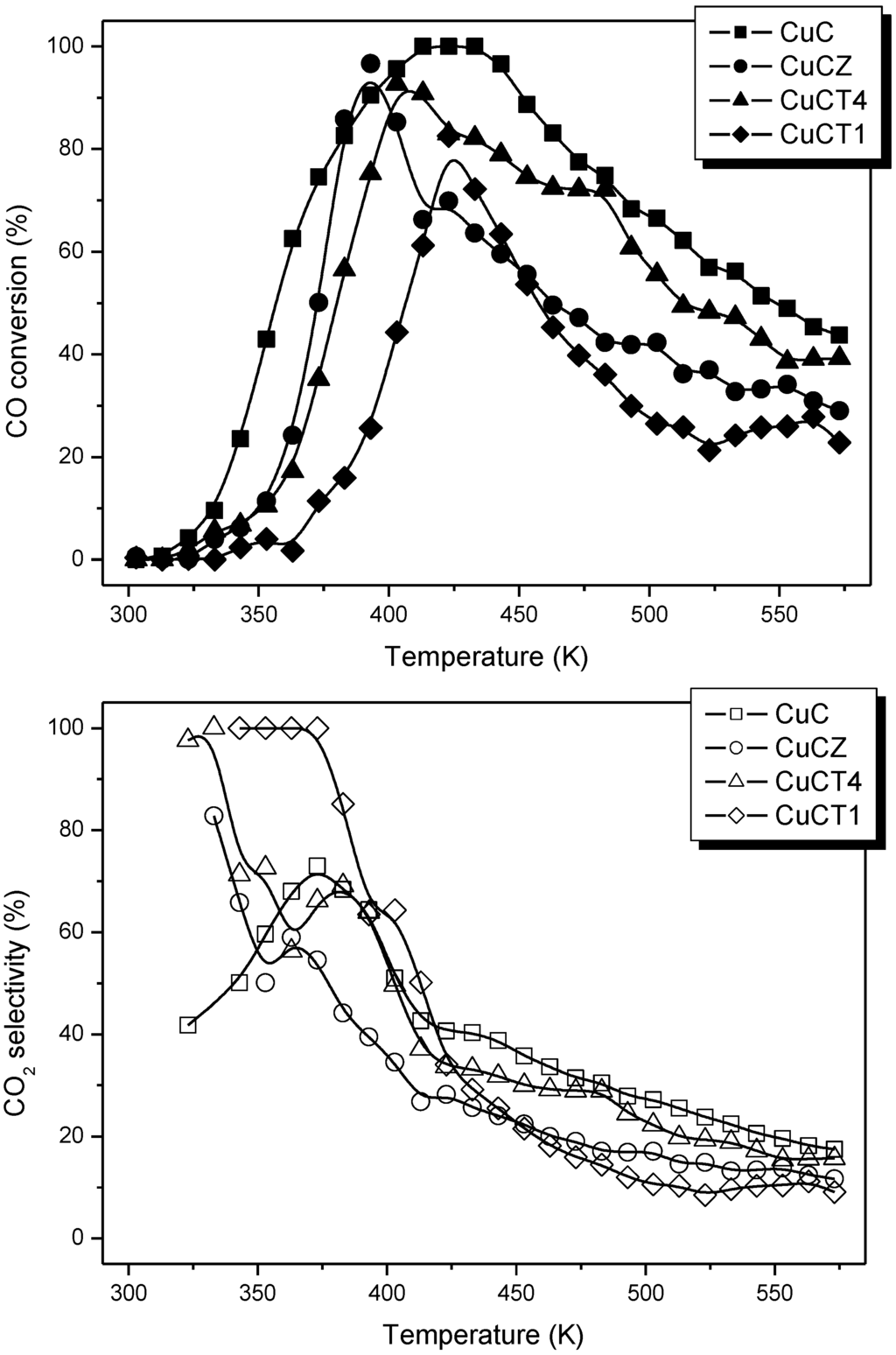
Figure 3.
Profiles of CO conversion (top) and selectivity for the CO-PROX process (bottom) obtained during catalytic tests under 1% CO + 1.25% O2 + 50% H2 (Ar balance) for the indicated catalysts. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
Figure 3.
Profiles of CO conversion (top) and selectivity for the CO-PROX process (bottom) obtained during catalytic tests under 1% CO + 1.25% O2 + 50% H2 (Ar balance) for the indicated catalysts. Note: This figure is adapted with permission from [18]. Copyright © 2005, Elsevier B.V.
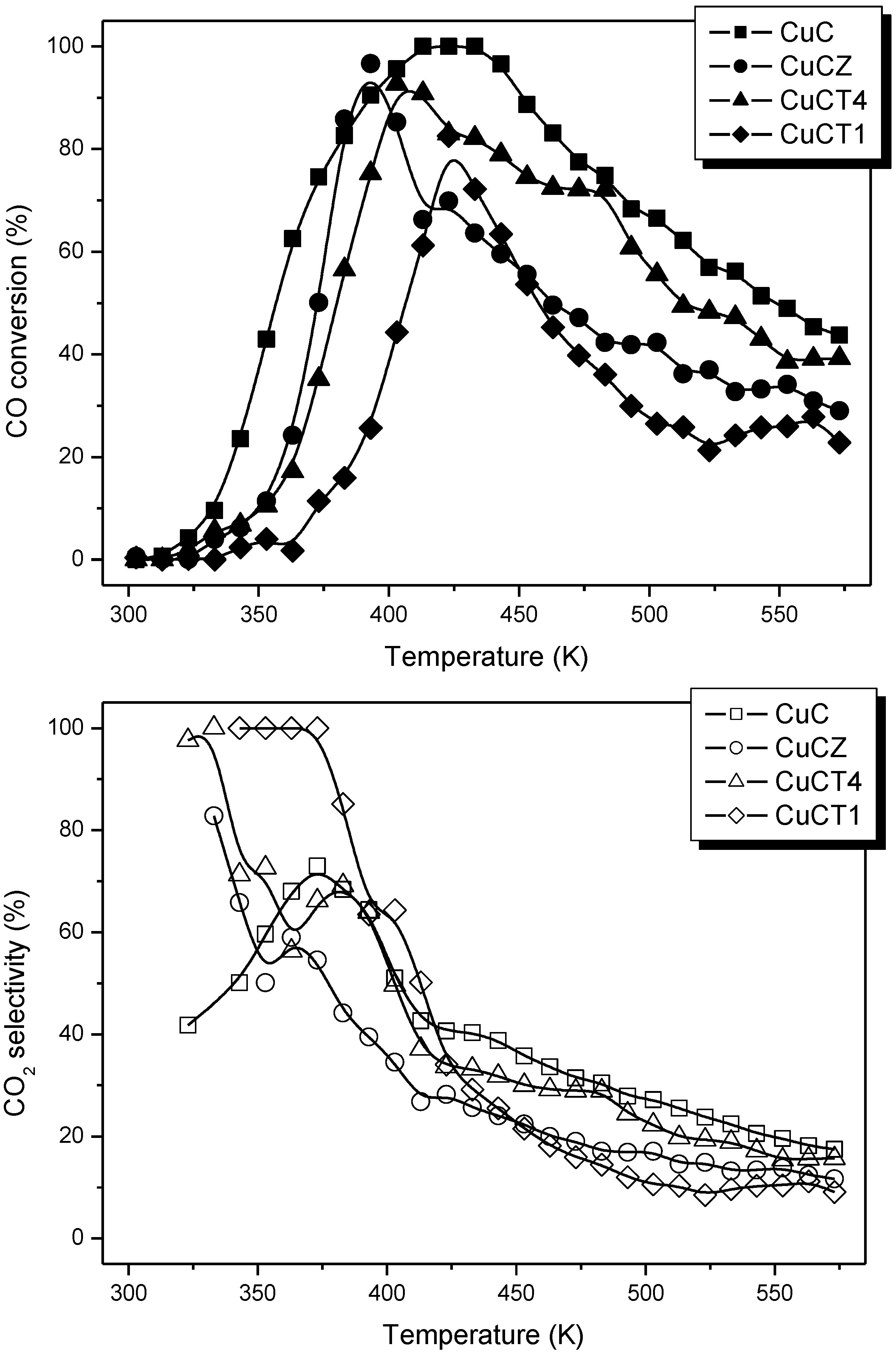
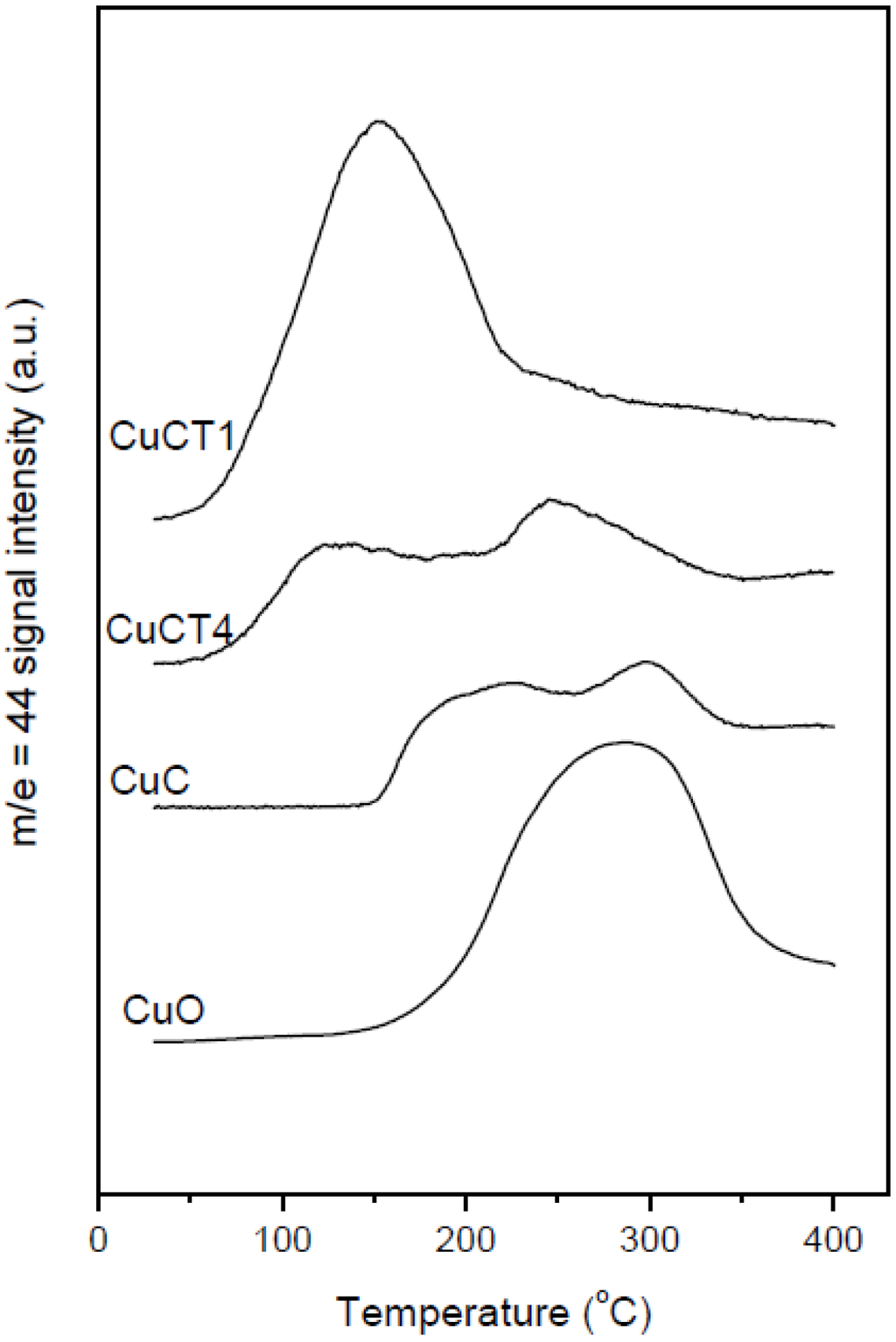
Figure 4.
CO2 production during the CO-TPR runs performed with a tubular reactor over the indicated samples. A CuO reference sample prepared by microemulsion has been included for comparative purpose. Note: This figure is reproduced with permission from [48]. Copyright © 2009, Elsevier Inc.
Figure 4.
CO2 production during the CO-TPR runs performed with a tubular reactor over the indicated samples. A CuO reference sample prepared by microemulsion has been included for comparative purpose. Note: This figure is reproduced with permission from [48]. Copyright © 2009, Elsevier Inc.
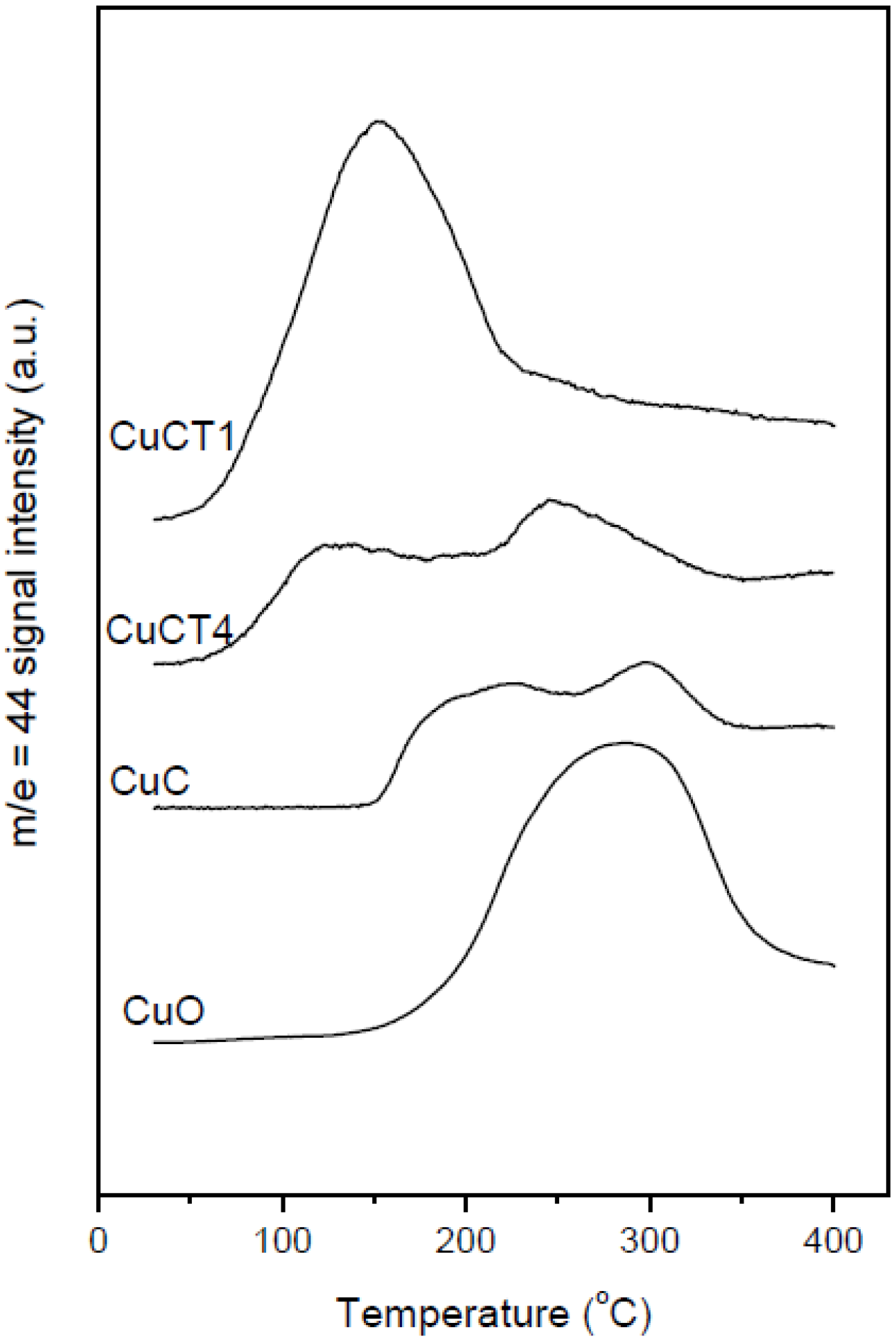
However, such apparently enhanced reduction of dispersed copper oxide entities upon Tb-doping of ceria (as inferred from interpretation of the CO-TPR profiles; Figure 4) does not correlate with the gradual decrease in CO oxidation activity under CO-PROX conditions (Figure 3). It has to be noted that such redox/catalytic correlations appear to be well established on the basis of both CO-TPR and H2-TPR experiments [26,32,33,34]. In conclusion, these results evidence that such a type of correlation is not fully valid when the support nature changes. This being the case, what could then be the reason for such discrepancies, taking also into account that correlation between copper oxide reducibility and CO oxidation activity has been widely demonstrated not only based on mentioned TPR experiments but also on the basis of various spectroscopic techniques [40,41]? The origin of the discrepancy can be related to the redox phenomena taking place at relatively low temperatures (even subambient) during the gas equilibration period carried out at room temperature prior to starting the recording of the CO-TPR data typically done after such gas equilibration when the heating ramp under diluted CO is launched. Indeed, a process of CO consumption, with no CO2 evolution, is detected during such equilibration period, in agreement also with previous experiments [48,51,52]. In this respect, amounts of CO consumed at 20 °C prior to launching the ramp are estimated as 230, 210 and 175 μmol/g for CuC, CuCT4 and CuCT1 samples, in good qualitative agreement with the aforementioned decrease of CO oxidation activity upon increasing the amount of terbium in the support (Figure 3). The respective CO oxidation activity is, in turn, reflected by the respective intensity of a Cu+-carbonyl species (attribution in accordance with previous works, including 12CO-13CO isotopic substitution experiments and analysis of carbonyls thermal stability, where full details can be found [27,29,48,49,51]) formed upon interaction of the calcined samples with CO at room temperature, Figure 5. Since the catalysts were shown to display a fully oxidized state of copper in the initial calcined sample, formation of such carbonyl species upon room temperature interaction evidences the existence of a low temperature copper oxide reduction process, also in agreement with concomitant formation of carbonate-type species upon such room temperature interaction with CO [49,51]. Such a room temperature copper oxide reduction process has been also demonstrated to take place upon interaction with the CO-PROX reactant mixture [18]. Therefore, the extent of support promotion of such a low temperature reduction process, rather than reduction processes occurring at higher temperatures that are the ones typically reflected by conventional TPR tests, appears to correlate with respective CO oxidation activity in this type of catalyst, as will be discussed in the following. It may be noted that additional evidence for this room temperature reduction process was obtained in previous work by XPS and EPR [29]. In turn, further demonstration of the involvement of a partially reduced state of copper on the CO oxidation activity was also recently achieved for this type of catalyst by operando-DRIFTS showing in turn the existence of an induction process related to the aforementioned reduction of copper oxide [53].
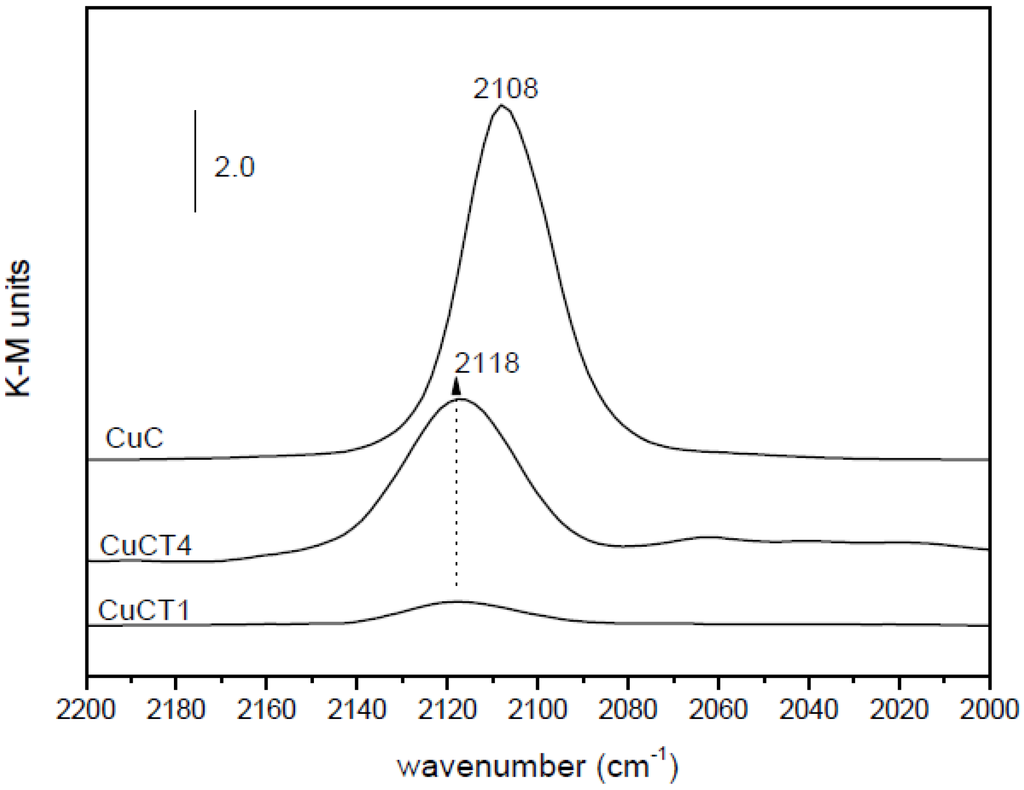
Figure 5.
DRIFTS spectra in the carbonyl stretching region following interaction of the indicated catalysts with a diluted CO flow at room temperature.
Figure 5.
DRIFTS spectra in the carbonyl stretching region following interaction of the indicated catalysts with a diluted CO flow at room temperature.
2.2. Nature of Active Sites/Entities and Further Hints on Catalytic/Redox Correlations
Considering the results exposed in the former section, a set of catalysts with most active configuration involving the combination of copper oxide and ceria were explored with regards to their CO-PROX properties by means of spectroscopic techniques under reaction conditions (the so-called operando techniques). Two different preparation methods (impregnation and microemulsion-coprecipitation) and different copper loadings have been used in order to attempt to cover the widest possible range of physicochemical characteristics in the oxide entities constituting the catalysts. The main characteristics of the initial calcined catalysts at their structural, morphological and electronic levels, on the basis of XRD, Raman, HRTEM-XEDS, SBET measurements, XANES, and XPS were reported in a former contribution, in which full details of catalytic activity results taken with a tubular reactor can also be found [40]. A brief summary of the characterization results is given in the following and some relevant data are collected in Table 1.

Table 1.
Main textural and structural characteristics of the indicated copper-ceria catalysts. The initial number in the samples prepared by impregnation reflects the respective Cu wt.%, while atomic amounts of copper and cerium are indicated in the names employed for the samples prepared by microemulsion-coprecipitation [40].
| Sample | Synthesis method | SBET (m2/g) | Lattice parameter a (Å) | Crystal size a (nm) | Phases detected b |
|---|---|---|---|---|---|
| 0.5CuO/CeO2 | impregnation | 116 | 5.410 | 7.6 | Fluorite CeO2 |
| 1CuO/CeO2 | impregnation | 107 | 5.410 | 7.8 | Fluorite CeO2 |
| 5CuO/CeO2 | impregnation | 101 | 5.413 | 8.1 | Fluorite CeO2, tenorite CuO |
| Ce0.95Cu0.05O2 | microemulsion coprecipitation | 130 | 5.410 | 7.0 | Fluorite Ce1− xCuxO2 |
| Ce0.8Cu0.2O2 | microemulsion coprecipitation | 151 | 5.413 | 6.6 | FluoriteCe1− xCuxO2 |
a For the fluorite phase; b Based on XRD and Raman [40].
Thus, XRD displayed only peaks corresponding to the fluorite structure of ceria, except for 5CuO/CeO2 for which weak and narrow peaks of tenorite CuO were detected, in agreement also with HRTEM, XANES and XPS results. As shown in Table 1, lattice parameters estimated from XRD analysis of the fluorite peaks are close to those expected for pure ceria for all the samples. It must be taken into account, however, that copper introduction into the ceria fluorite lattice is not expected to induce significant changes in this parameter [29,37]. Indeed, on the basis of analysis of lattice microstrain (determined from analysis of XRD results) and Raman results, significant differences between samples prepared by impregnation and microemulsion-coprecipitation were revealed [37,40]. These have been related to the fact that, as expected, all the copper remains essentially at the sample surface in the samples prepared by copper impregnation, while at least a part of the copper appears to be incorporated into the ceria fluorite lattice for those prepared by coprecipitation within microemulsion. In any case, it must be noted that the latter type of sample cannot be fully considered as pure Ce-Cu mixed oxides, since a certain copper surface segregation, increasing with the copper loading, is evident on the basis of mainly XEDS and Ar+-sputtering XPS analyses [40]. Accordingly, catalysts of the Ce1−xCuxO2 series (Table 1) can probably be better described as CuO/Ce1−zCuzO2−y (with undetermined z and y, and in any case with the amount of segregated CuO increasing with x [40]), since single solid solution of the copper is not fully achieved in any case (most particularly for Cu content >10 at.% [40]). In turn, although CuO-type clusters dispersed on the ceria support predominate for the catalysts prepared by impregnation, differences between catalysts of this series as a function of copper loading have been shown to be related, as noted in Table 1, to the presence of large crystalline CuO particles, in accordance also with electron microscopy investigation [40], in 5CuO/CeO2. This must be the result of having exceeded the copper oxide dispersion capacity of the CeO2 support above a certain copper oxide surface loading (in our case, detected for the sample with 5 wt.% Cu) [40]. On the other hand, the presence of copper induces some surface area decrease in the samples prepared by impregnation, probably due to some copper covering of interparticle pores (Table 1). In contrast, the surface area appreciably increases with the copper amount for the samples prepared by microemulsion–coprecipitation, in correlation with the aforementioned introduction of copper into the ceria lattice, which basically induces a decrease in primary particle size (Table 1). On the other hand, concerning the electronic state of copper, XANES (as also shown below) and XPS analyses revealed that the starting sample calcined under dilute O2 at 500 °C presents copper in a fully oxidized Cu2+ chemical state with relatively few differences (except for the crystalline CuO detected in 5CuO/CeO2) between the samples concerning copper electronic characteristics [37,40].
Concerning the catalytic/redox behavior of the series of samples collected in Table 1, DRIFTS experiments under CO-PROX conditions reveal the formation of bands of a similar nature for all catalysts upon contact with the reactant mixture at reaction temperatures between 303 and 523 K. These basically appear in three distinct spectral zones, as illustrated in Figure 6 for the 1CuO/CeO2 catalyst. The first zone displays bands corresponding mainly to hydroxyl species (isolated ones of various types giving sharp bands in the 3720–3600 cm−1 range and associated species giving a broad band extending from ca. 3800 to 3000 cm−1) [54,55]. A second spectral zone below 1700 cm−1 exhibits most intense bands of carbonate or related species [56,57,58]. Stretching vibrations of these species appearing in this region are ascribed to bidentate carbonates (bands at ca. 1583 and 1297; note this attribution was recently revisited and tridentate coordination was suggested for this species on the basis of combined IR-DFT analysis [59]); a combination band at ca. 2880 cm−1 (see the highest wavenumber zone at the left of Figure 6), particularly apparent in lower temperature spectra, is also attributed to these bidentate carbonate species. Polydentate carbonates showing the symmetric and antisymmetric stretching vibrations of the terminal CO bonds at ca. 1478 and 1356 cm−1 are also detected. The band at 1216 cm−1, along with that at 1399 cm−1 and a shoulder at ca. 1600 cm−1, are attributed to hydrogen carbonate species [57,58]; this is confirmed by the presence of a sharp OH stretching vibration at ca. 3618 cm−1 also belonging to these species; note that this type of carbonate must be formed upon interaction of CO with monodentate hydroxyls (giving rise to a band at ca. 3710 cm−1 in the spectrum of the original sample) [55,58], in accordance with observation by DRIFTS, Figure 6. The third spectral zone (at intermediate frequencies) shows the formation of CO2 (g), evolving in accordance with CO oxidation activity, and a carbonyl species (as discussed earlier, a Cu+-carbonyl giving rise to a band at ca. 2120–2110 cm−1 [27,29,49,51]. As discussed previously, the presence of these Cu+-CO species already upon initial contact at 303 K with the reactant mixture is consistent with the easy reduction of copper in the catalysts, considering that copper is fully oxidized in the initial calcined catalysts [40]. As also noted earlier, in the case of Cu/Ce1−xTbxO2−y catalysts, one of the main differences between the samples is related to the intensity of this Cu+-carbonyl band. Furthermore, as displayed in Figure 7 a correlation can be established between the intensity of such carbonyl species and the CO oxidation rate observed for every sample. Since such carbonyl species must be formed as a consequence of a support-promoted reduction process of the copper and taking also into account that the relatively low frequency of this band, with respect to those expected for this type of carbonyls [60], has been related to the interaction between the partially reduced copper centers and the underlying ceria [27,29,40,41,51], the correlation evidenced by Figure 7 reveals that the active species for CO oxidation under CO-PROX conditions must be related to surface-dispersed partially reduced copper oxide species interacting with the support, i.e., at CuO-support interfacial positions. This is consistent with the aforementioned results collected in previous reports [27,29,40,49], which show that these copper species are the most reducible upon contact with CO at low temperatures within a process whereby the ceria support that is in contact with the copper species can also become reduced [27,29,61,62]. Thus, differences between the CO oxidation activities under CO-PROX conditions for this type of catalyst is related to the extent of support promotion of such partial (to Cu+) CuO reduction at interfacial sites which is attained in each case. It is worth noting that the correlation between the intensity of the Cu+-carbonyl and CO oxidation activity over samples of this type was also independently demonstrated later by Baertsch et al. [63].
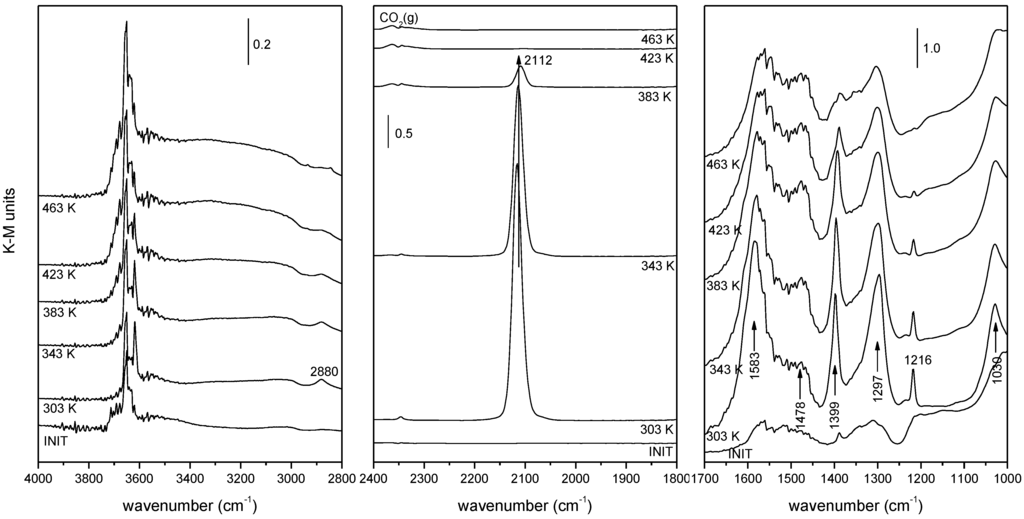
Figure 6.
DRIFTS spectra under CO-H2-O2 reactant mixture flow at the indicated temperatures for 1CuO/CeO2. The spectrum at the bottom corresponds to that recorded at 303 K after pretreatment under diluted O2 at 773 K, prior to contact with the reactant mixture. Note: This figure is adapted with permission from [40]. Copyright © 2007, American Chemical Society.
Figure 6.
DRIFTS spectra under CO-H2-O2 reactant mixture flow at the indicated temperatures for 1CuO/CeO2. The spectrum at the bottom corresponds to that recorded at 303 K after pretreatment under diluted O2 at 773 K, prior to contact with the reactant mixture. Note: This figure is adapted with permission from [40]. Copyright © 2007, American Chemical Society.
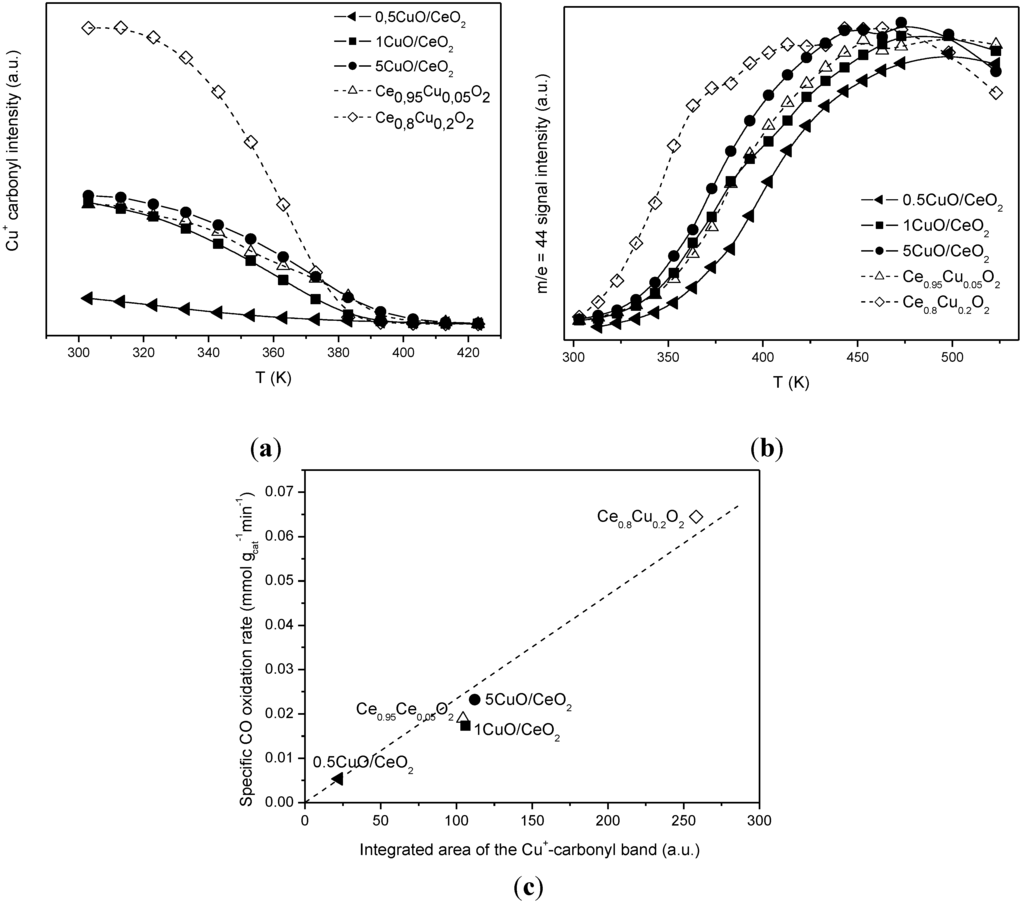
Figure 7.
(a) intensity of the Cu+-carbonyl as a function of the reaction temperature under CO-H2-O2 mixture for the indicated catalysts. (b) Intensity of the QMS m/e = 44 signal, corresponding to CO2, during the tests performed with the DRIFTS cell under CO-H2-O2 mixture. (c) correlation between the intensity of the Cu+-carbonyl just prior to CO oxidation onset and the initial CO oxidation activity. Note: This figure is adapted with permission from [40]. Copyright © 2007, American Chemical Society.
Figure 7.
(a) intensity of the Cu+-carbonyl as a function of the reaction temperature under CO-H2-O2 mixture for the indicated catalysts. (b) Intensity of the QMS m/e = 44 signal, corresponding to CO2, during the tests performed with the DRIFTS cell under CO-H2-O2 mixture. (c) correlation between the intensity of the Cu+-carbonyl just prior to CO oxidation onset and the initial CO oxidation activity. Note: This figure is adapted with permission from [40]. Copyright © 2007, American Chemical Society.
On the other hand, the reaction mechanism under which this type of catalyst operates for CO oxidation has been proposed to be of a redox, generally speaking, of the Mars-van Krevelen [64,65], type [6]. This has been, indeed, demonstrated for the 1CuO/CeO2 catalyst for which kinetic data are consistent with Mars-van Krevelen expressions [66]:
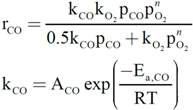
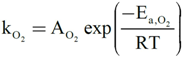 with ACO = 1.61 × 105 mol/g·s·bar, Ea,CO = 4.57 × 104 J/mol, AO2 = 2.87 × 103 mol/g·s·barn, Ea,O2 = 5.51 × 104 J/mol and n = 0.09 [66]. In this respect, it becomes pertinent to enquire as to the nature of the active oxygen species involved in the reaction mechanism. It must be recalled in this sense that different oxygen species (superoxide, peroxide, oxide) have been proposed to be involved as active sites in this type of system, which can certainly depend on the specific degree of hydroxylation present in each case [67,68]. To help gain perspective, Raman spectra were collected under reaction conditions for Cu0.2Ce0.8O2. They do not show the formation of peroxide or superoxide species under CO-PROX conditions; bands are expected at ca. 840 and 1125 cm−1, respectively [64,69] (Figure 8). The shift of the fluorite F2g mode band at ca. 460 cm−1 with increasing reaction temperature is in agreement with thermal expansion of the fluorite lattice (in accordance with spectra simultaneously recorded under the same conditions for CeO2) and reduction at the highest temperatures [64,70], while the decrease of the band at ca. 600 cm−1 (related to oxygen vacancies [37,64]) at the highest reaction temperatures is consistent with copper migration to the surface as a consequence of reduction to metallic copper [37], in agreement with XANES results described below. The partially reduced oxygen species (superoxide, peroxide) were proposed to be involved during CO-PROX processes in hydroxylated ceria-supported gold catalysts [70]. Their absence under CO-PROX conditions in Cu0.2Ce0.8O2 suggests that oxygen species involved in the redox processes can be related to oxide anions, in agreement with a recent investigation in which redox changes under CO/O2 in 1CuO/CeO2 were observed to occur without involvement of superoxides or peroxides even at 303 K [64], at which they can be stable [29].
with ACO = 1.61 × 105 mol/g·s·bar, Ea,CO = 4.57 × 104 J/mol, AO2 = 2.87 × 103 mol/g·s·barn, Ea,O2 = 5.51 × 104 J/mol and n = 0.09 [66]. In this respect, it becomes pertinent to enquire as to the nature of the active oxygen species involved in the reaction mechanism. It must be recalled in this sense that different oxygen species (superoxide, peroxide, oxide) have been proposed to be involved as active sites in this type of system, which can certainly depend on the specific degree of hydroxylation present in each case [67,68]. To help gain perspective, Raman spectra were collected under reaction conditions for Cu0.2Ce0.8O2. They do not show the formation of peroxide or superoxide species under CO-PROX conditions; bands are expected at ca. 840 and 1125 cm−1, respectively [64,69] (Figure 8). The shift of the fluorite F2g mode band at ca. 460 cm−1 with increasing reaction temperature is in agreement with thermal expansion of the fluorite lattice (in accordance with spectra simultaneously recorded under the same conditions for CeO2) and reduction at the highest temperatures [64,70], while the decrease of the band at ca. 600 cm−1 (related to oxygen vacancies [37,64]) at the highest reaction temperatures is consistent with copper migration to the surface as a consequence of reduction to metallic copper [37], in agreement with XANES results described below. The partially reduced oxygen species (superoxide, peroxide) were proposed to be involved during CO-PROX processes in hydroxylated ceria-supported gold catalysts [70]. Their absence under CO-PROX conditions in Cu0.2Ce0.8O2 suggests that oxygen species involved in the redox processes can be related to oxide anions, in agreement with a recent investigation in which redox changes under CO/O2 in 1CuO/CeO2 were observed to occur without involvement of superoxides or peroxides even at 303 K [64], at which they can be stable [29].
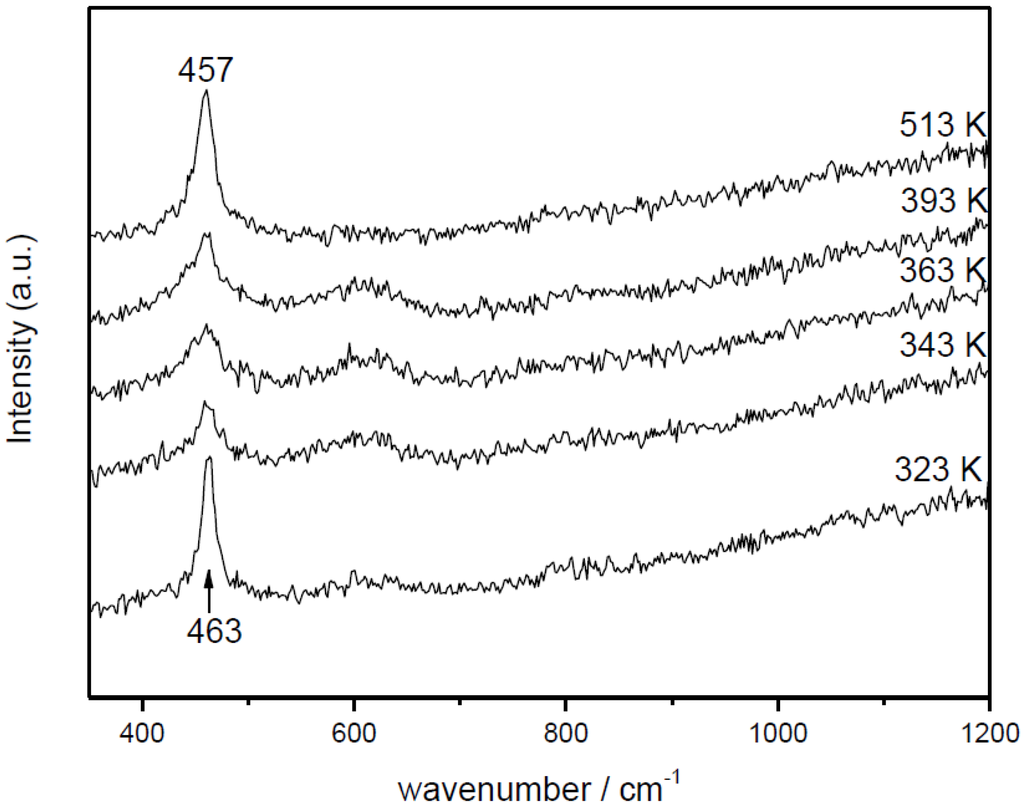
Figure 8.
Raman spectra obtained for Ce0.8Cu0.2O2 under CO-PROX conditions at the indicated temperatures. Note: This figure is adapted with permission from [41]. Copyright © 2007, American Chemical Society.
Figure 8.
Raman spectra obtained for Ce0.8Cu0.2O2 under CO-PROX conditions at the indicated temperatures. Note: This figure is adapted with permission from [41]. Copyright © 2007, American Chemical Society.
The analysis of the Cu K-edge XANES spectra of Cu0.2Ce0.8O2 and 5CuO/CeO2 under CO-PROX conditions indicates the presence of three different chemical species during the course of runs. The first one corresponds to a Cu2+ chemical state displaying geometry similar to that found in CuO, though it also displays some particularities as a consequence of interactions with the support, as discussed in more detail elsewhere [37]. The Cu2+ component predominates at low reaction temperature, as illustrated by Figure 9. Since DRIFTS results described earlier revealed that Cu+ species are formed at low temperature, it must be noted that the amount of Cu+-carbonyls formed must correspond to a relatively low portion of the whole copper present in the sample (note in this sense a maximal limit of ca. 10% as intrinsic error of the XANES technique/analysis). They must be exclusively related to interfacial sites in close interaction with the support which present the highest redox activity, in accordance with existence of a support promoting effect on the CuO reduction, as discussed previously [29]. A component corresponding to zero-valent Cu0, as identified from a comparison with a Cu foil reference, predominates at the end of the runs (Figure 9). Additionally, an intermediate species is detected during spectra analysis and attributed to a Cu+ state on the basis of its 1s → 4p/3d transition energy and spectral shape. Joint analysis of the evolutions of the various copper species and the gases evolving during the CO-PROX tests allow separating different relevant zones (Figure 9). The first one (zone I) at lowest reaction temperature involves exclusively activity for the CO oxidation reaction and has been discussed above on the basis of DRIFTS experiments. The second zone (II) displays a correlation between the onset of H2 oxidation and, at slightly lower temperature, the onset of massive copper reduction to Cu+. This correlation suggests the involvement of the latter species in H2 oxidation, in agreement with the high reactivity shown by partially reduced copper oxide towards hydrogen [71]. Note that as a difference from CO oxidation, H2 oxidation takes place only when the reduction is propagated to zones of the copper oxide nanoparticles far from the interface between the two oxide components. In this respect, H2 oxidation can be most dependent on the specific properties (size, shape [72]) of the dispersed copper oxide nanoparticles, as pointed out previously [18]. In contrast, CO oxidation properties are most likely governed by the characteristics of the CuO-support contacts, i.e., the interfacial properties [41]. A third zone (III) is detected at higher temperatures whereby the H2 oxidation reaction rate changes in coincidence with a sharp increase of the Cu+ contribution. This can be related to the formation of less active Cu2O and/or to sintering of the copper prior to generation of metallic copper [40,73]. This is detected at the highest reaction temperature and its formation fairly coincides with the formation of Ce3+ states (not shown; see [41] for details). It may be pointed out that the copper segregation produced by this reduction process can contribute to the deactivation observed in this type of system when maintained under the reactant mixture at relatively high temperature [7,20]. Validation of the correlations observed for Cu0.2Ce0.8O2 is provided by observation of similar ones for 5CuO/CeO2 [41].
The existence of different active sites (interfacial ones and on top of the reduced copper oxide particles) for each of the two reactions basically involved in the CO-PROX process over this type of catalyst (CO and H2 oxidation) is in agreement with kinetic analysis showing the absence of interference between CO and H2 during the two reactions (i.e., the rates of CO and H2 oxidation do not depend on partial pressure of H2 and CO, respectively) [39]. This has, however, been refuted recently by Kydd et al. [73], who suggest that some degree of competency between H2 and CO for the active sites may arise at relatively high reaction temperatures, in the non-selective region, when CO desorption from active reduced copper sites become favored, even though, to the best of our knowledge, no direct evidence for this is available yet [73].
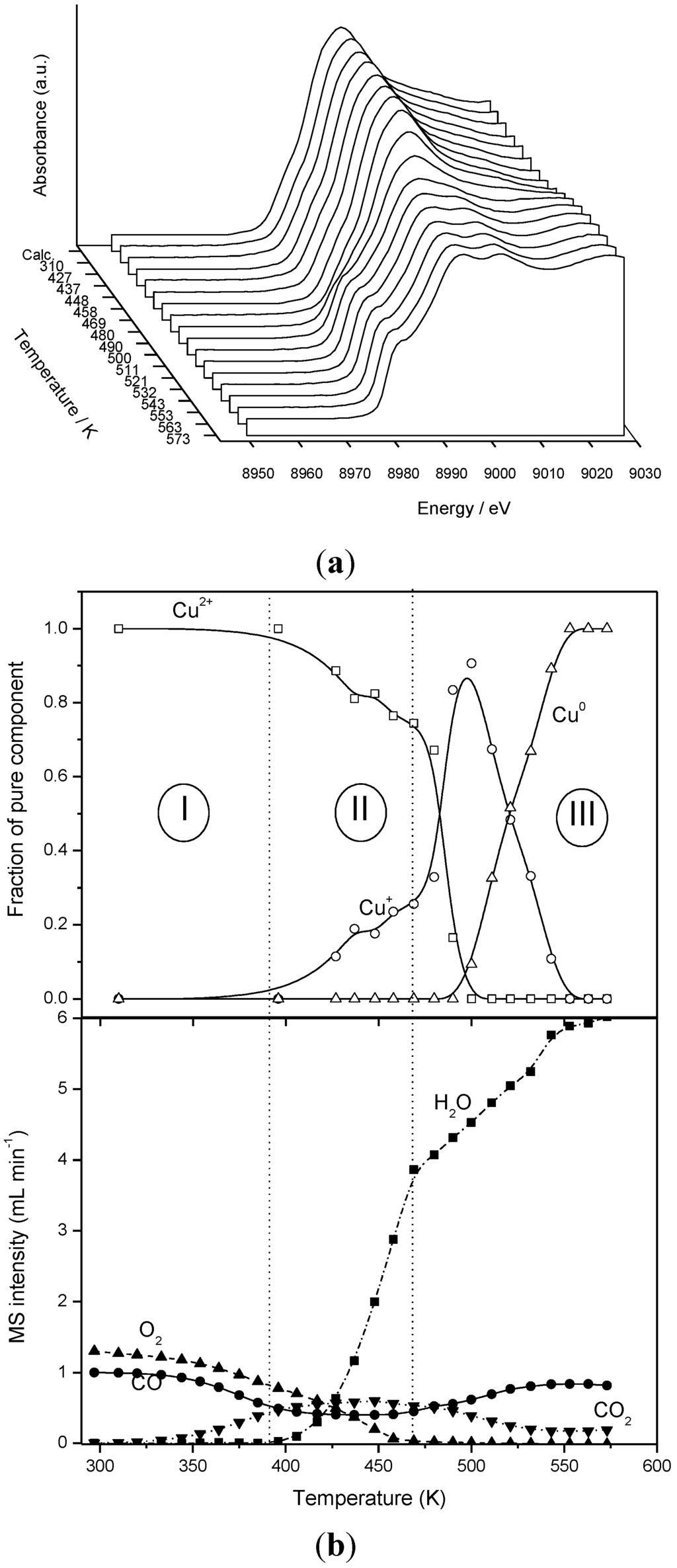
Figure 9.
Cu-K edge XANES spectra under CO-H2-O2 mixture over Ce0.8Cu0.2O2 (a). Evolution of chemical species extracted from spectra analysis and of the various gases detected in the course of the Operando test with the XAFS cell (b), quantified in accordance with calibration and appropriate gas interference corrections. Note: This figure is adapted with permission from [41]. Copyright © 2007, American Chemical Society.
Figure 9.
Cu-K edge XANES spectra under CO-H2-O2 mixture over Ce0.8Cu0.2O2 (a). Evolution of chemical species extracted from spectra analysis and of the various gases detected in the course of the Operando test with the XAFS cell (b), quantified in accordance with calibration and appropriate gas interference corrections. Note: This figure is adapted with permission from [41]. Copyright © 2007, American Chemical Society.
Another recent finding is related to a very recent study in which three samples of copper oxide supported by ceria with different surface morphological characteristics (nanocubes exposing (100) faces at their surface, nanorods exposing (110) and (100) faces and nanospheres with polyhedral shape) have been examined with respect to their CO-PROX performance [42]. An absence of correlation between CO oxidation activity and intensity of Cu+-carbonyls formed under CO-PROX reaction conditions (as explored by operando-DRIFTS) is observed in that case, which contrasts with the results exposed above (obtained with series of different copper loading samples supported on a ceria support with the same morphology or with samples prepared by copper-cerium microemulsion/coprecipitation.
This reveals differences in the catalytic properties of the dispersed copper oxide entities as a function of the surface characteristics of the ceria support with which they interact, i.e., a structural dependence of the CO oxidation reaction involved in the CO-PROX process. On the whole, the main conclusion of such a study can be that optimized CO-PROX catalysts can be obtained by using ceria nanocubes as support. This is based on the fact that such morphology could allow achieving maximum dispersion (i.e., avoiding copper oxide segregation into separated big copper oxide particles not interacting with the underlying support, as detected, for instance, when using ceria nanospheres as support; Table 1) in this case, and partly as a consequence of the relatively low specific surface area of ceria nanocubes [42] and larger particle size within homogeneously distributed dispersed copper oxide particles. Interaction of such dispersed copper oxide particles with the underlying (100) face of ceria can also be most relevant to explain the enhanced CO-PROX catalytic properties. The latter is related to the enhanced CO2 selectivity, which favorably balances the decrease of CO oxidation activity produced as a consequence of the stronger interaction with the support. The higher CO2 selectivity observed over the specimen supported on ceria nanocubes is shown to be related to difficulties in achieving full reduction of the dispersed copper oxide particles under CO-PROX conditions, on the basis of mainly operando-DRIFTS experiments, and also in agreement with arguments exposed above. On the other hand, other morphological details which could be relevant to this type of catalyst/process is related to control of the porous microstructure of the system, as shown in a recent study [74].
3. Summary and Main Conclusions
Catalysts constituted by combinations between copper oxide and ceria (or structurally related doped ceria) are amongst the most active catalysts within operative temperature ranges for the process of preferential oxidation of CO in H2-rich stream (CO-PROX). This is an economically and practically interesting procedure for the purification of hydrogen extracted from hydrocarbon sources, particularly focusing on its use as fuel of proton exchange membrane fuel cells in mobile or small-scale applications. Although general agreement exists concerning the necessity of establishing contacts between both components’ oxides, simply achievable upon dispersing at highest possible level copper oxide on high surface ceria, it is required to go a step further in the sense of understanding basic aspects of the CO-PROX process over this type of system in order to get optimized configurations, particularly considering that the main competitors within catalysts active for the process are very robust platinum-based catalysts. In this context, the present contribution basically revises work done in the authors’ laboratories focused on gaining basic insights in two interrelated aspects: (1) determining the nature of the sites active for the various reactions involved in the process; and (2) establishing catalytic/redox correlations. Concerning the first point, it must be noted that basically two reactions take place below about 200 °C (before formation of WGS-active metallic copper under CO-PROX reaction conditions) in the presence of a CO-PROX mixture over this type of catalyst: CO and H2 oxidation. In turn, concerning the second point, when starting from a fully oxidized catalyst, changes can be produced in the oxide components during the course of the interaction with the reactant mixture, i.e., considering the reducing nature of the mixture, reduction of the oxides components becomes thermodynamically favored. On this basis, operando-spectroscopic analysis of the catalysts has been carried out and allows concluding on the existence of a low temperature support promoting effect on the partial reduction (to Cu+) of interfacial sites of the dispersed CuO entities. Such interfacial partially reduced copper sites (Cu+ species) apparently constitute the most active sites for the CO oxidation reaction on the basis of the results obtained. This is mainly based on the finding of an interesting correlation between the intensity of Cu+-carbonyls formed under reaction conditions and the CO oxidation rate respectively observed. This has been recently shown to depend also on the specific surface morphology of the ceria support which evidently determines the CO oxidation activity of the active reduced copper entities. On the other hand, the extension of the reduction over the copper oxide entities (i.e., to sites out of the interface and accordingly most likely not so strongly affected by contact with the ceria support) upon interaction with the reactant mixture can provide the most active sites for the H2 oxidation reaction. The possible separation between the two types of the most active sites that can be involved during the two (H2 and CO) oxidation reactions taking place under CO-PROX conditions over this type of system (previous macroscopic kinetics experiments have also pointed out towards the presence of independent active sites for the two reactions [39]) can open the possibility to control their catalytic properties and design optimized systems for this important application. Indeed, this hypothesis has allowed the design of newly optimized catalysts of this type by using inverse CeO2/CuO configurations, as recently reported [75]. Nevertheless, it must be noted that other authors have argued for different active sites for each one of the two oxidation reactions; i.e., it has been proposed, although no direct demonstration is available to the best of our knowledge, that H2 and CO can compete, at least above a certain reaction temperature, for the same active sites. This hypothesis proposes that H2 oxidation starts as soon as the temperature is sufficiently high and CO desorption from copper-active sites becomes favored. We are presently working in our laboratory on understanding better how this functions.
On the other hand, the results point out some discrepancies as to the use of classical TPR runs for getting catalytic/redox correlations since redox processes of relevancy to explain CO oxidation can take place at relatively low temperature, typically during gases equilibration prior to data collection. In addition, as pointed out in previous works, the reaction mechanism under which the catalysts operate for CO oxidation can be of a redox Mars-van Krevelen type with oxide anions apparently constituting the active centers, according to Raman investigation. Other aspects of relevancy which have not been treated in detail in this contribution, are related to understanding the catalysts’ deactivation mechanisms [76,77], particularly in the presence of CO2 and H2O in the reactant mixture, which, as it occurs for nanogold systems, could limit the practical application of this type of catalyst. It must, however, be mentioned in this sense that monolithic forms of CO-PROX catalysts combining copper and ceria have displayed high performance for the process, even in the presence of important amounts of CO2 and H2O in the reactant stream [78]. On the other hand, copper segregation and formation of metallic copper is shown to occur at T > ca. 473 K under CO-H2-O2 mixture and can also contribute, in addition to formation of interfacial carbonate species and molecular water blocking effects [76,77], to deactivation observed under a simple CO-PROX mixture.
Acknowledgments
This work was funded by Ministerio de Ciencia e Innovación and Ministerio de Economía y Competitividad (Plan Nacional Projects CTQ2009-14527 and CTQ2012-32928) and Comunidad de Madrid (Project DIVERCEL, Ref.: S2009/ENE-1475). Support from EU COST CM1104 action is also acknowledged. Antonio López Cámara acknowledges a PhD grant from the CSIC JAE program.
Conflict of Interest
The authors declare no conflict of interest.
References
- Rostrup-Nielsen, J.R.; Sehested, J.; Norskov, J.K. Hydrogen and synthesis gas by steam- and CO2 reforming. Adv. Catal. 2002, 47, 65–139. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Papadopoulou, C.; Batista, J.; Hocevar, S.; Matralis, H.K. A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO-CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal. Today 2002, 75, 157–167. [Google Scholar]
- Oh, S.H.; Sinkevitch, R.M. Carbon monoxide removal from hydrogen-rich fuel cell feedstreams by selective catalytic oxidation. J. Catal. 1993, 142, 254–262. [Google Scholar] [CrossRef]
- Wang, J.B.; Lin, S.; Huang, T. Selective CO oxidation in rich hydrogen over CuO/samaria-doped ceria. Appl. Catal. A 2002, 232, 107–120. [Google Scholar] [CrossRef]
- Sedmak, G.; Hocevar, S.; Levec, J. Kinetics of selective CO oxidation in excess of H2 over the nanostructured Cu0.1Ce0.9O2−y catalyst. J. Catal. 2003, 213, 135–150. [Google Scholar] [CrossRef]
- Kim, D.H.; Cha, J.E. A CuO-CeO2 Mixed-oxide catalyst for CO clean-up by selective oxidation in hydrogen-rich mixtures. Catal. Lett. 2003, 86, 107–112. [Google Scholar]
- Korotkikh, O.; Farrauto, R. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Ghenciu, A.F. Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 389–399. [Google Scholar] [CrossRef]
- Haruta, M.; Date, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Schubert, M.M.; Venugopal, A.; Kahlich, M.J.; Plzak, V.; Behm, R.J. Influence of H2O and CO2 on the selective CO oxidation in H2-rich gases over Au/α-Fe2O3. J. Catal. 2004, 222, 32–40. [Google Scholar]
- Luengnaruemitchai, A.; Osuwan, S.; Gulari, E. Selective catalytic oxidation of CO in the presence of H2 over gold catalyst. Int. J. Hydrog. Energy 2004, 29, 429–435. [Google Scholar] [CrossRef]
- Deng, W.; de Jesus, J.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Low-content gold-ceria catalysts for the water-gas shift and preferential CO oxidation reactions. Appl. Catal. A 2005, 291, 126–135. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepcion, P.; Corma, A.; López, Nieto, J.M.; Puentes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Papavasiliou, J.; Tabakova, T.; Idakiev, V.; Ioannides, T. A comparative study of ceria-supported gold and copper oxide catalysts for preferential CO oxidation reaction. Chem. Eng. J. 2006, 124, 41–45. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Preferential oxidation of CO in H2 over CuO-CeO2 catalysts. Catal. Today 2004, 93–95, 241–246. [Google Scholar]
- Marbán, G.; Fuertes, A.B. Highly active and selective CuOx/CeO2 catalyst prepared by a single-step citrate method for preferential oxidation of carbon monoxide. Appl. Catal. B 2005, 57, 43–53. [Google Scholar]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Munuera, G. Preferential oxidation of CO in a H2-rich stream over CuO/CeO2 and CuO/(Ce,M)Ox (M = Zr, Tb) catalysts. J. Power Sour. 2005, 151, 32–42. [Google Scholar] [CrossRef]
- Mariño, F.; Descorme, C.; Duprez, D. Supported base metal catalysts for the preferential oxidation of carbon monoxide in the presence of excess hydrogen (PROX). Appl. Catal. B 2005, 58, 175–183. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Munuera, G.; Gamarra, D. Preferential oxidation of CO in rich H2 over CuO/CeO2: Details of selectivity and deactivation under the reactant stream. Appl. Catal. B 2006, 65, 207–216. [Google Scholar] [CrossRef]
- Park, J.-W.; Jeong, J.-H.; Yoon, W.-L.; Jung, H.; Lee, H.-T.; Lee, D.-K.; Park, Y.-K.; Rhee, Y.W. Activity and characterization of the Co-promoted CuO-CeO2/γ-Al2O3 catalyst for the selective oxidation of CO in excess hydrogen. Appl. Catal. A 2004, 274, 25–32. [Google Scholar] [CrossRef]
- Jobbagy, M.; Mariño, F.; Schönbrod, B.; Baronetti, G.; Laborde, M. Synthesis of copper-promoted CeO2 catalysts. Chem. Mater. 2006, 18, 1945–1950. [Google Scholar] [CrossRef]
- Moretti, E.; Lenarda, M.; Storaro, L.; Talon, A.; Montanari, T.; Busca, G.; Rodríguez-Castellón, E.; Jiménez-López, A.; Turco, M.; Bagnasco, G.; et al. One-step synthesis of a structurally organized mesoporous CuO-CeO2-Al2O3 system for the preferential CO oxidation. Appl. Catal. A 2008, 335, 46–55. [Google Scholar] [CrossRef]
- Gurbani, A.; Ayastuy, J.L.; González-Marcos, M.P.; Herrero, J.E.; Guil, J.M.; Gutiérrez-Ortiz, M.A. Comparative study of CuO-CeO2 catalysts prepared by wet impregnation and deposition-precipitation. Int. J. Hydrog. Energy 2009, 34, 547–553. [Google Scholar]
- Martínez-Arias, A.; Soria, J.; Cataluña, R.; Conesa, J.C.; Cortés Corberán, V. Influence of ceria dispersión on the catalytic performance of Cu/CeO2/Al2O3 catalysts for the CO oxidation reaction. Stud. Surf. Sci. Catal. 1998, 116, 591–600. [Google Scholar] [CrossRef]
- Liu, W.; Sarofim, A.F.; Flytzani-Stephanopoulos, M. Complete oxidation of carbon monoxide and methane over metal-promoted fluorite oxide catalysts. Chem. Eng. Sci. 1995, 49, 4871–4888. [Google Scholar]
- Martínez-Arias, A.; Fernández-García, M.; Gálvez, O.; Coronado, J.M.; Anderson, J.A.; Conesa, J.C.; Soria, J.; Munuera, G. Comparative study on redox properties and catalytic behavior for CO Oxidation of CuO/CeO2 and CuO/ZrCeO4 catalysts. J. Catal. 2000, 195, 207–216. [Google Scholar]
- Skårman, B.; Grandjean, D.; Benfield, R.E.; Hinz, A.; Andersson, A.; Wallenberg, L.R. Carbon monoxide oxidation on nanostructured CuOx/CeO2 composite particles characterized by HREM, XPS, XAS, and high-energy diffraction. J. Catal. 2002, 211, 119–133. [Google Scholar]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Munuera, G. Interfacial redox processes under CO/O2 in a nanoceria-supported copper oxide catalyst. J. Phys. Chem. B 2004, 108, 17983–17991. [Google Scholar]
- Ilichev, A.N.; Firsova, A.A.; Korchak, V.N. Mechanism of CO oxidation in excess H2 over CuO/CeO2 catalysts: ESR and TPD studies. Kinet. Catal. 2006, 47, 585–592. [Google Scholar] [CrossRef]
- Gamarra, D.; Hornés, A.; Koppány, Z.; Schay, Z.; Munuera, G.; Soria, J.; Martínez-Arias, A. Catalytic processes during preferential oxidation of CO in H2-rich streams over catalysts based on copper-ceria. J. Power Sour. 2007, 169, 110–116. [Google Scholar] [CrossRef]
- Luo, M.-F.; Song, Y.-P.; Lu, J.Q.; Wang, X.-Y.; Pu, Z.-Y. Identification of CuO species in high surface area CuO-CeO2 catalysts and their catalytic activities for CO oxidation. J. Phys. Chem. C 2007, 111, 12686–12692. [Google Scholar]
- Luo, M.-F.; Ma, J.-M.; Lu, J.-Q.; Song, Y.-P.; Wang, Y.-J. High-surface area CuO-CeO2 catalysts prepared by a surfactant-templated method for low-temperature CO oxidation. J. Catal. 2007, 246, 52–59. [Google Scholar]
- Pintar, A.; Batista, J.; Hocevar, S. TPR, TPO, and TPD examinations of Cu0.15Ce0.85O2−y mixed oxides prepared by co-precipitation, by the sol-gel peroxide route, and by citric acid-assisted synthesis. J. Coll. Interf. Sci. 2005, 285, 218–231. [Google Scholar] [CrossRef]
- Manzoli, M.; di Monte, R.; Boccuzzi, F.; Coluccia, S.; Kaspar, J. CO oxidation over CuOx-CeO2-ZrO2 catalysts: Transient behaviour and role of copper clusters in contact with ceria. Appl. Catal. B 2005, 61, 192–205. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H. Influence of the preparation method on the performance of CuO-CeO2 catalysts for the selective oxidation of CO. Appl. Catal. B 2005, 56, 87–93. [Google Scholar] [CrossRef]
- Wang, X.Q.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martínez-Arias, A.; Fernández-García, M. Unusual physical and chemical properties of Cu in Ce1−xCuxO2 oxides. J. Phys. Chem. B 2005, 109, 19595–19603. [Google Scholar]
- Wang, J.B.; Tsai, D.-H.; Huang, T.-J. Synergistic Catalysis of carbon monoxide oxidation over copper oxide supported on samaria-doped ceria. J. Catal. 2002, 208, 370–380. [Google Scholar] [CrossRef]
- Lee, H.C.; Kim, D.H. Kinetics of CO and H2 oxidation over CuO-CeO2 catalyst in H2 mixtures with CO2 and H2O. Catal. Today 2008, 132, 109–116. [Google Scholar] [CrossRef]
- Gamarra, D.; Munuera, G.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Midgley, P.A.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A.; Martínez-Arias, A. Structure-activity relationship in nanostructured copper-ceria-based preferential CO oxidation catalysts. J. Phys. Chem. C 2007, 111, 11026–11038. [Google Scholar] [CrossRef]
- Gamarra, D.; Belver, C.; Fernández-García, M.; Martínez-Arias, A. Selective CO oxidation in excess H2 over copper-ceria catalysts: Identification of active entities/species. J. Am. Chem. Soc. 2007, 129, 12064–12065. [Google Scholar] [CrossRef]
- Gamarra, D.; Lopez Camara, A.; Monte, M.; Rasmussen, S.B.; Chinchilla, L.E.; Hungria, A.B.; Munuera, G.; Gyorffy, N.; Schay, Z.; Corberan, V.C.; et al. Preferential oxidation of CO in excess H2 over CuO/CeO2 catalysts: Characterization and performance as a function of the exposed face present in the CeO2 support. Appl. Catal. B 2013, 130–131, 224–238. [Google Scholar]
- Bion, N.; Epron, F.; Moreno, M.; Mariño, F.; Duprez, D. Preferential oxidation of carbon monoxide in the presence of hydrogen (PROX) over noble metals and transition metal oxides: Advantages and drawbacks. Top. Catal. 2008, 51, 76–88. [Google Scholar] [CrossRef]
- López, I.; Valdés-Solís, T.; Marbán, G. An attempt to rank copper-based catalysts used in the CO-PROX reaction. Int. J. Hydrog. Energy 2008, 33, 197–205. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalysis by Ceria and Related Materials; Imperial College Press: London, UK, 2002. [Google Scholar]
- Hungría, A.B.; Martínez-Arias, A.; Fernández-García, M.; Iglesias-Juez, A.; Guerrero-Ruiz, A.; Calvino, J.J.; Conesa, J.C.; Soria, J. Structural, morphological, and oxygen handling properties of nanosized cerium-terbium mixed oxides prepared by microemulsion. Chem. Mater. 2003, 15, 4309–4316. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Hungría, A.B.; Conesa, J.C.; Munuera, G. Spectroscopic characterization of heterogeneity and redox effects in zirconium-cerium (1:1) mixed oxides prepared by microemulsion methods. J. Phys. Chem. B 2003, 107, 2667–2677. [Google Scholar]
- Hornés, A.; Bera, P.; López Cámara, A.; Gamarra, D.; Munuera, G.; Martínez-Arias, A. CO-TPR-DRIFTS-MS in situ study of CuO/Ce1−xTb2O2−y (x = 0, 0.2 axnd 0.5) catalysts: Support effects on redox properties and CO oxidation catalysis. J. Catal. 2009, 268, 367–375. [Google Scholar] [CrossRef]
- Bera, P.; López Cámara, A.; Hornés, A.; Martínez-Arias, A. Comparative in situ DRIFTS-MS study of 12CO- and 13CO-TPR on CuO/CeO2 catalyst. J. Phys. Chem. C 2009, 113, 10689–10695. [Google Scholar]
- Wang, J.B.; Shih, W.-H.; Huang, T.-J. Study of Sm2O3-doped CeO2/Al2O3-supported copper catalyst for CO oxidation. Appl. Catal. A 2000, 203, 191–199. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Soria, J.; Conesa, J.C. Spectroscopic study of a Cu/CeO2 catalyst subjected to redox treatments in carbon monoxide and oxygen. J. Catal. 1999, 182, 367–377. [Google Scholar]
- Caputo, T.; Lisi, L.; Pirone, R.; Russo, G. On the role of redox properties of CuO/CeO2 catalysts in the preferential oxidation of CO in H2-rich gases. Appl. Catal. A 2008, 348, 42–53. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Iglesias-Juez, A.; Soria, J.; Conesa, J.C.; Anderson, J.A.; Munuera, G. Operando DRIFTS study of the redox and catalytic properties of CuO/Ce1−xTbxO2−d (x = 0–0.5) catalysts: Evidence of an induction step during CO oxidation. Phys. Chem. Chem. Phys. 2012, 14, 2144–2151. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. An FTIR study of surface ceria hydroxy groups during a redox process with H2. J. Chem. Soc. Faraday Trans. 1996, 92, 4669–4673. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by fourier-transform infrared spectroscopy. Part 1.—Formation of carbonate species on dehydroxylated CeO2, at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 929–943. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Adsorption of carbon monoxide and carbon dioxide on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 2.—Formation of formate species on partially reduced CeO2 at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 1451–1461. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Kröhnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.C.; Knop-Gericke, A.; Paál, Z.; et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions. J. Catal. 2006, 237, 1–16. [Google Scholar] [CrossRef]
- Vayssilov, N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. reassignment of the vibrational spectra of carbonates, formates, and related surface species on ceria: A combined density functional and infrared spectroscopy investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Scarano, D.; Bordiga, S.; Lamberti, C.; Spoto, G.; Ricchiardi, G.; Zecchina, A.; Otero Areán, C. FTIR study of the interaction of CO with pure and silica-supported copper(I) oxide. Surf. Sci. 1998, 411, 272–285. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Hungría, A.B.; Iglesias-Juez, A.; Gálvez, O.; Anderson, J.A.; Conesa, J.C.; Soria, J.; Munuera, G. Redox interplay at copper oxide-(Ce,Zr)Ox interfaces: Influence of the presence of NO on the catalytic activity for CO oxidation over CuO/CeZrO4. J. Catal. 2003, 214, 261–272. [Google Scholar]
- Martínez-Arias, A.; Gamarra, D.; Fernández-García, M.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A. Comparative study on redox properties of nanosized CeO2 and CuO/CeO2 under CO/O2. J. Catal. 2006, 240, 1–7. [Google Scholar] [CrossRef]
- Polster, C.S.; Nair, H.; Baertsch, C.D. Study of active sites and mechanism responsible for highly selective CO oxidation in H2 rich atmospheres on a mixed Cu and Ce oxide catalysts. J. Catal. 2009, 266, 308–319. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Vannice, M.A. An analysis of the Mars-van Krevelen rate expression. Catal. Today 2007, 123, 18–22. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Gamarra, D.; Fernández-García, M.; Hornés, A.; Belver, C. Spectroscopic study on the nature of active entities in copper-ceria CO-PROX catalysts. Top. Catal. 2009, 52, 1425–1432. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Cataluña, R.; Conesa, J.C.; Soria, J. Effect of copper-ceria interactions on copper reduction in a Cu/CeO2/Al2O3 catalyst subjected to thermal treatments in CO. J. Phys. Chem. B 1998, 102, 809–817. [Google Scholar] [CrossRef]
- Guzman, J.; Carrettin, S.; Corma, A. spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef]
- Pushkarev, V.V.; Kovalchuk, V.I.; d’Itri, J.L. probing defect sites on the ceo2 surface with dioxygen. J. Phys. Chem. B 2004, 108, 5341–5348. [Google Scholar] [CrossRef]
- Spanier, J.E.; Robinson, R.D.; Zhang, F.; Chan, S.-W.; Herman, I.P. Size-dependent properties of CeO2−y nanoparticles as studied by Raman scattering. Phys. Rev. B 2001, 64, 245407. [Google Scholar] [CrossRef]
- Kim, J.Y.; Rodriguez, J.A.; Hanson, J.C.; Frenkel, A.I.; Lee, P.L. Reduction of CuO and Cu2O with H2: H embedding and kinetic effects in the formation of suboxides. J. Am. Chem. Soc. 2003, 125, 10684–10692. [Google Scholar]
- Fernández-García, M.; Martínez-Arias, A.; Hanson, J.C.; Rodriguez, J.A. Nanostructured oxides in chemistry: Characterization and properties. Chem. Rev. 2004, 104, 4063–4104. [Google Scholar] [CrossRef]
- Kydd, R.; Ferri, D.; Hug, P.; Scott, J.; Teoh, W.Y.; Amal, R. Temperature-induced evolution of reaction sites and mechanisms during preferential oxidation of CO. J. Catal. 2011, 277, 64–71. [Google Scholar] [CrossRef]
- Yen, H.; Seo, Y.; Kaliaguine, S.; Kleitz, F. Tailored mesostructured copper/ceria catalysts with enhanced performance for preferential oxidation of CO at low temperature. Angew. Chem. Int. Ed. 2012, 51, 12032–12035. [Google Scholar] [CrossRef]
- Hornés, A.; Hungría, A.B.; Bera, P.; López Cámara, A.; Fernández-García, M.; Martínez-Arias, A.; Barrio, L.; Estrella, M.; Zhou, G.; Fonseca, J.J.; et al. Inverse CeO2/CuO catalyst as an alternative to classical direct configurations for preferential oxidation of CO in hydrogen-rich stream. J. Am. Chem. Soc. 2010, 132, 34–35. [Google Scholar] [CrossRef]
- Gamarra, D.; Martínez-Arias, A. Preferential oxidation of CO in rich H2 over CuO/CeO2: Operando-DRIFTS analysis of deactivating effect of CO2 and H2O. J. Catal. 2009, 263, 189–195. [Google Scholar] [CrossRef]
- Gamarra, D.; Fernández-García, M.; Belver, C.; Martínez-Arias, A. Operando DRIFTS and XANES study of deactivating effect of CO2 on a Ce0.8Cu0.2O2 CO-PROX catalyst. J. Phys. Chem. C 2010, 114, 18576–18582. [Google Scholar]
- Zeng, S.H.; Liu, Y.; Wang, Y.Q. CuO-CeO2/Al2O3 /FeCrAl monolithic catalysts prepared by sol-pyrolysis method for preferential oxidation of carbon monoxide. Catal. Lett. 2007, 117, 119–125. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).