Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers

 ,
,
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Instruments
2.3. Synthesis
3. Characterizations
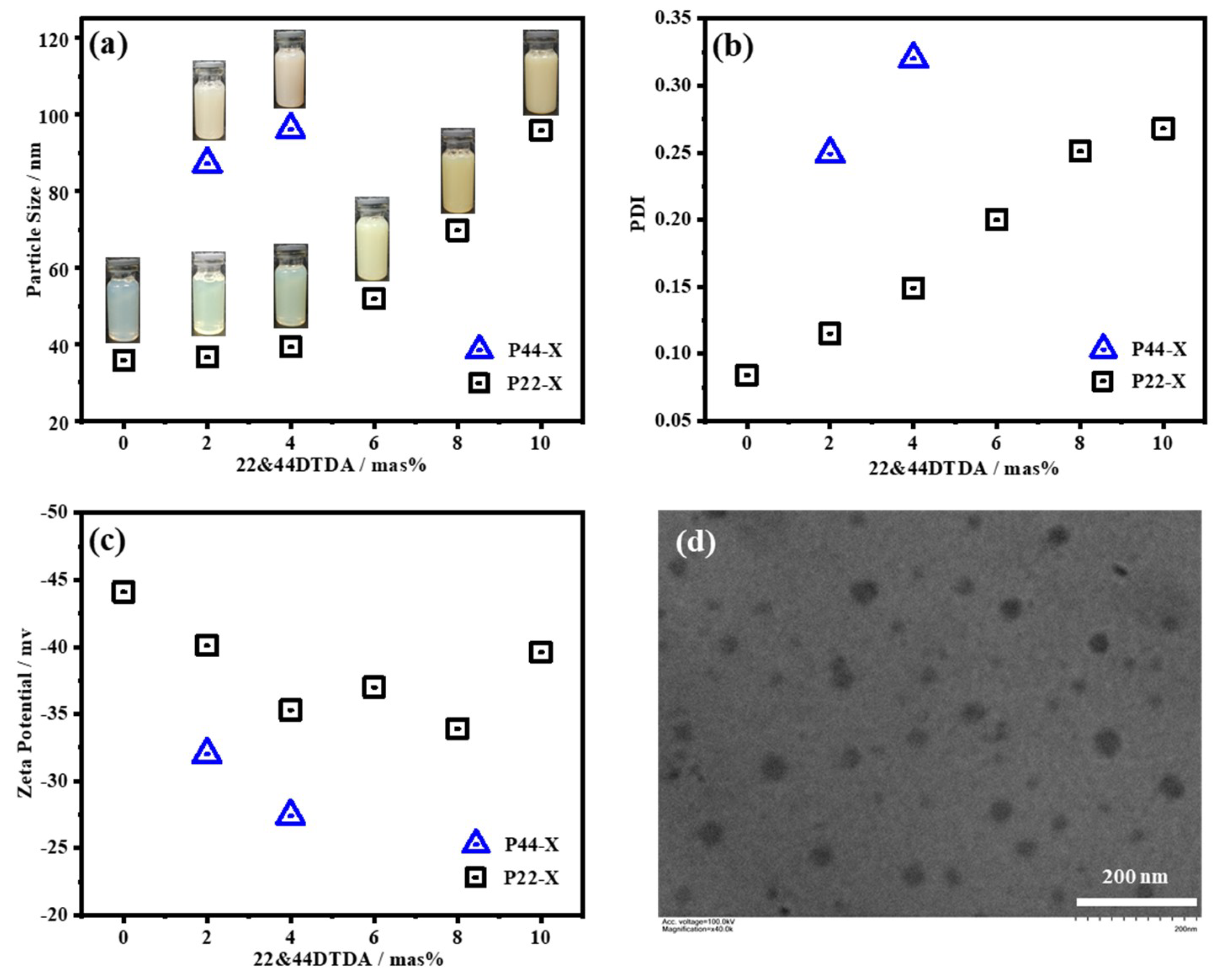
3.1. Dynamic Light Scattering (DLS)
3.2. Nuclear Magnetic Resonance (NMR)
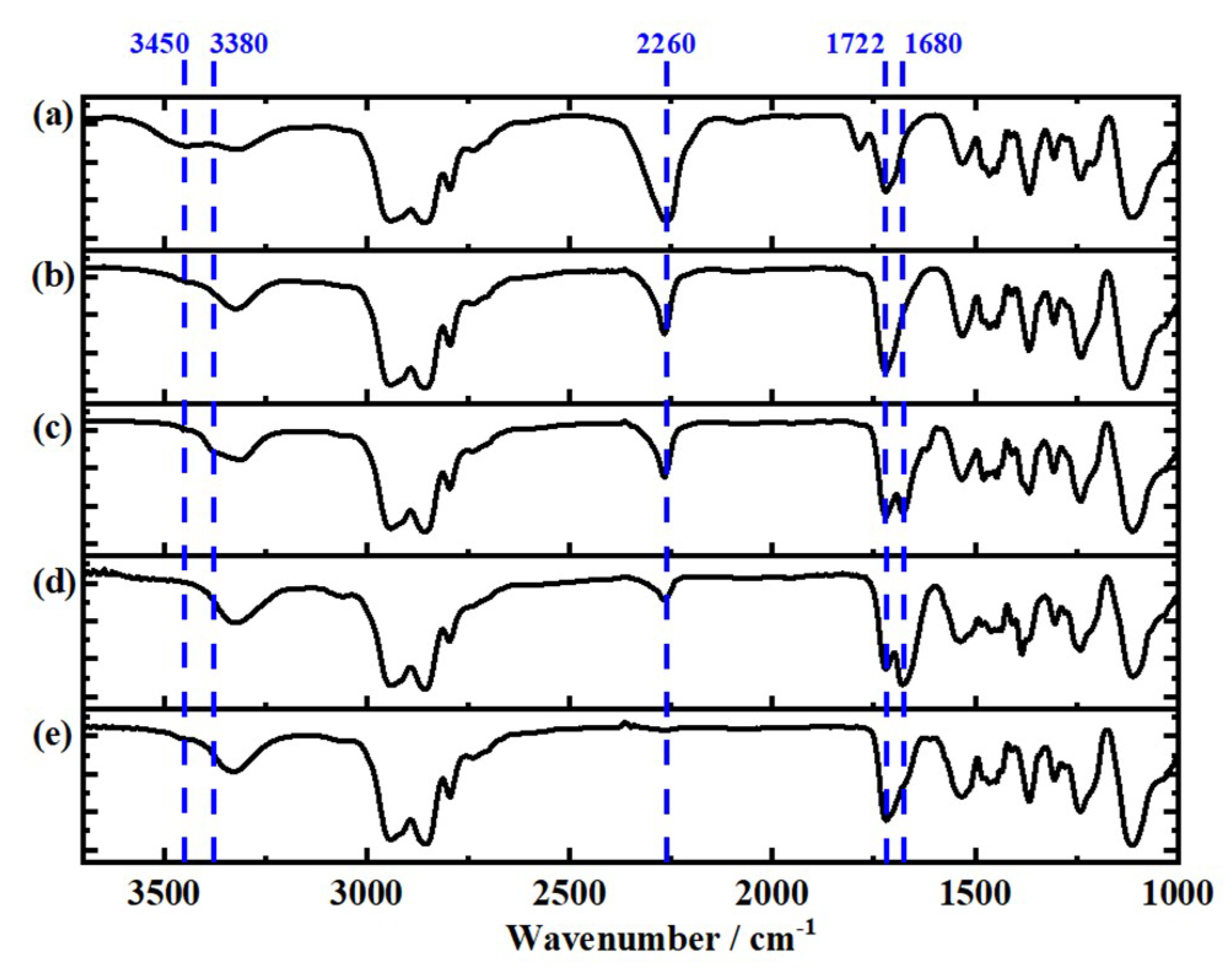
3.3. Infrared Spectroscopy
3.4. Gel Permeation Chromatography (GPC)
3.5. Tensile Tests
3.6. Self-healing and Reprocessing Tests
3.7. Molecular Simulation
3.8. Dynamic Mechanical Analysis (DMA)
3.9. Atomic Force Microscopy (AFM)
3.10. Transmission Electron Microscopy (TEM)
3.11. Scanning Electron Microscopy (SEM)
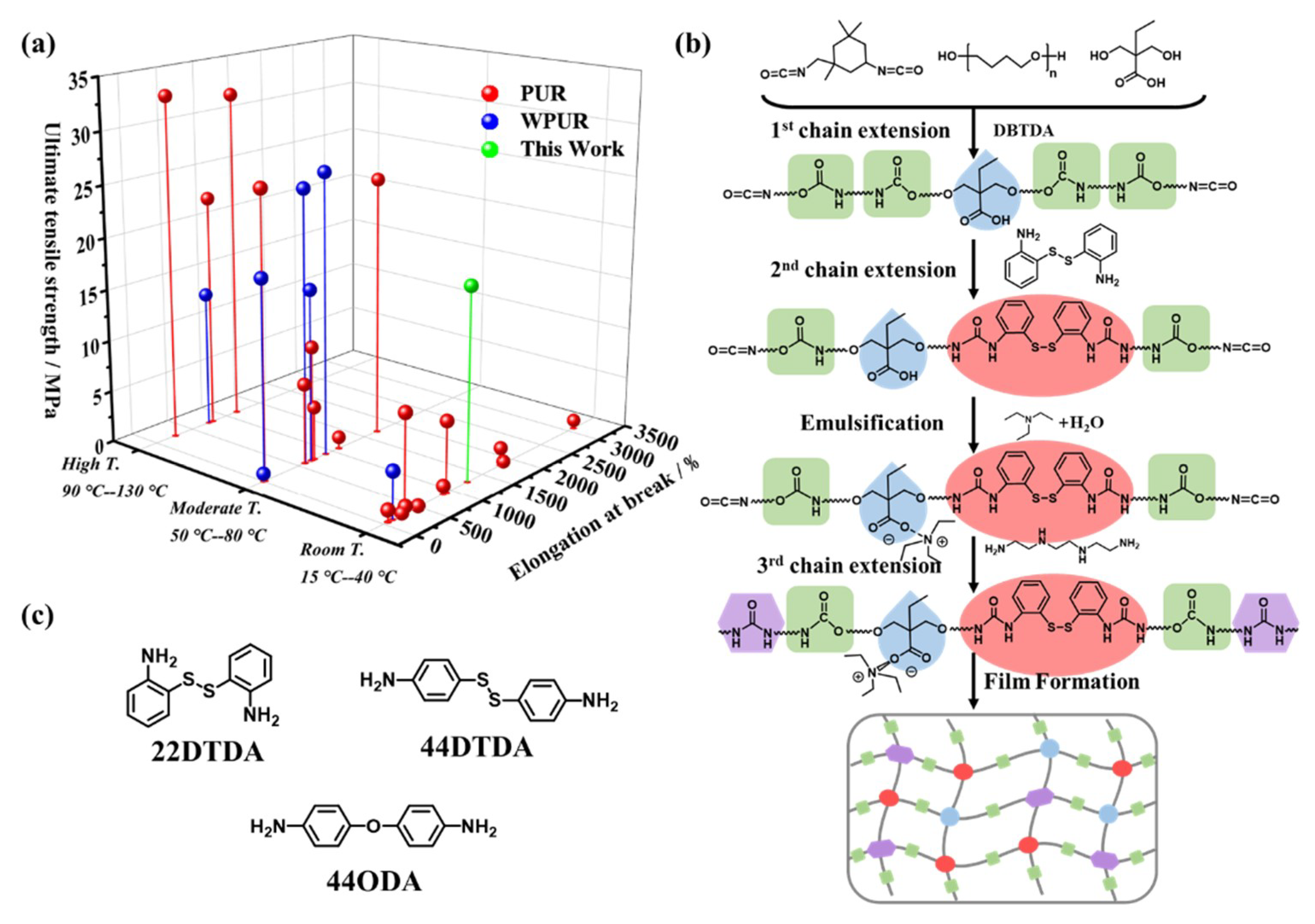
4. Results and Discussion
4.1. Synthesis of the WPUR Dispersions
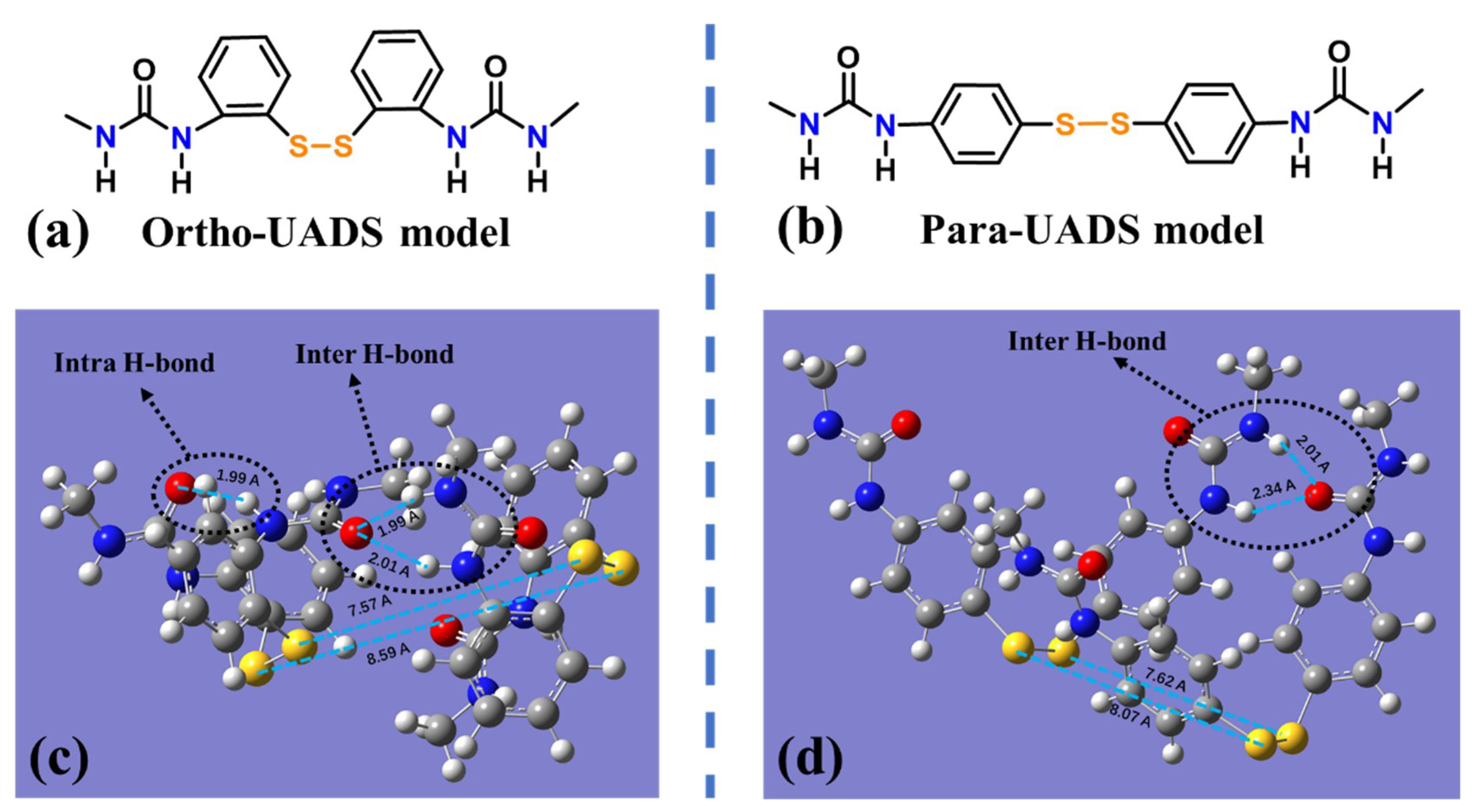
4.2. Molecular Simulation
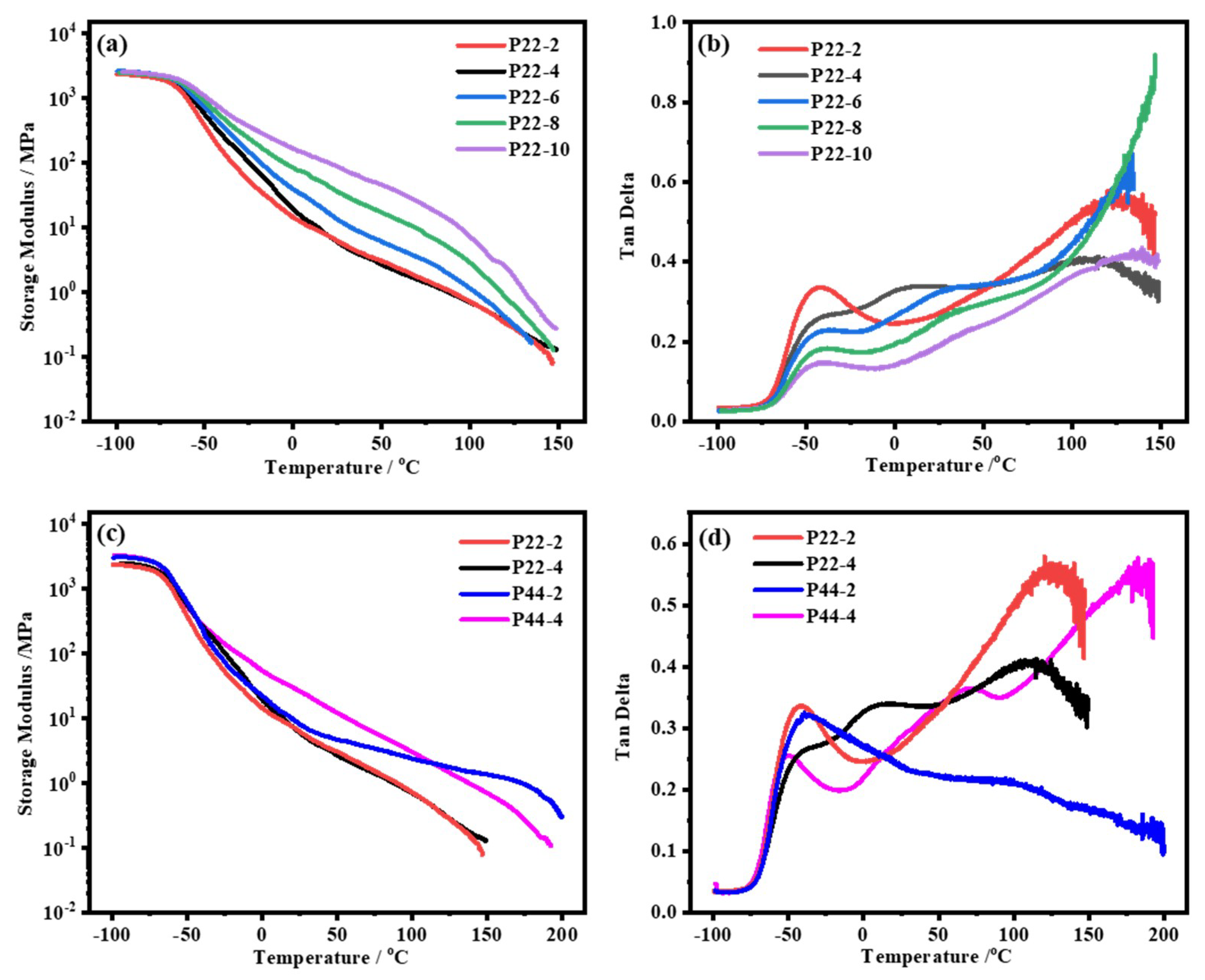
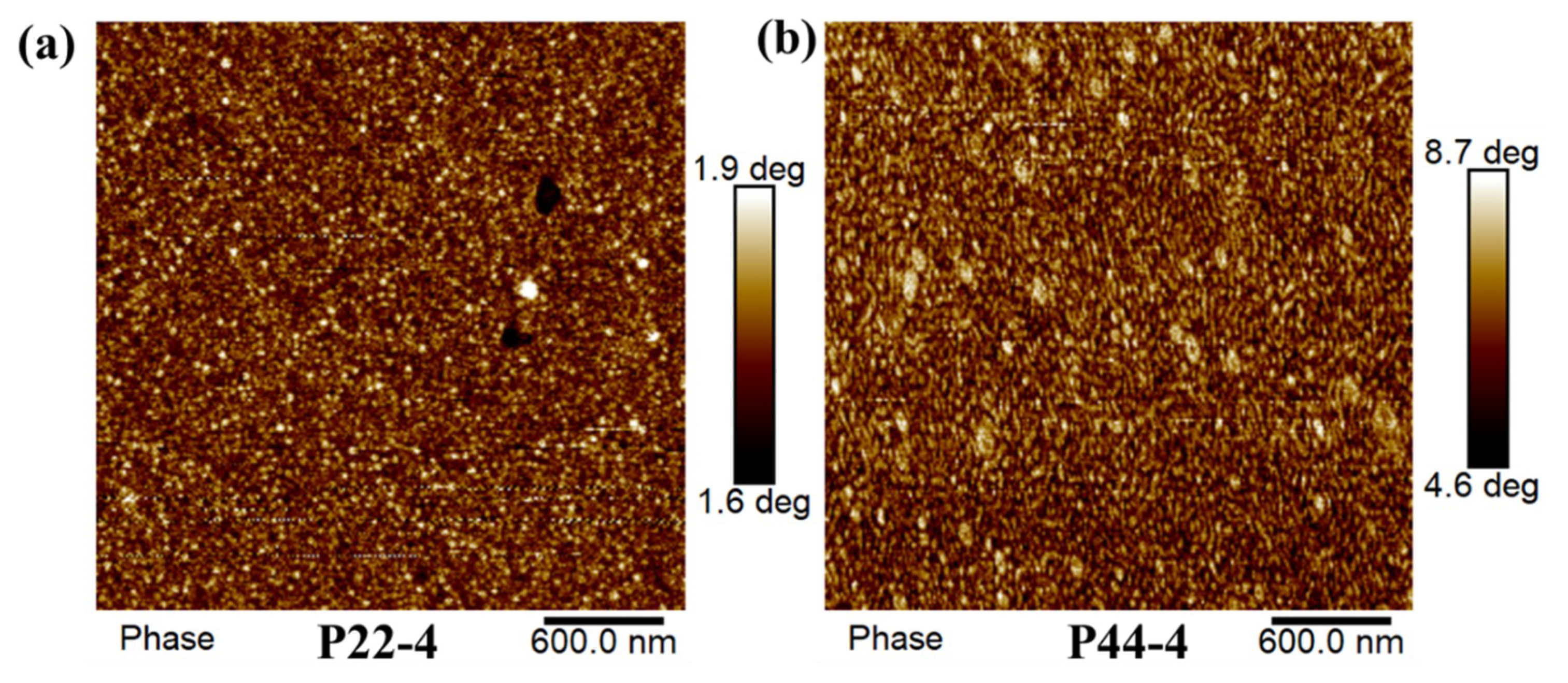
4.3. Microphase Separation of WPUR Films
4.4. Self-healing Ability of WPUR Films
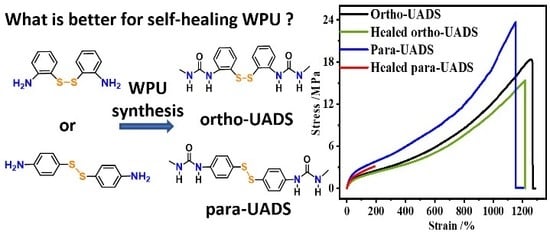
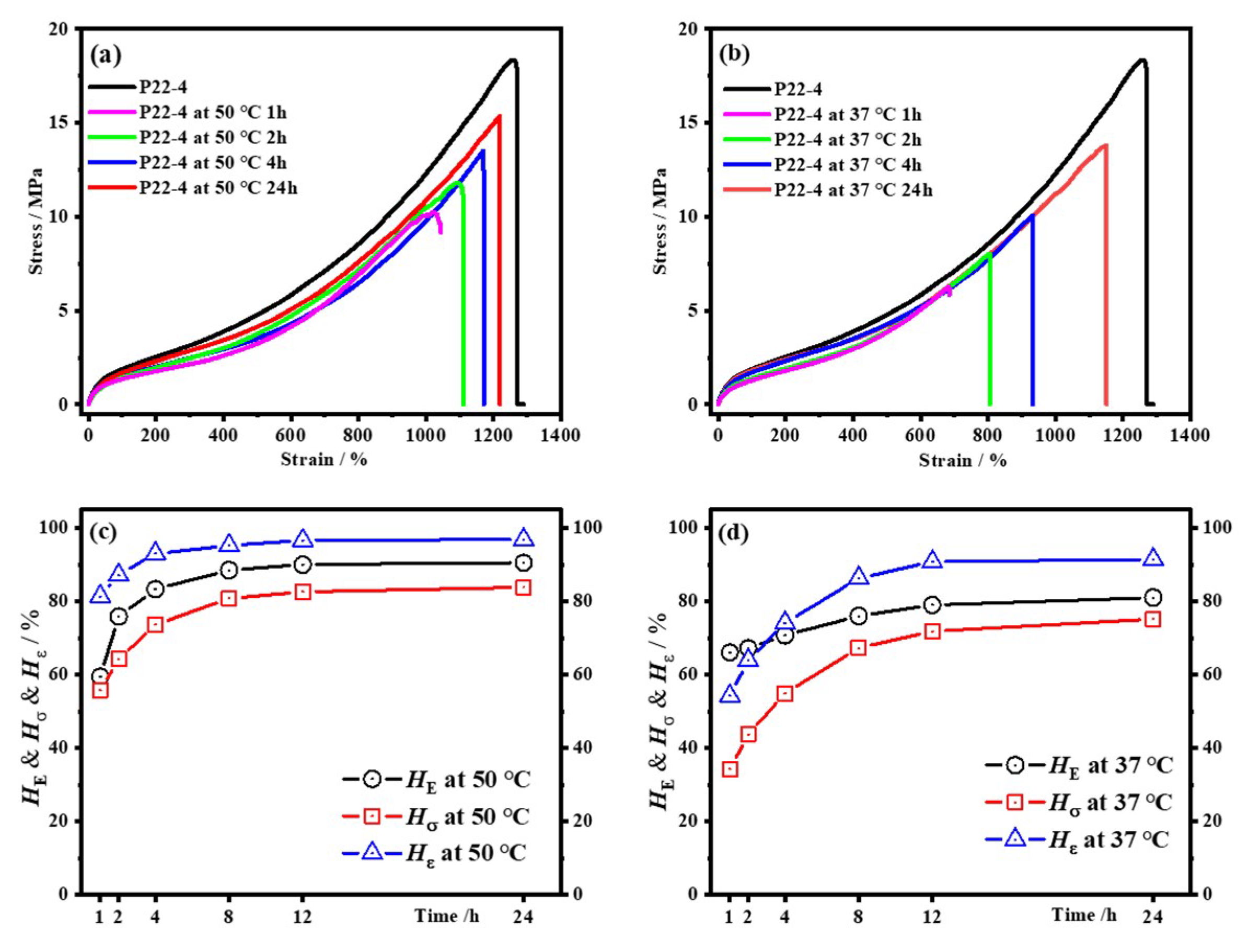
4.4.1. Influence of Substitution Pattern on Healing Efficiency
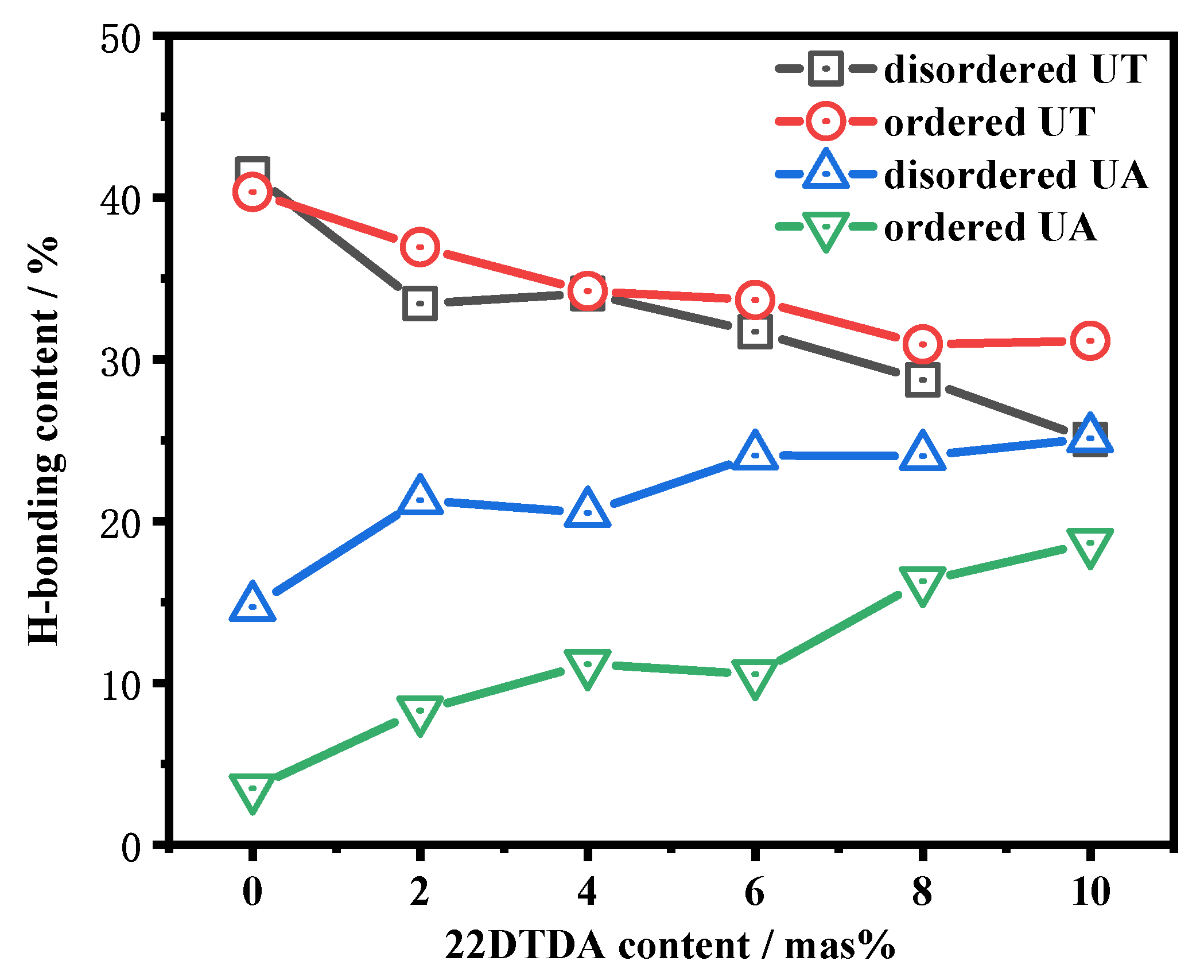
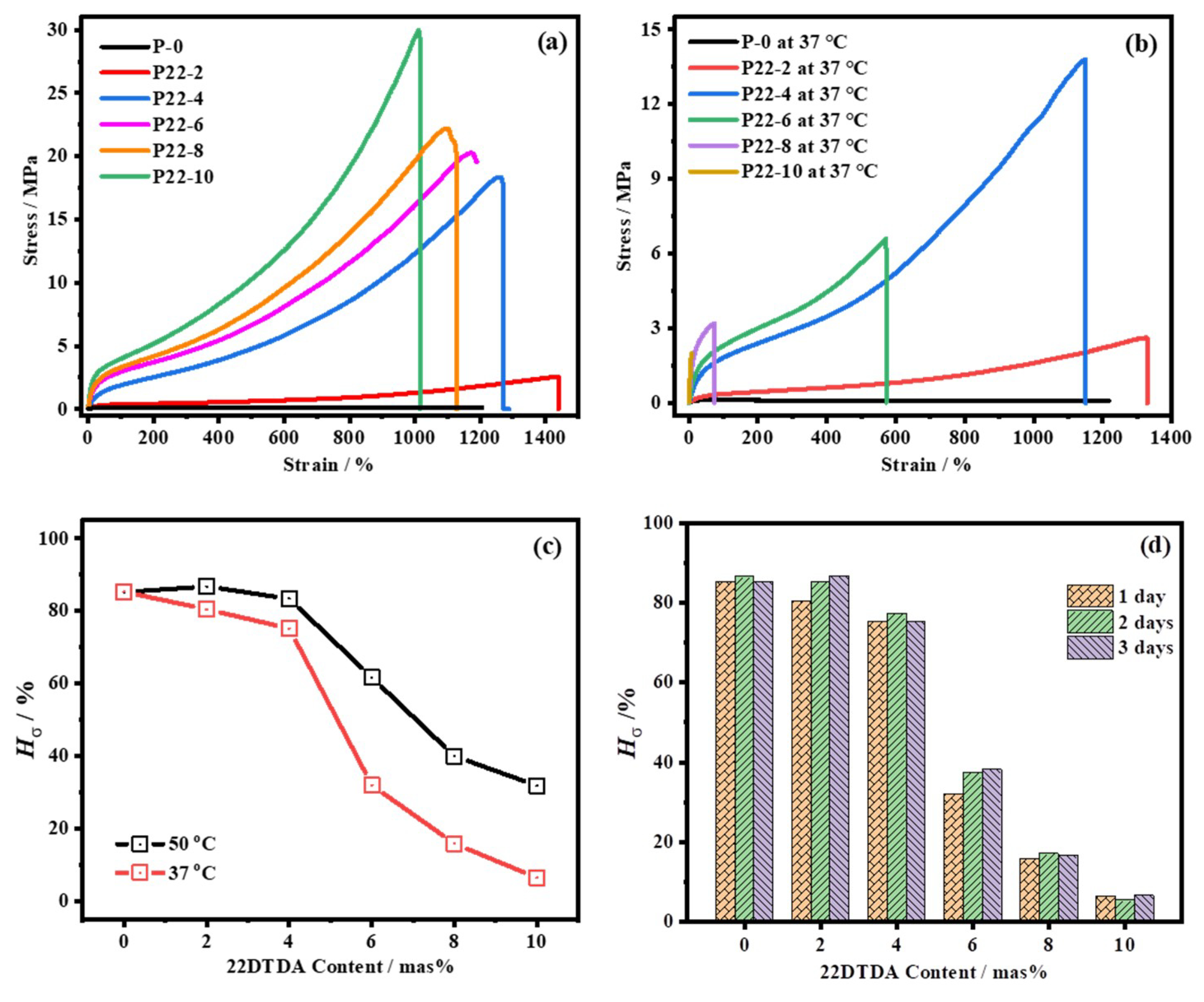
4.4.2. Influence of the 22DTDA Content on the Healing Efficiency
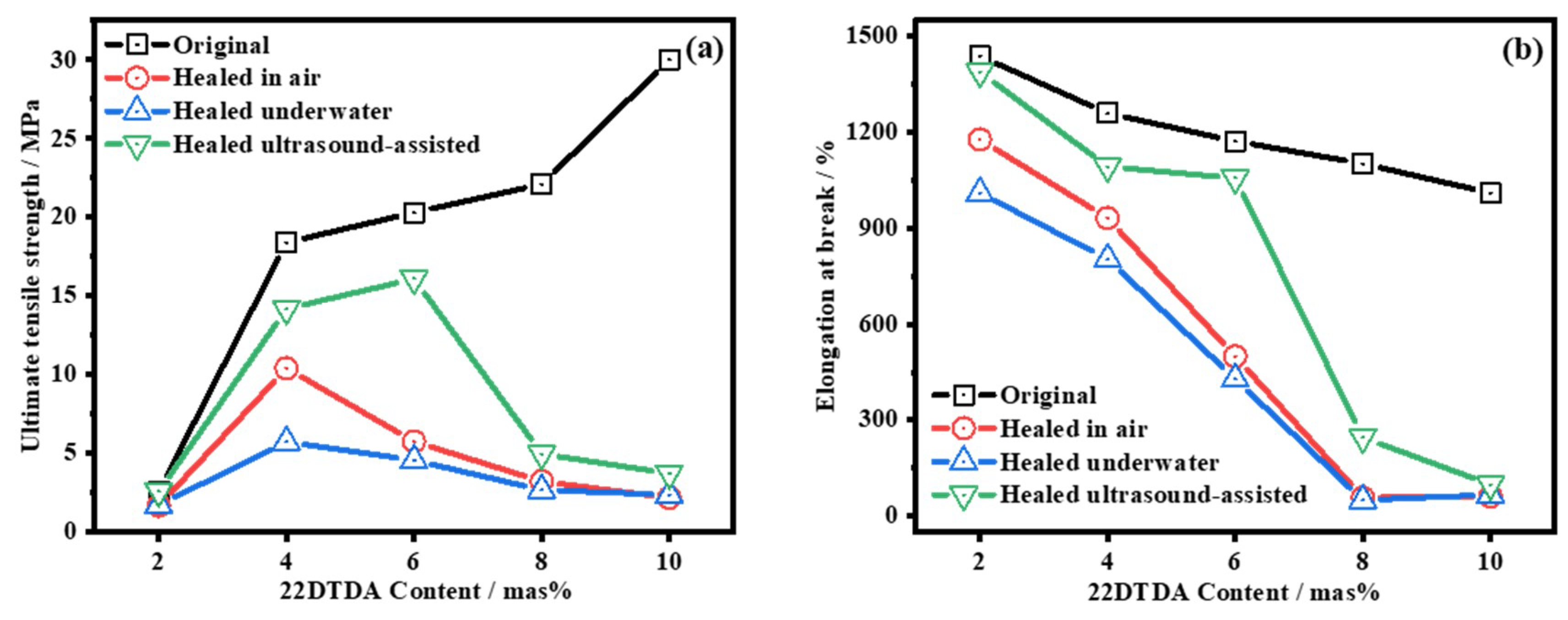
4.4.3. Ultrasound-Induced Self-Healing
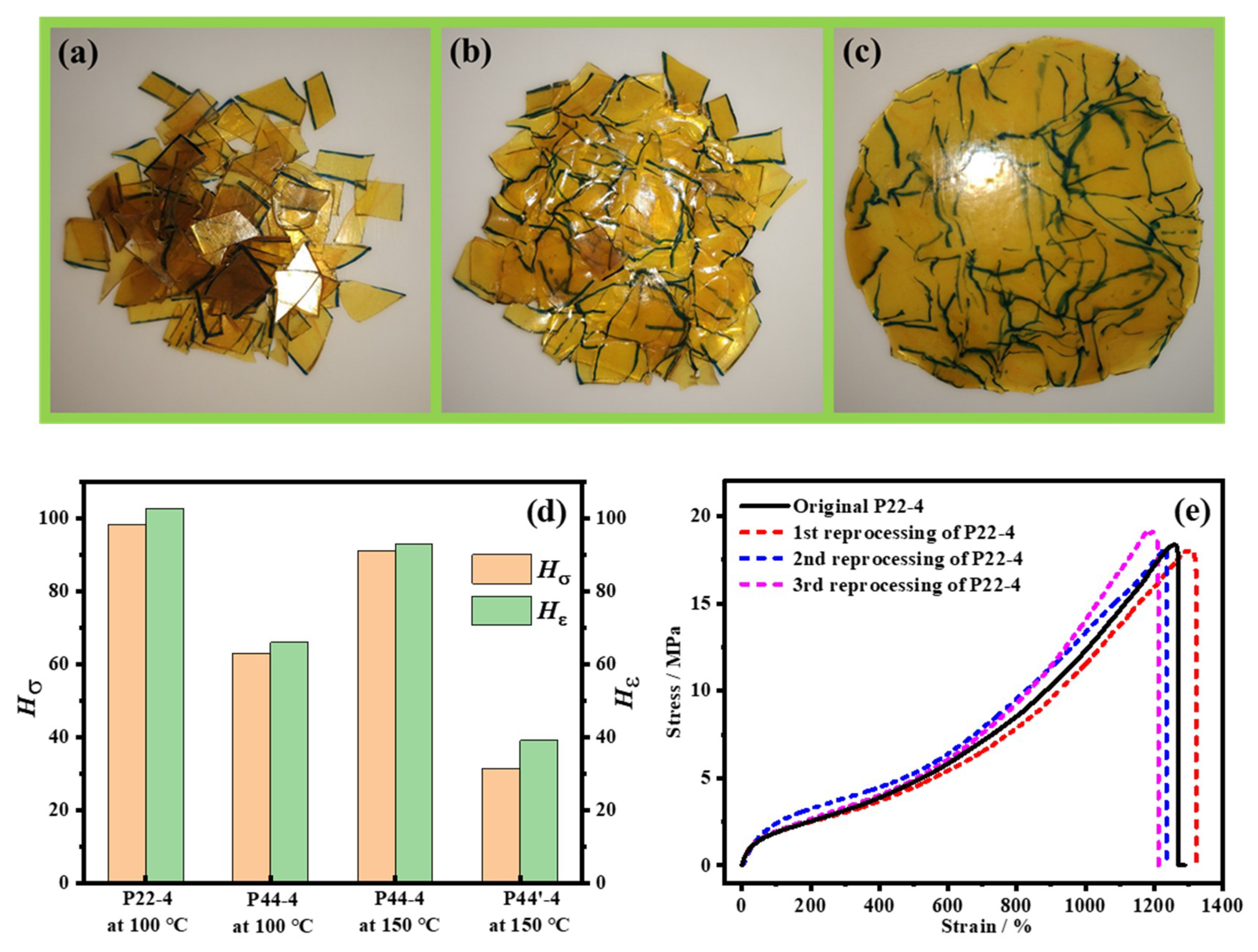
4.5. Reprocessability of WPUR Films
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Ding, X.C.; Urban, M.W. Chemical and physical aspects of self-healing materials. Prog. Polym. Sci. 2015, 49–50, 34–59. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Rong, M.Z. Intrinsic self-healing of covalent polymers through bond reconnection towards strength restoration. Polym. Chem. 2013, 4, 4878–4884. [Google Scholar] [CrossRef]
- Billiet, S.; Hillewaere, X.K.; Teixeira, R.F.; Du Prez, F.E. Chemistry of crosslinking processes for self-healing polymers. Macromol. Rapid Commun. 2013, 34, 290–309. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Mangun, C.L.; Griffin, A.S.; Moore, J.S.; Sottos, N.R.; White, S.R. Thermally stable autonomic healing in epoxy using a dual-microcapsule system. Adv. Mater. 2014, 26, 282–287. [Google Scholar] [CrossRef] [PubMed]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Toohey, K.S.; Sottos, N.R.; Lewis, J.A.; Moore, J.S.; White, S.R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hao, C.; Wang, L.W.; Li, Y.Z.; Liu, W.C.; Xin, J.N.; Zhang, J.W. Eugenol-Derived Biobased Epoxy: Shape Memory, Repairing, and Recyclability. Macromolecules 2017, 50, 8588–8597. [Google Scholar] [CrossRef]
- Banerjee, S.; Tripathy, R.; Cozzens, D.; Nagy, T.; Keki, S.; Zsuga, M.; Faust, R. Photoinduced smart, self-healing polymer sealant for photovoltaics. ACS Appl. Mater. Interfaces 2015, 7, 2064–2072. [Google Scholar] [CrossRef]
- Kaur, G.; Johnston, P.; Saito, K. Photo-reversible dimerisation reactions and their applications in polymeric systems. Polym. Chem. 2014, 5, 2171–2186. [Google Scholar] [CrossRef]
- Xia, N.N.; Rong, M.Z.; Zhang, M.Q. Stabilization of catechol–boronic ester bonds for underwater self-healing and recycling of lipophilic bulk polymer in wider pH range. J. Mater. Chem. A 2016, 4, 14122–14131. [Google Scholar] [CrossRef]
- Deng, G.; Li, F.; Yu, H.; Liu, F.; Liu, C.; Sun, W.; Jiang, H.; Chen, Y. Dynamic Hydrogels with an Environmental Adaptive Self-Healing Ability and Dual Responsive Sol–Gel Transitions. ACS Macro. Lett. 2012, 1, 275–279. [Google Scholar] [CrossRef]
- Ghosh, S.K. Self-healing Materials: Fundamentals, Resign Strategies and Applications; Wiley-VCH: Hoboken, NJ, USA, 2009. [Google Scholar]
- Feula, A.; Pethybridge, A.; Giannakopoulos, I.; Tang, X.; Chippindale, A.; Siviour, C.R.; Buckley, C.P.; Hamley, I.W.; Hayes, W. A Thermoreversible Supramolecular Polyurethane with Excellent Healing Ability at 45 °C. Macromolecules 2015, 48, 6132–6141. [Google Scholar] [CrossRef]
- Kang, J.; Son, D.; Wang, G.N.; Liu, Y.; Lopez, J.; Kim, Y.; Oh, J.Y.; Katsumata, T.; Mun, J.; Lee, Y.; et al. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Mater. 2018, 30, 1706846–1706854. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Nan, Y.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2018, 359, 72–76. [Google Scholar] [CrossRef]
- Chen, Y.; Kushner, A.M.; Williams, G.A.; Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef]
- Yang, J.X.; Long, Y.Y.; Pan, L.; Men, Y.F.; Li, Y.S. Spontaneously Healable Thermoplastic Elastomers Achieved through One-Pot Living Ring-Opening Metathesis Copolymerization of Well-Designed Bulky Monomers. ACS Appl. Mater. Interfaces 2016, 8, 12445–12455. [Google Scholar] [CrossRef]
- Xia, N.N.; Xiong, X.M.; Wang, J.; Rong, M.Z.; Zhang, M.Q. A seawater triggered dynamic coordinate bond and its application for underwater self-healing and reclaiming of lipophilic polymer. Chem. Sci. 2016, 7, 2736–2742. [Google Scholar] [CrossRef]
- Xia, N.N.; Xiong, X.M.; Rong, M.Z.; Zhang, M.Q.; Kong, F. Self-Healing of Polymer in Acidic Water toward Strength Restoration through the Synergistic Effect of Hydrophilic and Hydrophobic Interactions. ACS Appl. Mater. Interfaces 2017, 9, 37300–37309. [Google Scholar] [CrossRef]
- Li, C.H.; Wang, C.; Keplinger, C.; Zuo, J.L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C.; et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.L.; Chortos, A.; Pfattner, R.; Lissel, F.; Chiu, Y.C.; Feig, V.; Xu, J.; Kurosawa, T.; Gu, X.; Wang, C.; et al. Stretchable Self-Healing Polymeric Dielectrics Cross-Linked Through Metal-Ligand Coordination. J. Am. Chem. Soc. 2016, 138, 6020–6027. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cao, L.; Lin, B.; Liang, X.; Chen, Y. Design of Self-Healing Supramolecular Rubbers by Introducing Ionic Cross-Links into Natural Rubber via a Controlled Vulcanization. ACS Appl. Mater. Interfaces 2016, 8, 17728–17737. [Google Scholar] [CrossRef]
- Liu, J.; Liu, J.; Wang, S.; Huang, J.; Wu, S.W.; Tang, Z.H.; Guo, B.C.; Zhang, L.Q. An advanced elastomer with an unprecedented combination of excellent mechanical properties and high self-healing capability. J. Mater. Chem. A 2017, 5, 25660–25671. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Xie, P.; Rong, M.Z.; Zhang, M.Q. Catalyst-free dynamic exchange of aromatic Schiff base bonds and its application to self-healing and remolding of crosslinked polymers. J. Mater. Chem. A 2015, 3, 19662–19668. [Google Scholar] [CrossRef]
- Imato, K.; Nishihara, M.; Kanehara, T.; Amamoto, Y.; Takahara, A.; Otsuka, H. Self-healing of chemical gels cross-linked by diarylbibenzofuranone-based trigger-free dynamic covalent bonds at room temperature. Angew. Chem. Int. Ed. Engl. 2012, 51, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Imato, K.; Takahara, A.; Otsuka, H. Self-Healing of a Cross-Linked Polymer with Dynamic Covalent Linkages at Mild Temperature and Evaluation at Macroscopic and Molecular Levels. Macromolecules 2015, 48, 5632–5639. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Cromwell, O.R.; Chung, J.; Guan, Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137, 6492–6495. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Das, A.; Sallat, A.; Bohme, F.; Suckow, M.; Basu, D.; Wiessner, S.; Stockelhuber, K.W.; Voit, B.; Heinrich, G. Ionic Modification Turns Commercial Rubber into a Self-Healing Material. ACS Appl. Mater. Interfaces 2015, 7, 20623–20630. [Google Scholar] [CrossRef]
- Qiu, T.; Wang, X.J.; Lin, X.Y.; Zhu, Z.Q.; Li, X.Y.; Guo, L.H. Emulsion polymerization to synthesize self-healing films toward healing on fractures: A feasible strategy. J. Polym. Sci. Pol. Chem. 2016, 54, 3071–3078. [Google Scholar] [CrossRef]
- Lafont, U.; van Zeijl, H.; van der Zwaag, S. Influence of cross-linkers on the cohesive and adhesive self-healing ability of polysulfide-based thermosets. ACS Appl. Mater. Interfaces 2012, 4, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qiu, T.; Zhu, Z.; Guo, L.; Li, X. Self-Healing Polycaprolactone Networks through Thermo-Induced Reversible Disulfide Bond Formation. Macromol. Rapid. Commun. 2018, 39, 1800121–1800125. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, L.; Zhao, X.; Hou, S.; Guo, B.; Ma, P.X. Self-healing supramolecular bioelastomers with shape memory property as a multifunctional platform for biomedical applications via modular assembly. Biomaterials 2016, 104, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1705145–1705153. [Google Scholar] [CrossRef]
- Li, K.; Xu, Z.P.; Zhao, S.F.; Meng, X.Y.; Zhang, R.L.; Li, J.H.; Leng, J.F.; Zhang, G.P.; Cao, D.X.; Sun, R. Biomimetic, recyclable, highly stretchable and self-healing conductors enabled by dual reversible bonds. Chem. Eng. J. 2019, 371, 203–212. [Google Scholar] [CrossRef]
- Deng, J.; Kuang, X.; Liu, R.; Ding, W.; Wang, A.C.; Lai, Y.C.; Dong, K.; Wen, Z.; Wang, Y.; Wang, L.; et al. Vitrimer Elastomer-Based Jigsaw Puzzle-Like Healable Triboelectric Nanogenerator for Self-Powered Wearable Electronics. Adv. Mater. 2018, 30, 1705918–1705927. [Google Scholar] [CrossRef]
- Honarkar, H. Waterborne polyurethanes: A review. J. Dispers. Sci. Technol. 2017, 39, 507–516. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 3218–3226. [Google Scholar] [CrossRef]
- Rivero, G.; Nguyen, L.T.T.; Hillewaere, X.K.D.; Du Prez, F.E. One-Pot Thermo-Remendable Shape Memory Polyurethanes. Macromolecules 2014, 47, 2010–2018. [Google Scholar] [CrossRef]
- Nguyen, L.T.T.; Truong, T.T.; Nguyen, H.T.; Le, L.; Nguyen, V.Q.; Van Le, T.; Luu, A.T. Healable shape memory (thio)urethane thermosets. Polym. Chem. 2015, 6, 3143–3154. [Google Scholar] [CrossRef]
- Fang, Y.L.; Du, X.S.; Jiang, Y.X.; Du, Z.L.; Pan, P.T.; Cheng, X.; Wang, H.B. Thermal-Driven Self-Healing and Recyclable Waterborne Polyurethane Films Based on Reversible Covalent Interaction. ACS Sustain. Chem. Eng. 2018, 6, 14490–14500. [Google Scholar] [CrossRef]
- Ling, J.; Rong, M.Z.; Zhang, M.Q. Photo-stimulated self-healing polyurethane containing dihydroxyl coumarin derivatives. Polymer 2012, 53, 2691–2698. [Google Scholar] [CrossRef]
- Aguirresarobe, R.H.; Martin, L.; Aramburu, N.; Irusta, L.; Fernandez-Berridi, M.J. Coumarin based light responsive healable waterborne polyurethanes. Prog. Org. Coat. 2016, 99, 314–321. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Visible-Light-Induced Self-Healing Diselenide-Containing Polyurethane Elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef]
- Pepels, M.; Filot, I.; Klumperman, B.; Goossens, H. Self-healing systems based on disulfide–thiol exchange reactions. Polym. Chem. 2013, 4, 4955–4965. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; de Luzuriaga, A.R.; Cabanero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Xu, Y.R.; Chen, D.J. A Novel Self-Healing Polyurethane Based on Disulfide Bonds. Macromol. Chem. Phys. 2016, 217, 1191–1196. [Google Scholar] [CrossRef]
- Yang, Y.L.; Lu, X.; Wang, W.W. A tough polyurethane elastomer with self-healing ability. Mater. Design 2017, 127, 30–36. [Google Scholar] [CrossRef]
- Jian, X.X.; Hu, Y.W.; Zhou, W.L.; Xiao, L.Q. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Lai, Y.; Kuang, X.; Zhu, P.; Huang, M.; Dong, X.; Wang, D. Colorless, Transparent, Robust, and Fast Scratch-Self-Healing Elastomers via a Phase-Locked Dynamic Bonds Design. Adv. Mater. 2018, 30, 1802556–1802563. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Li, J.H.; Zhang, G.P.; Sun, R.; Wong, C.P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of a robust, transparent and yellowing-resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Nevejans, S.; Ballard, N.; Rivilla, I.; Fernandez, M.; Santamaria, A.; Reck, B.; Asua, J.M. Synthesis of mechanically strong waterborne poly(urethane-urea)s capable of self-healing at elevated temperatures. Eur. Polym. J. 2019, 112, 411–422. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, F.; Luo, Y. Self-Healing Mechanism of Microcracks on Waterborne Polyurethane with Tunable Disulfide Bond Contents. ACS Omega 2019, 4, 1703–1714. [Google Scholar] [CrossRef]
- Wan, T.; Chen, D.J. Mechanical enhancement of self-healing waterborne polyurethane by graphene oxide. Prog. Org. Coatings 2018, 121, 73–79. [Google Scholar] [CrossRef]
- Wan, T.; Chen, D. Preparation of β-cyclodextrin reinforced waterborne polyurethane nanocomposites with excellent mechanical and self-healing property. Compos. Sci. Technol. 2018, 168, 55–62. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; de Luzuriaga, A.R.; Cabanero, G.; Grande, H.J.; Odriozola, I. The processability of a poly(urea-urethane) elastomer reversibly crosslinked with aromatic disulfide bridges. J. Mater. Chem. A 2014, 2, 5710–5715. [Google Scholar] [CrossRef]
- Formoso, E.; Asua, J.M.; Matxain, J.M.; Ruiperez, F. The role of non-covalent interactions in the self-healing mechanism of disulfide-based polymers. Phys. Chem. Chem. Phys. 2017, 19, 18461–18470. [Google Scholar] [CrossRef] [PubMed]
- Nevejans, S.; Ballard, N.; Miranda, J.I.; Reck, B.; Asua, J.M. The underlying mechanisms for self-healing of poly(disulfide)s. Phys. Chem. Chem. Phys. 2016, 18, 27577–27583. [Google Scholar] [CrossRef] [PubMed]
- Matxain, J.M.; Asua, J.M.; Ruiperez, F. Design of new disulfide-based organic compounds for the improvement of self-healing materials. Phys. Chem. Chem. Phys. 2016, 18, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.W. DLVO Theory; Springer: Berlin/Heidelberg, Germany, 2011; p. 238. [Google Scholar] [CrossRef]
- He, Y.; Xie, D.; Zhang, X. The structure, microphase-separated morphology, and property of polyurethanes and polyureas. J. Mater. Sci. 2014, 49, 7339–7352. [Google Scholar] [CrossRef]
- Aguirresarobe, R.H.; Martin, L.; Fernandez-Berridi, M.J.; Irusta, L. Autonomic healable waterborne organic-inorganic polyurethane hybrids based on aromatic disulfide moieties. Express Polym. Lett. 2017, 11, 266–277. [Google Scholar] [CrossRef]
- Fritze, U.F.; von Delius, M. Dynamic disulfide metathesis induced by ultrasound. Chem. Commun. (Camb) 2016, 52, 6363–6366. [Google Scholar] [CrossRef]
- Engle, L.P.; Wagener, K.B. A Review of Thermally Controlled Covalent Bond Formation in Polymer Chemistry. J. Macromol. Sci. Part C 1993, 33, 239–257. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IPDI | PTMEG | DMBA | 22DTDA(a) | DBTDA | TEA | Water | TETA |
|---|---|---|---|---|---|---|---|---|
| /g (mmol) | /g (mmol) | /g (mmol) | /g (mmol) | /g (mmol) | /g (mmol) | /g (mol) | /g (mmol) | |
| P-0 | 3 (13.5) | 12.77 (6.39) | 0.87 (5.88) | 0 (0) | 0.09 (0.14) | 0.59 (5.88) | 52.24 (2.9) | 0.09 (0.62) |
| P22-2 | 3 (13.5) | 11.16 (5.88) | 0.80 (5.41) | 0.32 (1.29) | 0.08 (0.13) | 0.55 (5.41) | 47.97 (2.67) | 0.09 (0.62) |
| P44-2 | 3 (13.5) | 11.16 (5.88) | 0.80 (5.41) | 0.32 (1.29) | 0.08 (0.13) | 0.55 (5.41) | 47.97 (2.67) | 0.09 (0.62) |
| P22-4 | 3 (13.5) | 9.79 (4.9) | 0.74 (5) | 0.59 (2.38) | 0.07 (0.11) | 0.50 (5) | 44.35 (2.46) | 0.09 (0.62) |
| P44-4 | 3 (13.5) | 9.79 (4.9) | 0.74 (5) | 0.59 (2.38) | 0.07 (0.11) | 0.50 (5) | 44.35 (2.46) | 0.09 (0.62) |
| P44′-4 | 3 (13.5) | 9.79 (4.9) | 0.74 (5) | 0.59 (2.94) | 0.07 (0.11) | 0.50 (5) | 44.35 (2.46) | 0.09 (0.62) |
| P22-6 | 3 (13.5) | 8.61 (4.31) | 0.69 (4.66) | 0.83 (3.34) | 0.07 (0.11) | 0.47 (4.66) | 41.24 (2.29) | 0.09 (0.62) |
| P22-8 | 3 (13.5) | 7.58 (3.79) | 0.64 (4.32) | 1.03 (4.15) | 0.06 (0.1) | 0.44 (4.32) | 38.53 (2.14) | 0.09 (0.62) |
| P22-10 | 3 (13.5) | 6.69 (3.35) | 0.60 (4.05) | 1.21 (4.87) | 0.06 (0.1) | 0.41 (4.05) | 36.16 (2) | 0.09 (0.62) |
| No. | E0 /MPa | σ0 /MPa | ε0 /% | σ50 /MPa | ε50 /% | Hσ50 /% | Hε50 /% | σ37 /MPa | ε37 /% | Hσ37 /% | Hε37 /% |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P-0 | 0.57 | 0.16 | 1205 | 0.14 | 1154 | 85.2 | 95.8 | 0.14 | 1142 | 85.3 | 94.8 |
| P22-2 | 1.74 | 2.58 | 1439 | 2.24 | 1393 | 86.8 | 96.8 | 2.08 | 1386 | 80.5 | 96.3 |
| P22-4 | 6.51 | 18.4 | 1260 | 15.4 | 1215 | 83.4 | 96.4 | 13.8 | 1150 | 75.2 | 91.3 |
| P22-6 | 12.1 | 20.3 | 1173 | 12.5 | 917 | 61.8 | 78.2 | 6.60 | 572 | 32.0 | 50.8 |
| P22-8 | 15.0 | 22.1 | 1102 | 8.84 | 479 | 40.0 | 43.5 | 3.51 | 71 | 15.9 | 6.4 |
| P22-10 | 24.9 | 30.0 | 1011 | 9.57 | 304 | 31.9 | 30.1 | 1.95 | 9 | 6.5 | 0.9 |
| P44-2 | 2.90 | 8.06 | 1449 | 2.97 | 514 | 36.8 | 35.5 | 1.93 | 290 | 23.9 | 20.0 |
| P44-4 | 9.59 | 23.7 | 1155 | 3.10 | 133 | 13.1 | 11.5 | 2.39 | 135 | 10.1 | 11.7 |
| P44′-4 | 9.8 | 26.9 | 1053 | 1.69 | 29 | 6.3 | 2.7 | 0 | 0 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Qiu, T.; Sun, X.; Guo, L.; He, L.; Ye, J.; Li, X. Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers. Polymers 2020, 12, 989. https://doi.org/10.3390/polym12040989
Zhang L, Qiu T, Sun X, Guo L, He L, Ye J, Li X. Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers. Polymers. 2020; 12(4):989. https://doi.org/10.3390/polym12040989
Chicago/Turabian StyleZhang, Liangdong, Teng Qiu, Xiting Sun, Longhai Guo, Lifan He, Jun Ye, and Xiaoyu Li. 2020. "Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers" Polymers 12, no. 4: 989. https://doi.org/10.3390/polym12040989
APA StyleZhang, L., Qiu, T., Sun, X., Guo, L., He, L., Ye, J., & Li, X. (2020). Achievement of Both Mechanical Properties and Intrinsic Self-Healing under Body Temperature in Polyurethane Elastomers: A Synthesis Strategy from Waterborne Polymers. Polymers, 12(4), 989. https://doi.org/10.3390/polym12040989