
X-ray Diffraction, Spectroscopy, Optical Properties, NPA, NBO, FMO, and Hirshfeld Surface Analyses of Two Newly Synthesized Piperidinium Ionic Liquids

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Experimental
2.1. Materials and Methods
2.2. Synthesis
2.2.1. Synthesis of Piperidinium [4-(4-chlorophenyl)-3-cyano-5-(ethoxycarbonyl)-6-phenylpyridin-2-yl]sulfanide (Scheme 1; 4a)

2.2.2. Synthesis of Piperidinium [3-cyano-5-(ethoxycarbonyl)-6-methyl-4-[(E)-2-phenylethenyl]pyridin-2-yl]sulfanide (Scheme 1; 4b)
2.3. Crystal Structure Measurement
2.4. Computational Details
3. Results and Discussion
3.1. X-ray Diffraction Studies
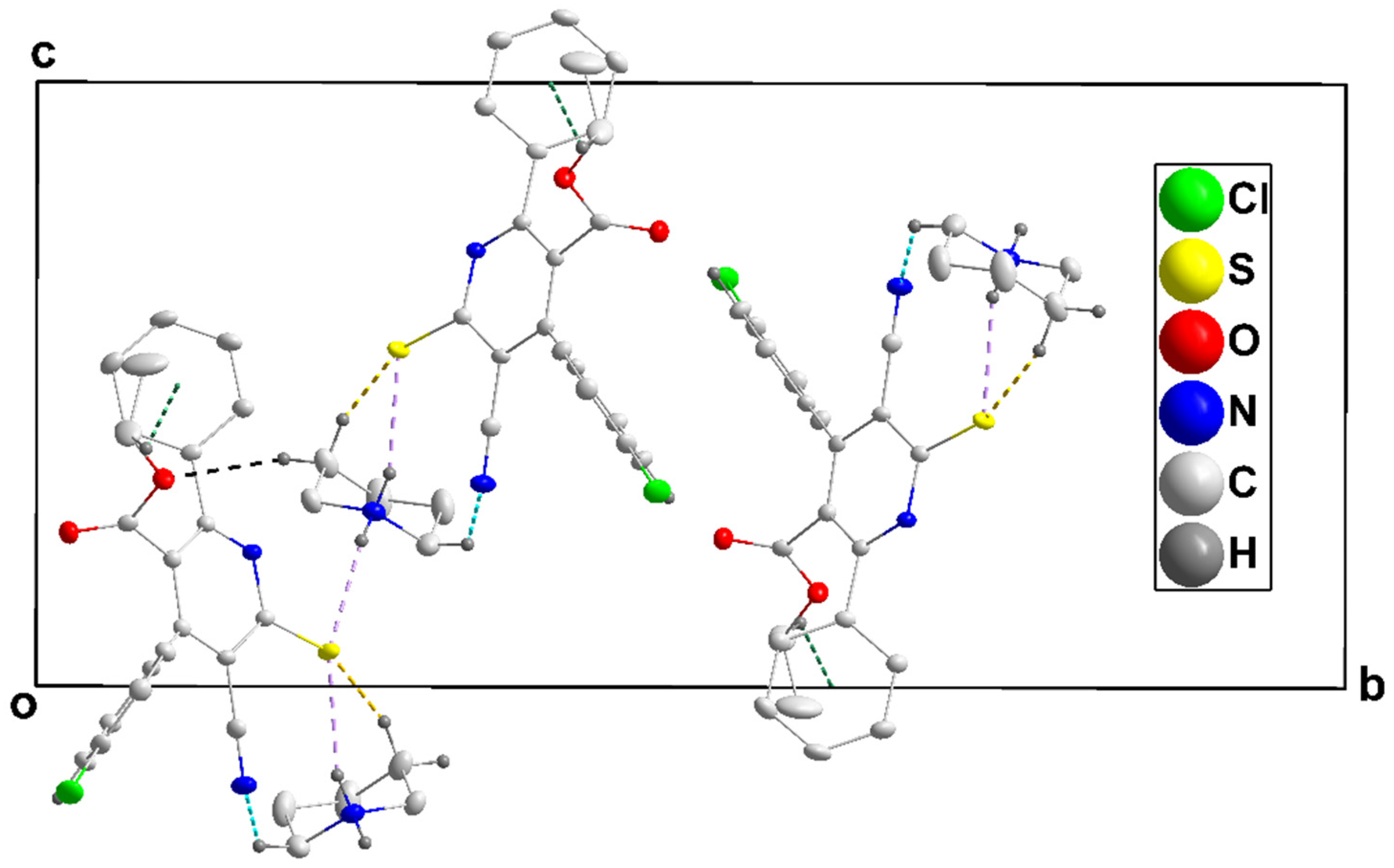
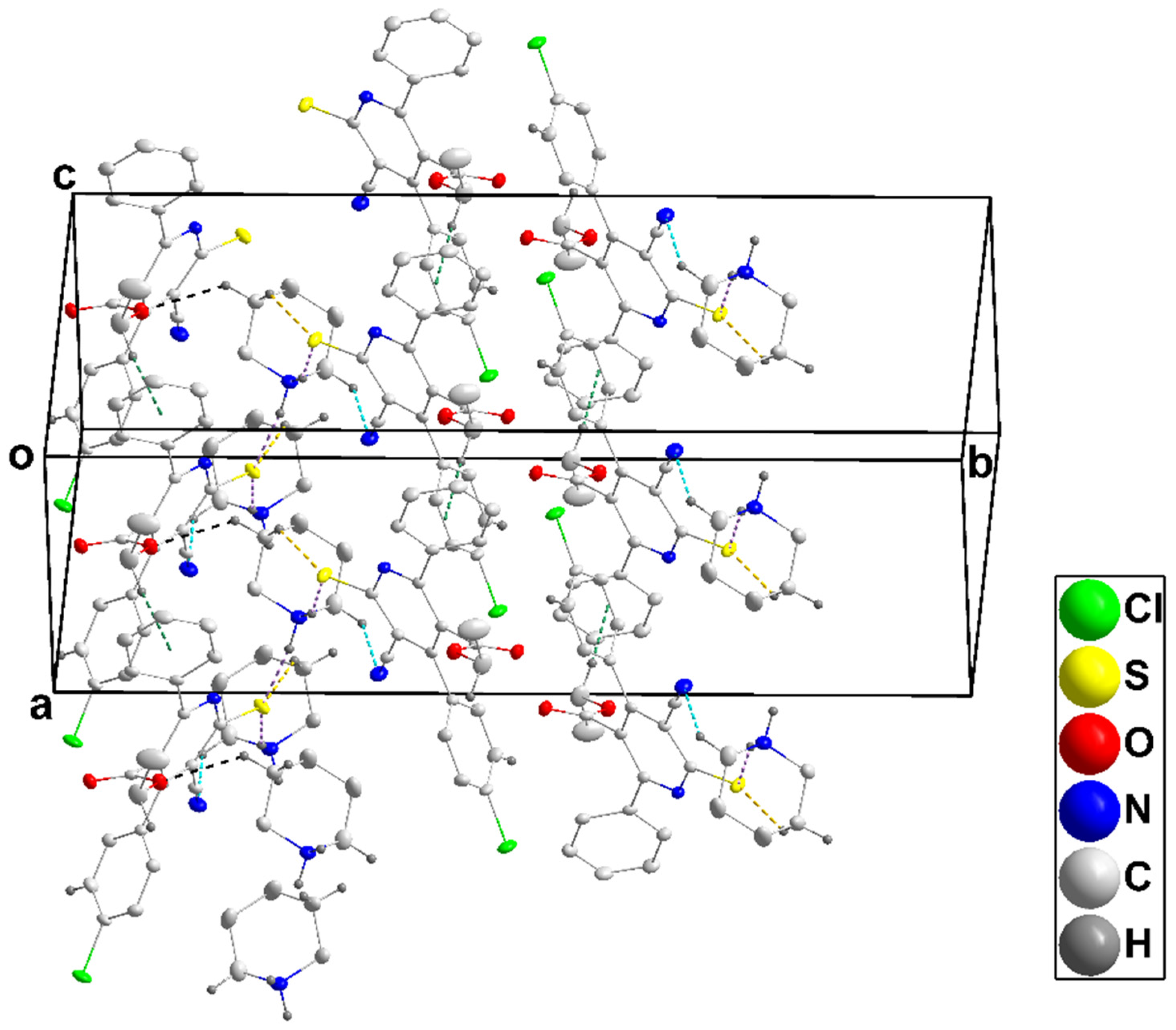
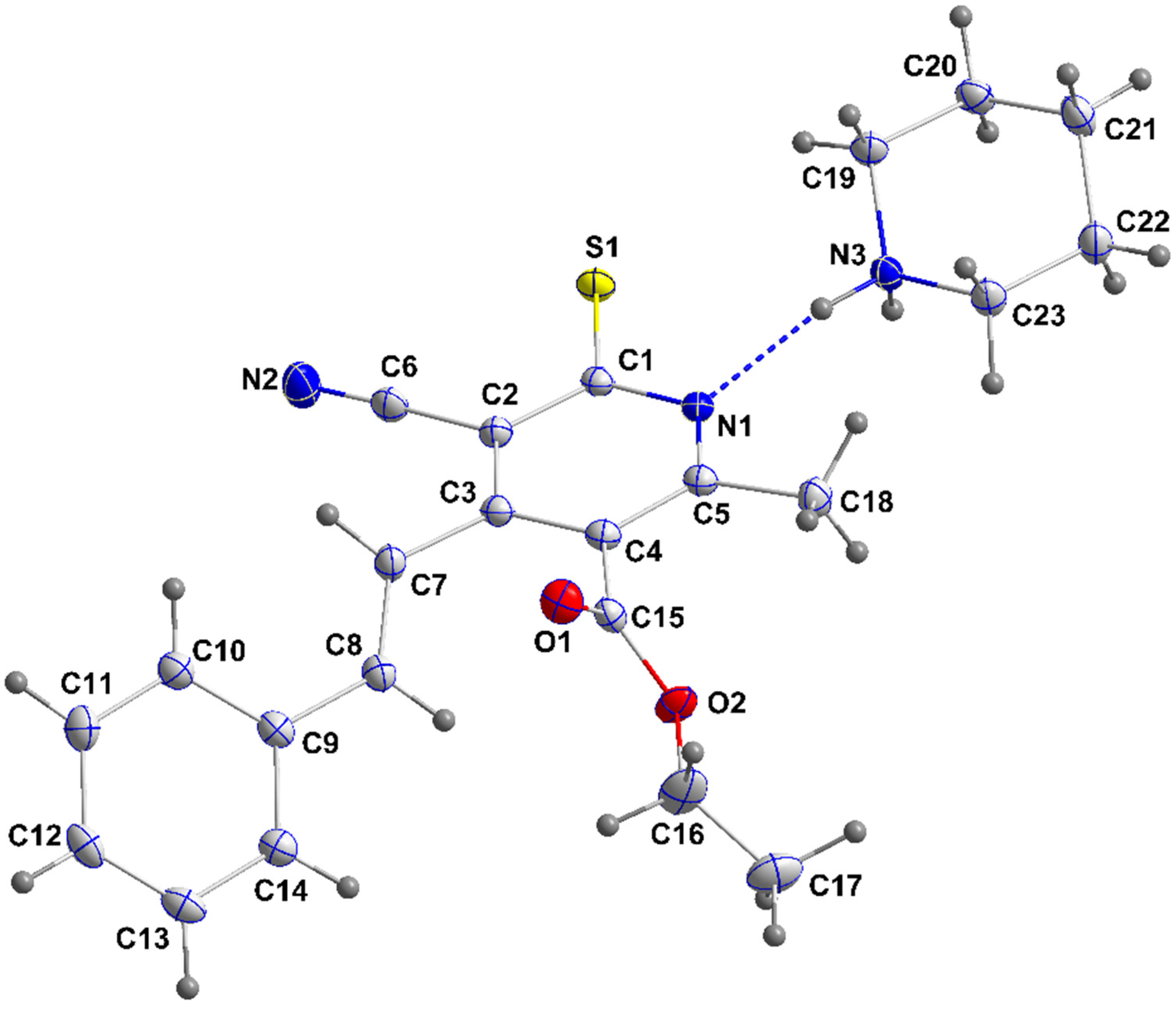
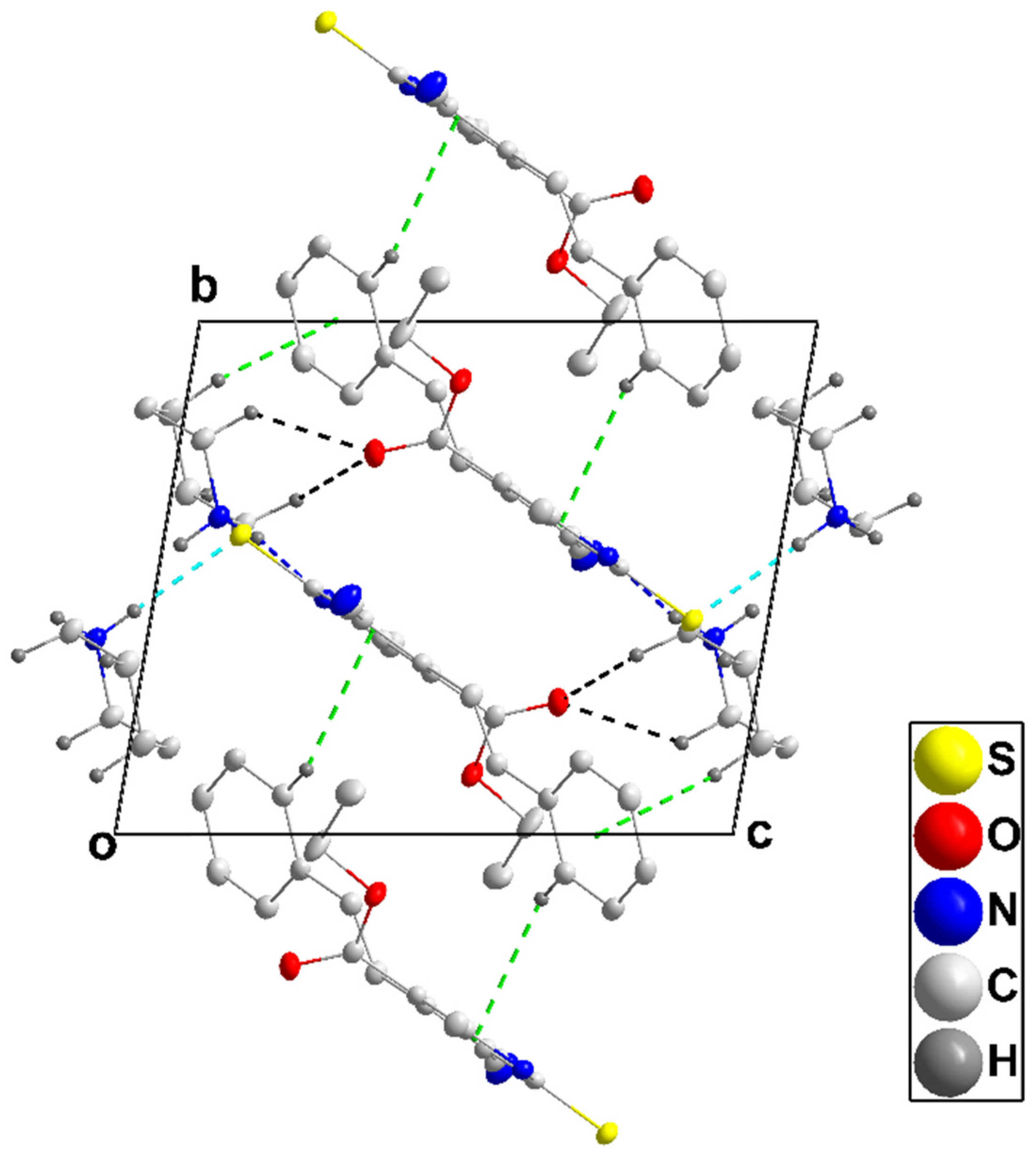
3.1.1. Crystal Structure of 4a
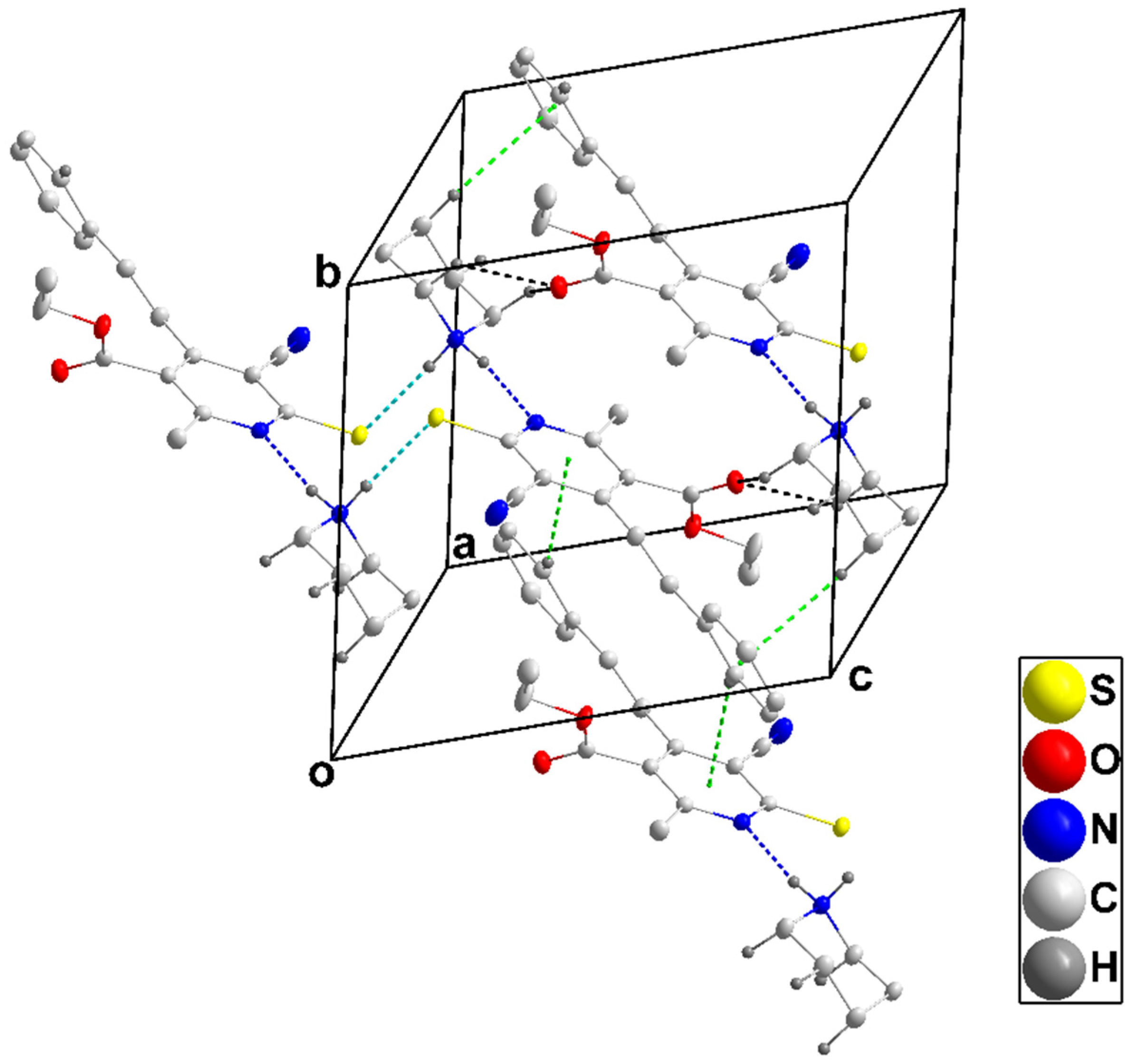
3.1.2. Crystal Structure of 4b
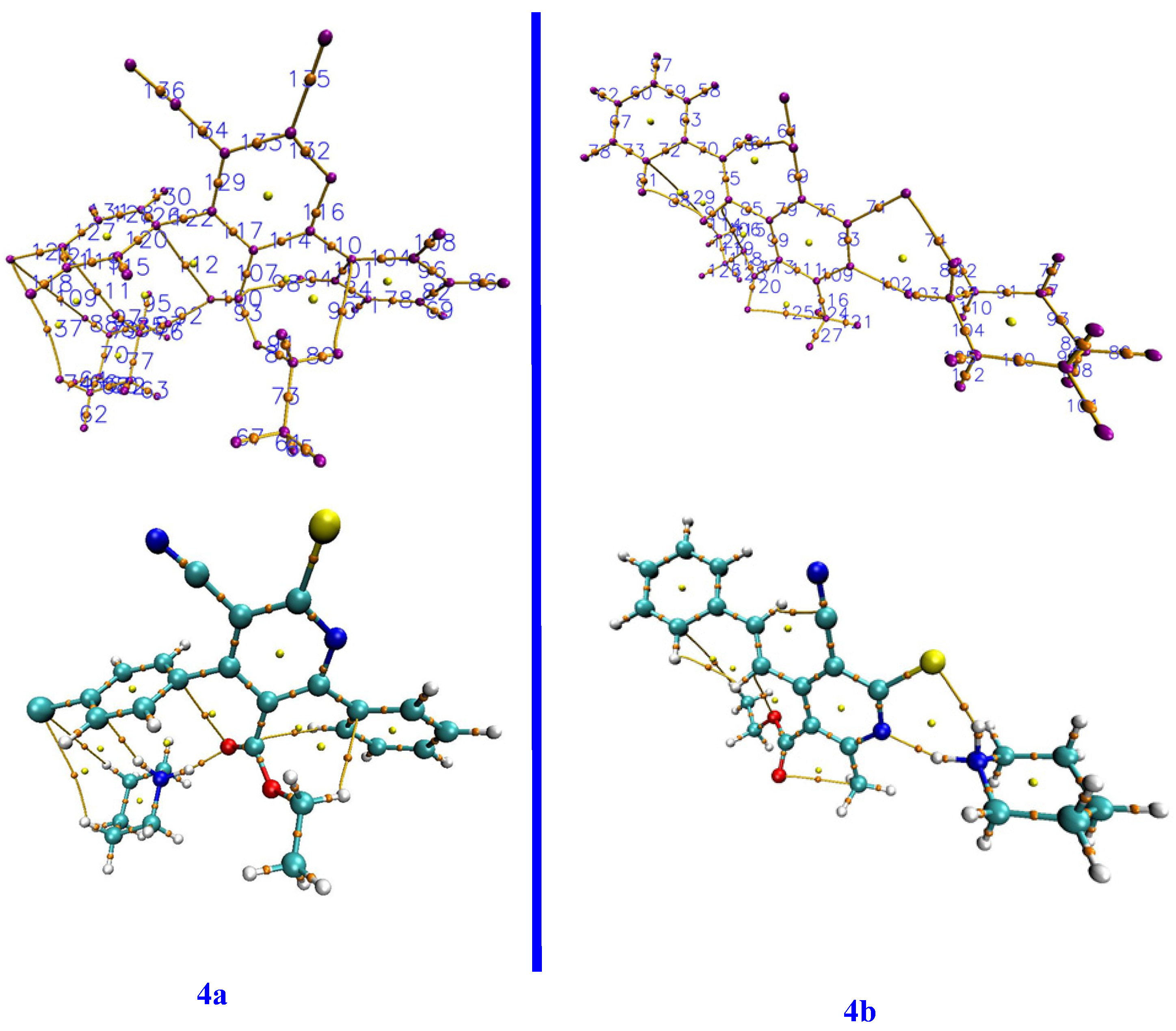
3.2. Optimized Geometries and Stability of Ionic Liquids
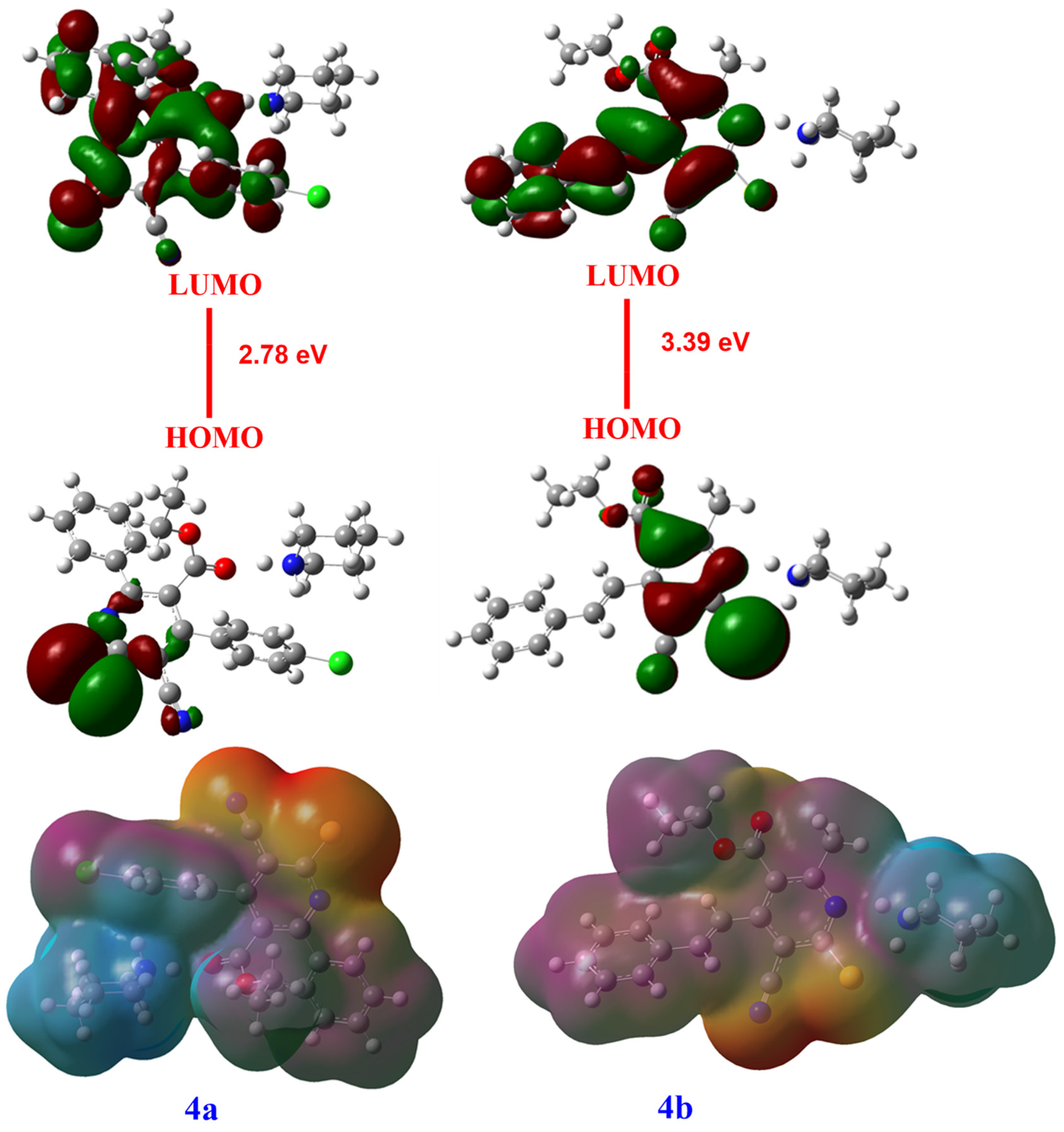
3.3. FMOS Analysis and NBO Charge Study
3.4. Molecular Electrostatic Potential (MEP)
3.5. Electronic Properties and Global Reactivity of 4a and 4b
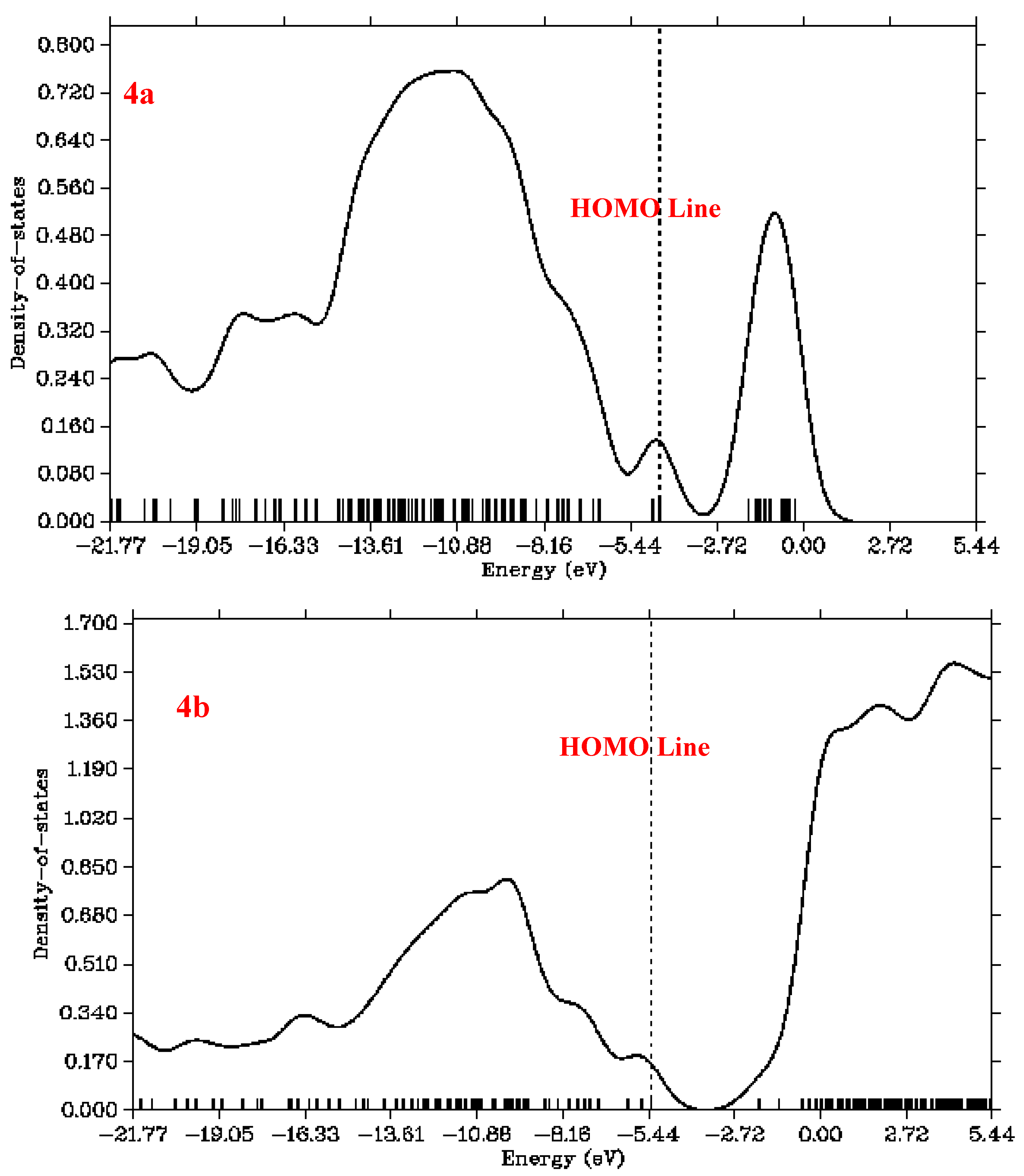
3.6. Total Density of States (TDOS) Analysis
3.7. Optical and Nonlinear Optical (NLO) Properties
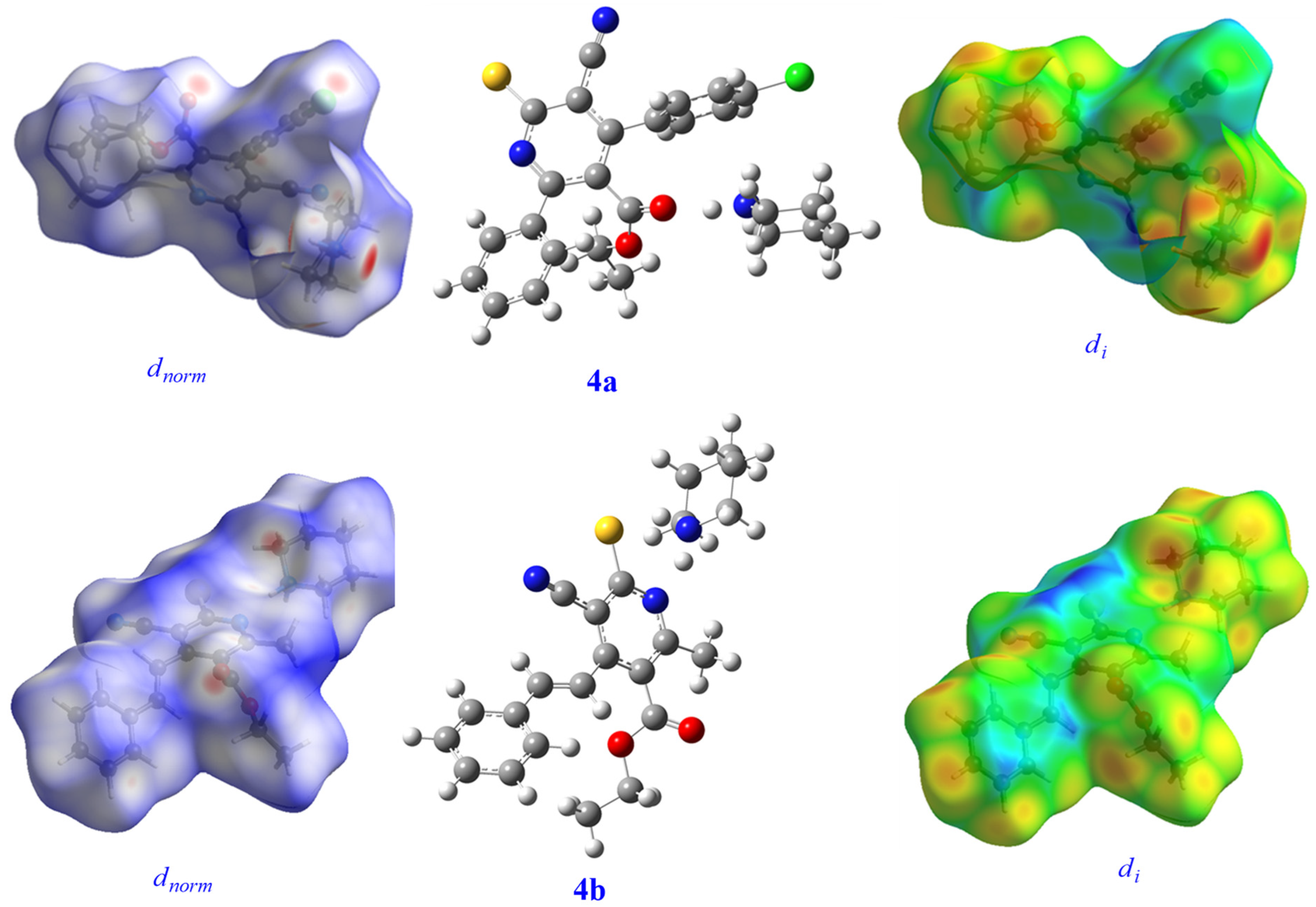
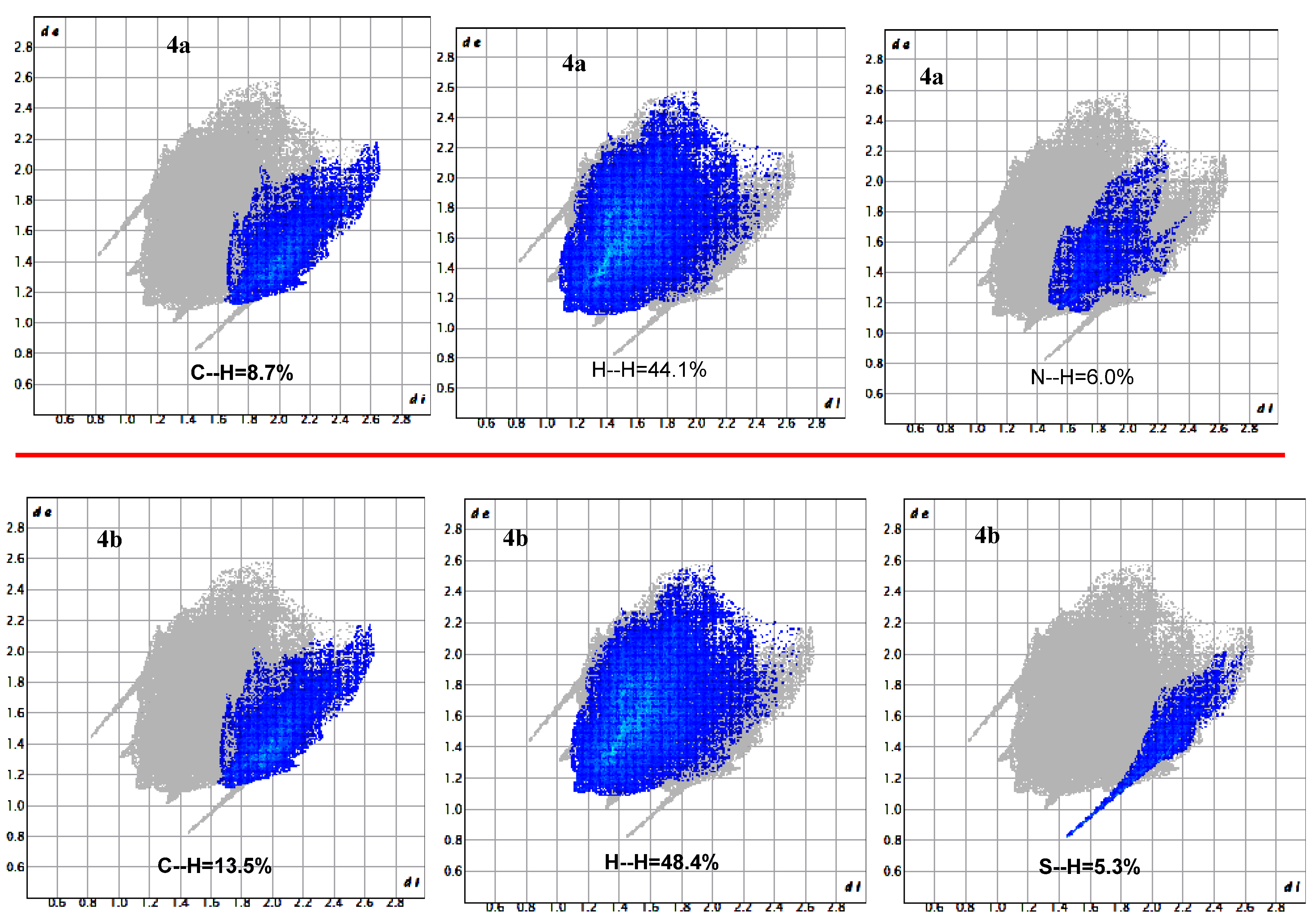
3.8. Hirschfeld Surface Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henry, G.D. De novo synthesis of substituted pyridines. Tetrahedron 2004, 29, 6043–6061. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals: Miniperspective. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Bakhite, E.A.; Abd-Ella, A.A.; El-Sayed, M.E.A.; Abdel-Raheem, S.A.A. Pyridine derivatives as insecticides. Part 1: Synthesis and toxicity of some pyridine derivatives against Cowpea Aphid, Aphis craccivora Koch (Homoptera: Aphididae). J. Agric. Food Chem. 2014, 62, 9982–9986. [Google Scholar] [CrossRef] [PubMed]
- Abbas, I.; Gomha, S.; Elaasser, M.; Bauomi, M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk. J. Chem. 2015, 39, 334–346. [Google Scholar] [CrossRef]
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem 2015, 1, 1–11. [Google Scholar]
- Shamroukh, A.H.; Kotb, E.R.; Anwar, M.M.; Sharaf, M. A Review on the Chemistry of Nicotinonitriles and Their Applications. Egypt. J. Chem. 2021, 64, 4509–4529. [Google Scholar] [CrossRef]
- He, D.; Wang, B.; Duan, K.; Zhou, Y.; Li, M.; Jiang, H.; Wu, W. Synthesis of Densely Substituted Pyridine Derivatives from 1-Methyl-1, 3-(ar) enynes and Nitriles by a Formal [4 + 2] Cycloaddition Reaction. Org. Lett. 2022, 24, 1292–1297. [Google Scholar] [CrossRef]
- Manna, F.; Chimenti, F.; Bolasco, A.; Filippelli, A.; Palla, A.; Filippelli, W.; Lampa, E.; Mercantini, R. Anti-inflammatory, analgesic and antipyretic 4, 6-disubstituted 3-cyanopyridine-2-ones and 3-cyano-2-aminopyridines. Eur. J. Med. Chem. 1992, 27, 627–632. [Google Scholar] [CrossRef]
- Baldwin, J.J.; Engelhardt, E.L.; Hirschmann, R.; Ponticello, G.S.; Atkinson, J.G.; Wasson, B.K.; Sweet, C.S.; Scriabine, A. Heterocyclic analogs of the antihypertensive. beta.-adrenergic blocking agent (S)-2-[3-(tert-butylamino)-2-hydroxypropoxy]-3-cyanopyridine. J. Med. Chem. 1980, 23, 65–70. [Google Scholar] [CrossRef]
- Alaa, A.-M.; El-Subbagh, H.I.; Kunieda, T. Lewis acid-promoted transformation of 2-alkoxypyridines into 2-aminopyridines and their antibacterial activity. Part 2: Remarkably facile C–N bond formation. Bioorg. Med. Chem. 2005, 13, 4929–4935. [Google Scholar]
- Bekhit, A.A.; Baraka, A.M. Novel milrinone analogs of pyridine-3-carbonitrile derivatives as promising cardiotonic agents. Eur. J. Med. Chem. 2005, 40, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, H.A.; Moustafa, A.H.; Haikal, A.E.-F.Z.; Abu-El-Halawa, R.; El Sayed, H. Synthesis, antitumor and antimicrobial activities of 4-(4-chlorophenyl)-3-cyano-2-(β-O-glycosyloxy)-6-(thien-2-yl)-nicotinonitrile. Eur. J. Med. Chem. 2011, 46, 2948–2954. [Google Scholar] [CrossRef]
- Malki, A.; Mohsen, M.; Aziz, H.; Rizk, O.; Shaaban, O.; El-Sayed, M.; Sherif, Z.A.; Ashour, H. New 3-Cyano-2-substituted pyridines induce apoptosis in MCF 7 breast cancer cells. Molecules 2016, 21, 230. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.M.; Gomha, S.M.; El-Sayed, A.A.; Mabkhot, Y.N.; Alsayari, A.; Muhsinah, A. Bin Facile synthesis and antiproliferative activity of new 3-cyanopyridines. BMC Chem. 2019, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, A.; Pandeya, S.N. Pyridine a versatile nucleuse in pharmaceutical field. Asian J. Pharm. Clin. Res 2011, 4, 5–8. [Google Scholar]
- Ueda, M.; Matsumura, S.; Masui, M.; Matsuura, E.; Kawakami, M.; Fujitomo, H.; Umeda, T.; Kagawa, H.; Hirohata, S.; Shima, K. Pharmacological studies on a new dihydrothienopyridine calcium antagonist. 3rd communication: Antihypertensive effects of S-(+)-methyl-4, 7-dihydro-3-isobutyl-6-methyl-4-(3-nitrophenyl) thieno [2, 3-b] pyridine-5-carboxylate in hypertensive rats and dogs. Arzneimittel-forschung 1993, 43, 1282–1290. [Google Scholar] [PubMed]
- Al-Trawneh, S.A.; El-Abadelah, M.M.; Zahra, J.A.; Al-Taweel, S.A.; Zani, F.; Incerti, M.; Cavazzoni, A.; Vicini, P. Synthesis and biological evaluation of tetracyclic thienopyridones as antibacterial and antitumor agents. Bioorg. Med. Chem. 2011, 19, 2541–2548. [Google Scholar] [CrossRef]
- Madhusudana, K.; Shireesha, B.; Naidu, V.G.M.; Ramakrishna, S.; Narsaiah, B.; Rao, A.R.; Diwan, P. V Anti-inflammatory potential of thienopyridines as possible alternative to NSAIDs. Eur. J. Pharmacol. 2012, 678, 48–54. [Google Scholar] [CrossRef]
- Bousquet, E.; Guerrera, F.; Siracusa, M.A.; Caruso, A.; Amico-Roxas, M. Synthesis and pharmacological activity of 3-substituted pyrido [3′, 2′: 4, 5] thieno [3, 2-d] pyrimidin-4 (3H)-ones. Farmaco. Sci. 1984, 39, 110–119. [Google Scholar] [CrossRef]
- Bousquet, E.; Romeo, G.; Guerrera, F.; Caruso, A.; Amico-Roxas, M. [Synthesis and analgesic activity of 3-substituted derivatives of pyrido [3′, 2′: 4, 5] thieno [3, 2-d] pyrimidin-4 (3H)-ones]. Farmaco. Sci. 1985, 40, 869–874. [Google Scholar]
- Hussein, A.M.; Abu-Shanab, F.A.; Ishak, E.A. The multi-component condensation in the synthesis of the substituted 2-alkylsulphanil-4, 6-diarilpyridine-3-carbonitriles and their derivatives. Phosphorus Sulfur. Silicon. Relat. Elem. 2000, 159, 55–68. [Google Scholar] [CrossRef]
- Quintela, J.M.; Peinador, C.; Veiga, C.; González, L.; Botana, L.M.; Alfonso, A.; Riguera, R. Synthesis and antiallergic activity of pyridothienopyrimidines. Bioorg. Med. Chem. 1998, 6, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Madding, G.D.; Thompson, M.D. Regioselective syntheses of 2-amino-4, 5-dialkylthiophene-3-carboxylates and their conversion to 3, 4-dihydro-4-oxothieno [2, 3-d] pyrimidine-2-carboxylates. J. Heterocycl. Chem. 1987, 24, 581–587. [Google Scholar] [CrossRef]
- Böhm, N.; Krasselt, U.; Leistner, S.; Wagner, G. Reactions of 4-oxo-4H-pyrido (3′, 2′: 4, 5) thieno (3, 2-d)-1, 3-oxazines with amines. Pharmazie 1992, 47, 897–901. [Google Scholar]
- Wagner, G.; Leistner, S.; Vieweg, H.; Krasselt, U.; Prantz, J. Synthesis of thieno (2, 3-b) pyridines with oxalamidic acid or an oxalamidic alkylester residues and of 4-alkoxy-pyrido (3′, 2′: 4, 5) thieno (3, 2-d) pyridine-2-carboxylic acid derivates. Pharmazie 1993, 48, 342–346. [Google Scholar]
- Dai, X.-Y.; Zeng, X.-X.; Peng, F.; Han, Y.-Y.; Lin, H.-J.; Xu, Y.-Z.; Zhou, T.; Xie, G.; Deng, Y.; Mao, Y.-Q. A novel anticancer agent, SKLB70359, inhibits human hepatic carcinoma cells proliferation via G0/G1 cell cycle arrest and apoptosis induction. Cell. Physiol. Biochem. 2012, 29, 281–290. [Google Scholar] [CrossRef]
- Lockman, J.W.; Reeder, M.D.; Suzuki, K.; Ostanin, K.; Hoff, R.; Bhoite, L.; Austin, H.; Baichwal, V.; Willardsen, J.A. Inhibition of eEF2-K by thieno [2, 3-b] pyridine analogues. Bioorg. Med. Chem. Lett. 2010, 20, 2283–2286. [Google Scholar] [CrossRef]
- Wu, J.-P.; Fleck, R.; Brickwood, J.; Capolino, A.; Catron, K.; Chen, Z.; Cywin, C.; Emeigh, J.; Foerst, M.; Ginn, J. The discovery of thienopyridine analogues as potent IκB kinase β inhibitors. Part II. Bioorg. Med. Chem. Lett. 2009, 19, 5547–5551. [Google Scholar] [CrossRef]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine. Chem.Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef]
- Singh, S.K. Solubility of lignin and chitin in ionic liquids and their biomedical applications. Int. J. Biol. Macromol. 2019, 132, 265–277. [Google Scholar] [CrossRef]
- Singh, S.K.; Savoy, A.W. Ionic liquids Synthesis and applications: An overview. J. Mol. Liq. 2020, 297, 112038–112060. [Google Scholar] [CrossRef]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.M.; Awad, W.H.; Gilman, J.W.; Maupin, P.H.; De Long, H.C.; Trulove, P.C. Flammability, thermal stability, and phase change characteristics of several trialkylimidazolium salts. Green Chem. 2003, 5, 724–727. [Google Scholar] [CrossRef]
- Zang, H.; Feng, Y.; Lou, J.; Wang, K.; Wu, C.; Liu, Z.; Zhu, X. Synthesis and performance of piperidinium-based ionic liquids as catalyst for biomass conversion into 3-acetamido-5-acetylfuran. J. Mol. Liq. 2022, 366, 120281. [Google Scholar] [CrossRef]
- Bruker. APEX3, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2016; p. 2014. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Ahsin, A.; Ayub, K. Superalkali-based alkalides Li3O@[12-crown-4]M (where M = Li, Na, and K) with remarkable static and dynamic NLO properties; A DFT study. Mater. Sci. Semicond. Process. 2022, 138, 106254. [Google Scholar] [CrossRef]
- Caricato, M.; Frisch, M.J.; Hiscocks, J.; Frisch, M.J. Gaussian 09: IOps Reference; Gaussian Inc.: Wallingford, CT, USA, 2009; ISBN 0972718788. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 5; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 4a | 4b |
|---|---|---|
| CCDC Deposit Number | 2237196 | 2237198 |
| Chemical formula | C21H14ClN2O2S·C5H12N | C18H15N2O2S·C5H12N |
| Mr | 480.01 | 409.53 |
| Crystal system, space group | Monoclinic, P21/c | Triclinic, P |
| Temperature (K) | 120 | 120 |
| a, b, c (Å) | 7.6105 (4), 26.1975 (14), 12.2488 (7) | 10.0768 (4), 10.4188 (4), 11.3759 (5) |
| α, β, γ (°) | β (°) = 99.357 (1) | 78.519 (1), 82.891 (1), 65.707 (1) |
| V (Å3) | 2409.6 (2) | 1065.63 (8) |
| Z | 4 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.27 | 0.18 |
| Crystal size (mm) | 0.21 × 0.20 × 0.09 | 0.30 × 0.27 × 0.17 |
| Data collection | ||
| Diffractometer | Bruker Smart APEX CCD | Bruker Smart APEX CCD |
| Tmin, Tmax | 0.92, 0.97 | 0.82, 0.90 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 22901, 6129, 4967 | 20787, 5673, 4716 |
| Rint | 0.028 | 0.025 |
| (sin θ/λ)max (Å−1) | 0.685 | 0.686 |
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.037, 0.105, 1.09 | 0.039, 0.115, 1.11 |
| No. of reflections | 6129 | 5673 |
| No. of parameters | 391 | 370 |
| H-atom treatment | All H-atom parameters refined | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.42, −0.22 | 0.49, −0.22 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N3—H3A···S1i | 0.875(19) | 2.486(19) | 3.3011(12) | 155.3(16) |
| N3—H3B···S1ii | 0.953(18) | 2.406(18) | 3.2504(12) | 147.6(14) |
| C13—H13A···Cg2i | 1.01(2) | 2.87(2) | 3.6702(17) | 136.0(14) |
| C19—H19···O1iii | 0.934(17) | 2.552(17) | 3.2713(17) | 134.2(13) |
| C22—H22B···N2i | 1.000(17) | 2.597(17) | 3.3280(19) | 129.8(12) |
| C25—H25A···O2iv | 0.983(18) | 2.518(18) | 3.2771(18) | 133.9(13) |
| C25—H25B···S1i | 0.954(17) | 2.870(18) | 3.6474(16) | 139.4(13) |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N3—H3A···S1i | 0.883(15) | 2.393(15) | 3.2702(10) | 172.1(12) |
| N3—H3B···N1 | 0.980(16) | 1.980(17) | 2.9479(12) | 169.0(14) |
| C14—H14···Cg1ii | 0.971(16) | 2.848(15) | 3.6112(13) | 136.2(13) |
| C19—H19B···O1iii | 1.001(15) | 2.387(15) | 3.1508(14) | 132.4(11) |
| C21—H21B···Cg2iii | 0.948(16) | 2.972(16) | 3.9189(14) | 151.4(13) |
| C23—H23B···O1iii | 1.068(14) | 2.592(14) | 3.3617(14) | 128.4(10) |
| Parameters | 4a | 4b |
|---|---|---|
| Symmetry | C1 | C1 |
| Total energy | −2179.57 | −1605.68 |
| Dipole moment | 22.923 | 9.46 |
| Polarizability (αo) | 402.47 | 368.105 |
| Isotropic average polarizability volume | 59.64 | 54.547 |
| Hyperpolarizability (βo) | 2400.72 | 2722.63 |
| βvec | 2382.71 | 2660.48 |
| Parameters | 4a | 4b |
|---|---|---|
| EHOMO | −4.511 | −5.35 |
| ELUMO | −1.72 | −1.95 |
| EH-L | 2.78 | 3.39 |
| Q(N/O/S) | −0.67/−0.71/−0.15 | −0.72/−0.61/−0.274 |
| Q(Cl) | −0.002 | − |
| Ionization potential (IP) | 4.51 | 5.35 |
| Electron affinity (EA) | 1.72 | 1.95 |
| Chemical hardness (Ƞ) | 1.39 | 1.71 |
| Chemical potential (µ) | −3.11 | −3.65 |
| Electronegativity (χ) | 3.11 | 3.65 |
| Electrophilicity index (ω) | 3.47 | 3.89 |
| 4a | ||||||
| BCP | Interactions | ρ (r) | ∇2 (r) | G (r) | V (r) | H (r) |
| 112 | O15-O21 | 0.01242 | 0.04261 | 0.00960 | −0.00853 | 0.0010661 |
| 111 | H49-C23 | 0.01082 | 0.03143 | 0.00681 | −0.00576 | 0.0010481 |
| 137 | H52-Cl27 | 0.001360 | 0.003735 | 0.0007138 | −0.0004936 | 0.0002201 |
| 109 | H51-Cl27 | 0.0021867 | 0.00616756 | 0.0011639 | −0.0007859 | 0.00037798 |
| 92 | H48-O15 | 0.066432 | 0.0017422 | 0.051628 | −0.59701 | −0.008073 |
| 90 | H34-O8 | 0.009801 | 0.032557 | 0.0065859 | −0.00503255 | 0.0015534 |
| 98 | C14-H28 | 0.0096181 | 0.0390092 | 0.0073554 | −0.0049584 | 0.0023969 |
| 136 | C19-N20 | 0.46213 | 0.2099 | 0.8788 | −0.1705 | −0.82634 |
| 4b | ||||||
| 71 | C2-S1 | 0.20319 | −0.382452 | 0.11288 | −0.32139 | −0.208505 |
| 74 | H46-S1 | 0.042270 | 0.0500240 | 0.019821 | −0.0271371 | −0.0073155 |
| 102 | H45-S1 | 0.0512185 | 0.101639 | 0.03064215 | −0.0358745 | −0.0052323 |
| 125 | O18-C16 | 0.012298 | 0.0501636 | 0.0108061 | −0.00907131 | 0.017348 |
| 64 | C22-H25 | 0.0110728 | 0.044180 | 0.0085618 | −0.00607857 | 0.00248324 |
| 88 | H30-H38 | 0.004022 | 0.039976 | 0.0075161 | −0.005038 | 0.00247811 |
| 4a | |||
| Minimum | Mean | Maximum | |
| di | 0.8622 | 1.6990 | 2.7256 |
| de | 0.8633 | 1.6983 | 2.7694 |
| dnorm | −0.3775 | 0.4886 | 1.6265 |
| Shapeindex | −0.9972 | 0.1955 | 0.9965 |
| curvedness | −4.0821 | −0.9298 | 0.4266 |
| 4b | |||
| Minimum | Mean | Maximum | |
| di | 0.8219 | 1.6588 | 2.6637 |
| de | 0.8220 | 1.6608 | 2.5890 |
| dnorm | −0.4416 | 0.4854 | 1.3567 |
| Shapeindex | −0.9991 | 0.2039 | 0.9987 |
| curvedness | −3.2614 | −0.9192 | 0.3724 |
| 4a | |||||||
| Inside Atom | Outside Atom | ||||||
| Cl | S | O | N | C | H | Total % | |
| C | 0.9 | - | - | - | 0.2 | 8.7 | 9.7 |
| Cl | - | - | 0.9 | 0.1 | 0.8 | 4.3 | 6.1 |
| H | 2.5 | 4.5 | 3.2 | 5.3 | 7.4 | 44.1 | 67.0 |
| N | 0.1 | - | - | - | - | 6.0 | 6.1 |
| O | 0.7 | - | - | - | - | 3.4 | 4.1 |
| S | - | - | - | - | - | 6.9 | 6.9 |
| 4b | |||||||
| Inside atom | Outside atom | ||||||
| S | O | N | C | H | Total % | ||
| C | - | 0.5 | - | 0.3 | 13.5 | 14.3 | |
| H | 3.1 | 2.9 | 5.2 | 9.9 | 48.4 | 69.6 | |
| N | - | 0.5 | - | - | 6.3 | 6.8 | |
| O | - | - | 0.5 | 0.5 | 3.1 | 4.0 | |
| S | - | - | - | - | 5.3 | 5.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Bakri, Y.; Mohamed, S.K.; Ahsin, A.; Bakhite, E.A.; Marae, I.S.; A. H. Al-waleedy, S.; Mague, J.T.; Al-Salahi, R. X-ray Diffraction, Spectroscopy, Optical Properties, NPA, NBO, FMO, and Hirshfeld Surface Analyses of Two Newly Synthesized Piperidinium Ionic Liquids. Crystals 2023, 13, 1583. https://doi.org/10.3390/cryst13111583
El Bakri Y, Mohamed SK, Ahsin A, Bakhite EA, Marae IS, A. H. Al-waleedy S, Mague JT, Al-Salahi R. X-ray Diffraction, Spectroscopy, Optical Properties, NPA, NBO, FMO, and Hirshfeld Surface Analyses of Two Newly Synthesized Piperidinium Ionic Liquids. Crystals. 2023; 13(11):1583. https://doi.org/10.3390/cryst13111583
Chicago/Turabian StyleEl Bakri, Youness, Shaaban K. Mohamed, Atazaz Ahsin, Etify A. Bakhite, Islam S. Marae, Safiyyah A. H. Al-waleedy, Joel T. Mague, and Rashad Al-Salahi. 2023. "X-ray Diffraction, Spectroscopy, Optical Properties, NPA, NBO, FMO, and Hirshfeld Surface Analyses of Two Newly Synthesized Piperidinium Ionic Liquids" Crystals 13, no. 11: 1583. https://doi.org/10.3390/cryst13111583
APA StyleEl Bakri, Y., Mohamed, S. K., Ahsin, A., Bakhite, E. A., Marae, I. S., A. H. Al-waleedy, S., Mague, J. T., & Al-Salahi, R. (2023). X-ray Diffraction, Spectroscopy, Optical Properties, NPA, NBO, FMO, and Hirshfeld Surface Analyses of Two Newly Synthesized Piperidinium Ionic Liquids. Crystals, 13(11), 1583. https://doi.org/10.3390/cryst13111583