
Highly Stretchable Fully Biomass Autonomic Self-Healing Polyamide Elastomers and Their Foam for Selective Oil Absorption



Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
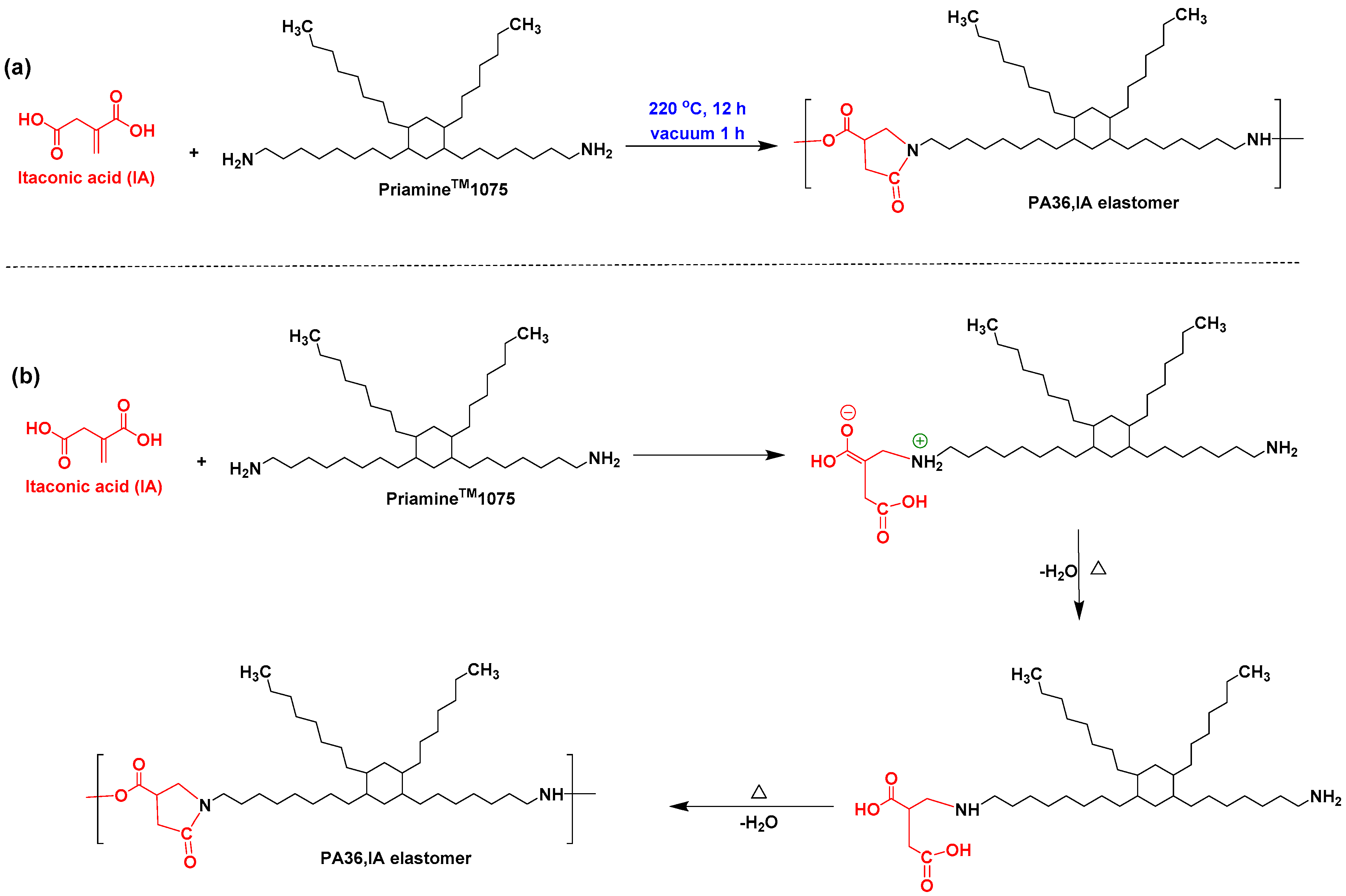
2.2. Synthesis of PA36,IA Thermoplastic Elastomer
2.3. Supercritical Carbon Dioxide (scCO2) Batch Foaming Process
2.4. Characterization
3. Results and Discussion
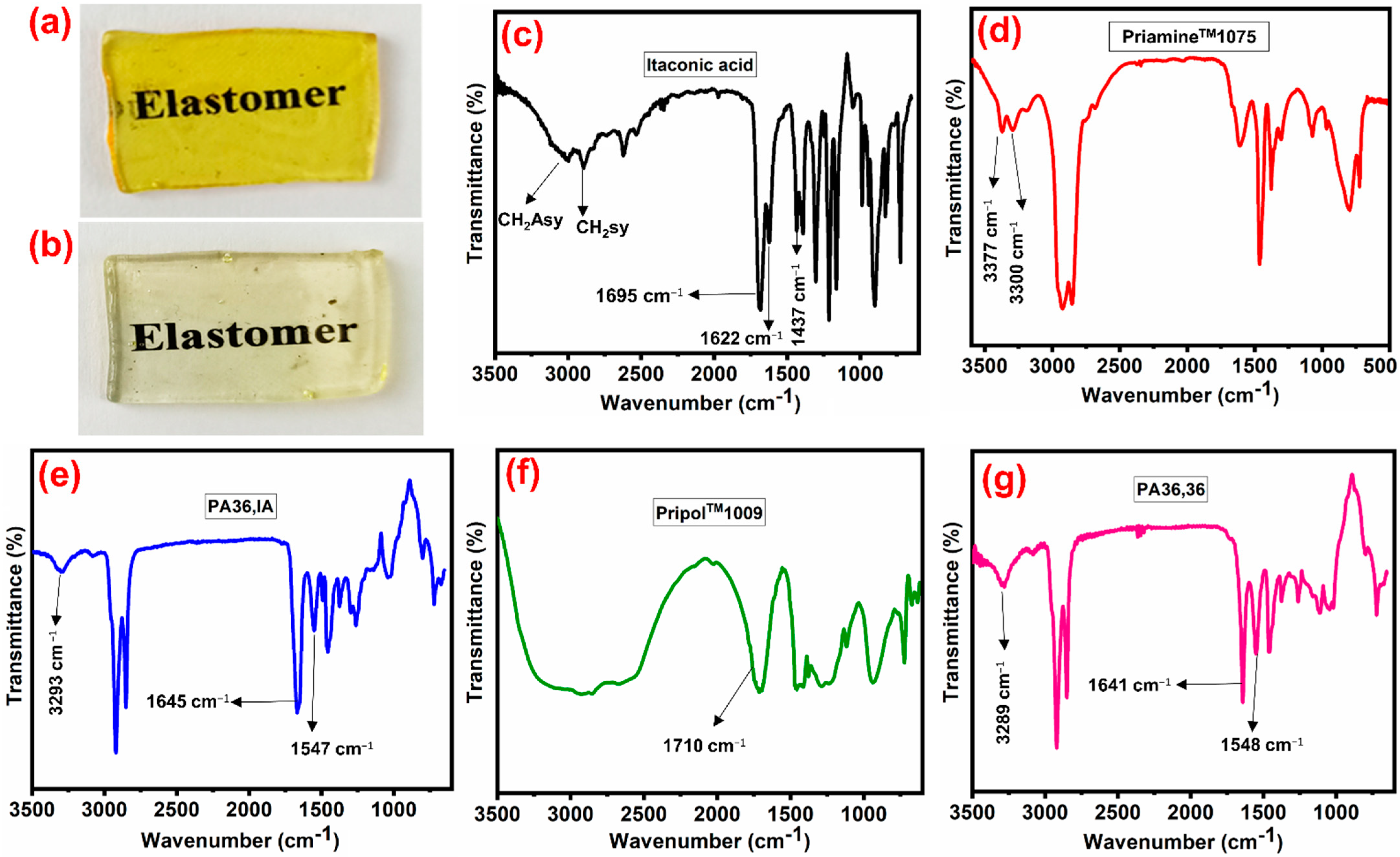
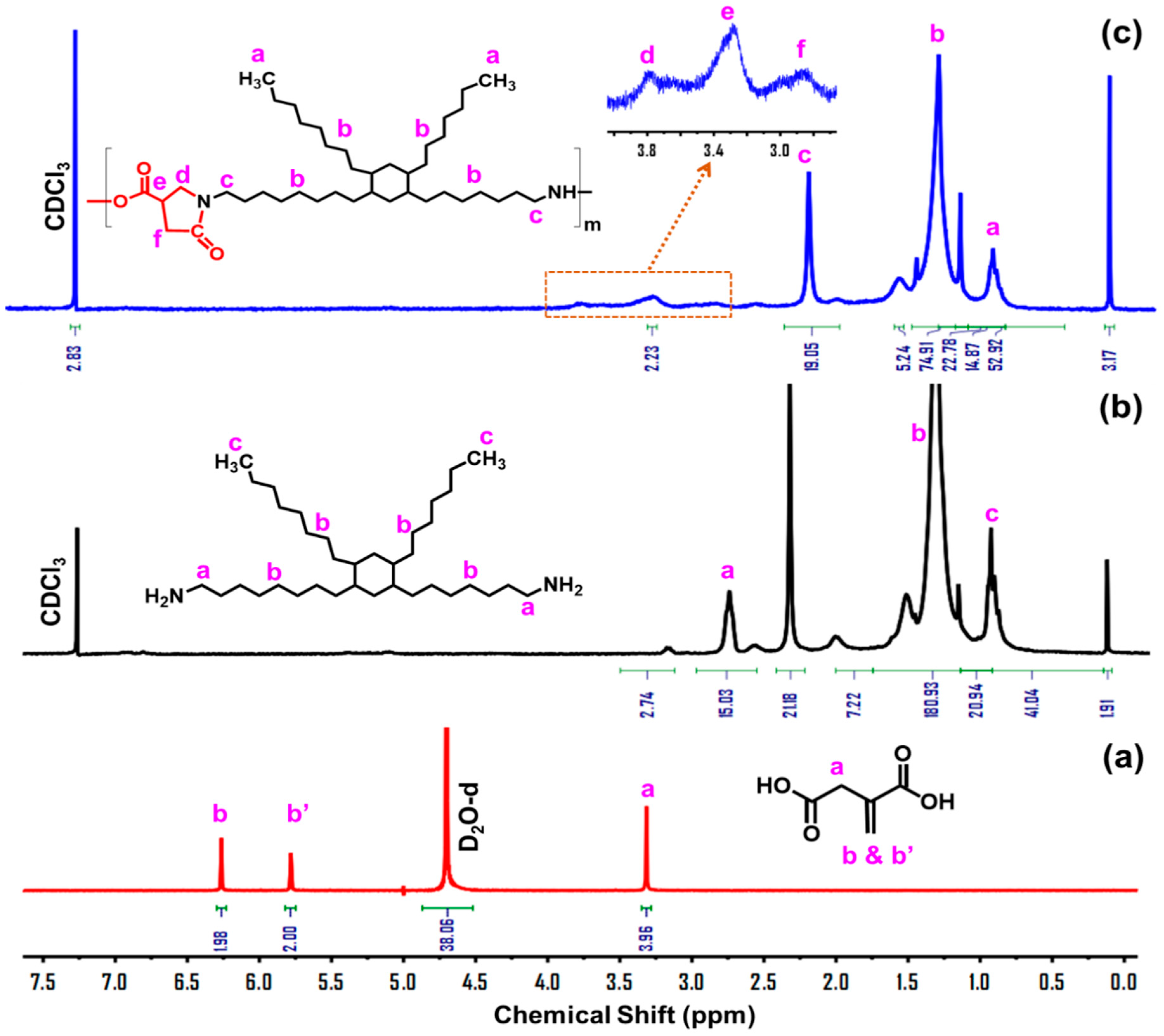
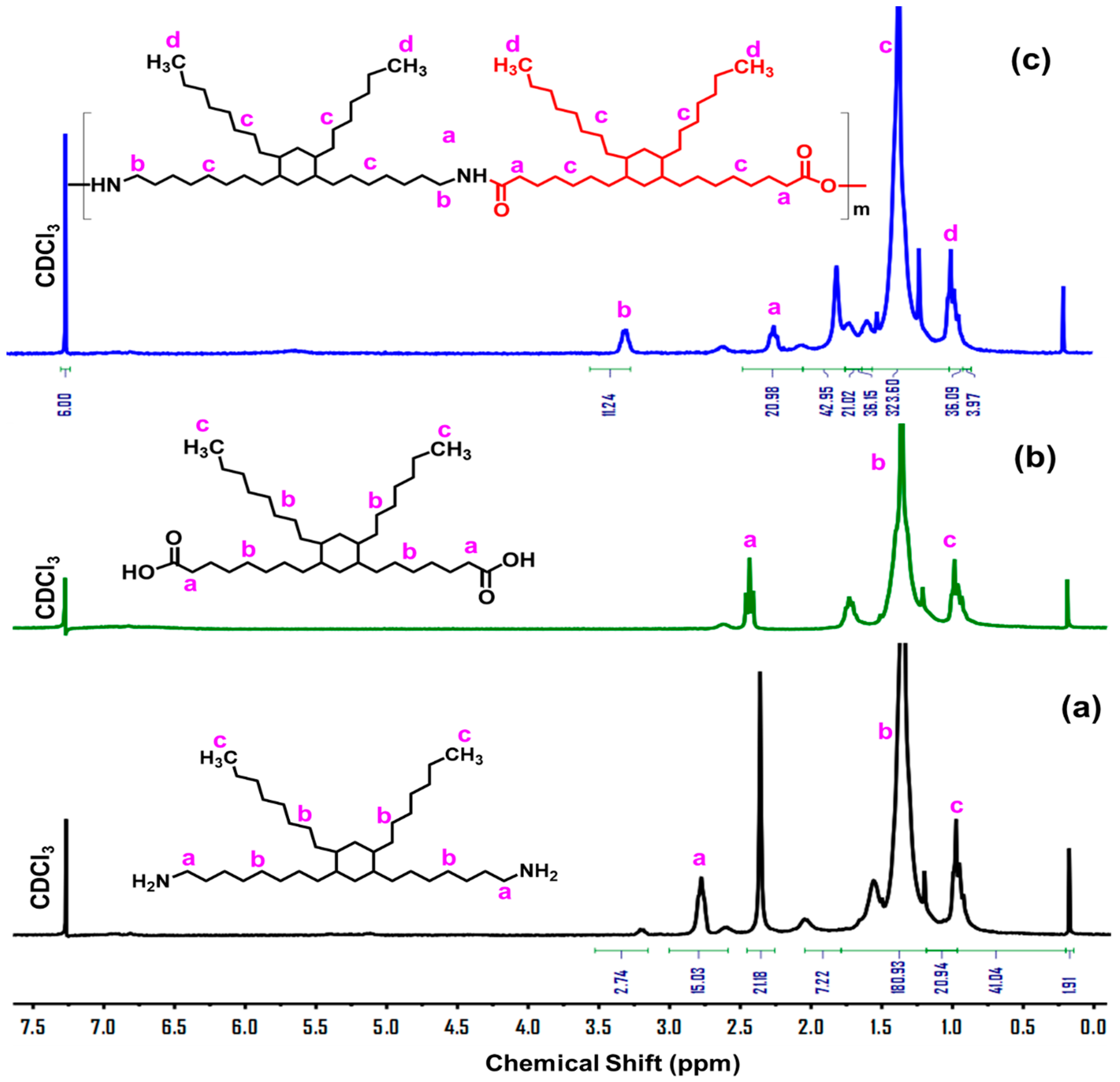
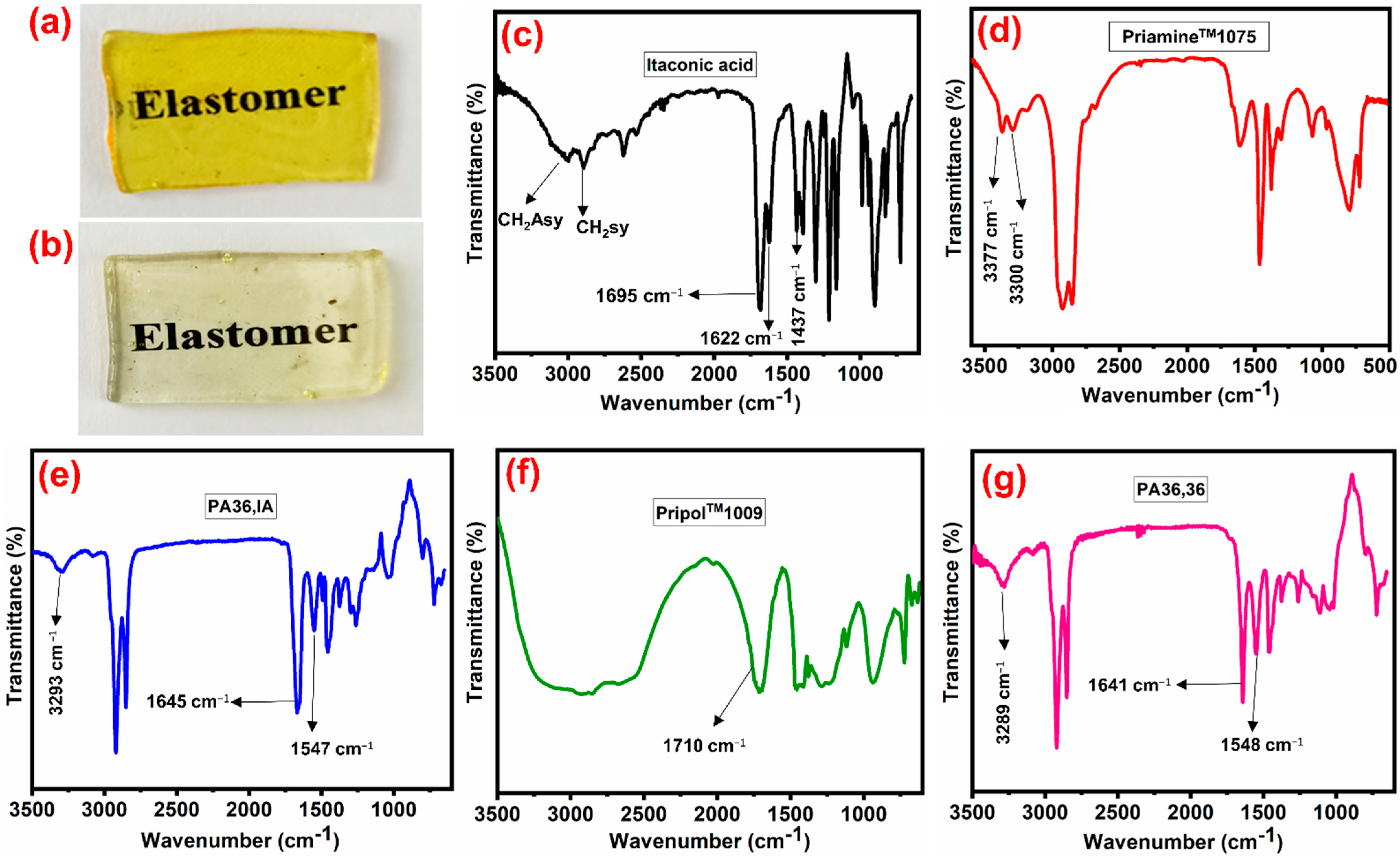
3.1. Design, Synthesis, and Structure Characterization of the Bio-Based Thermoplastic PA Elastomers
3.2. Thermal Properties
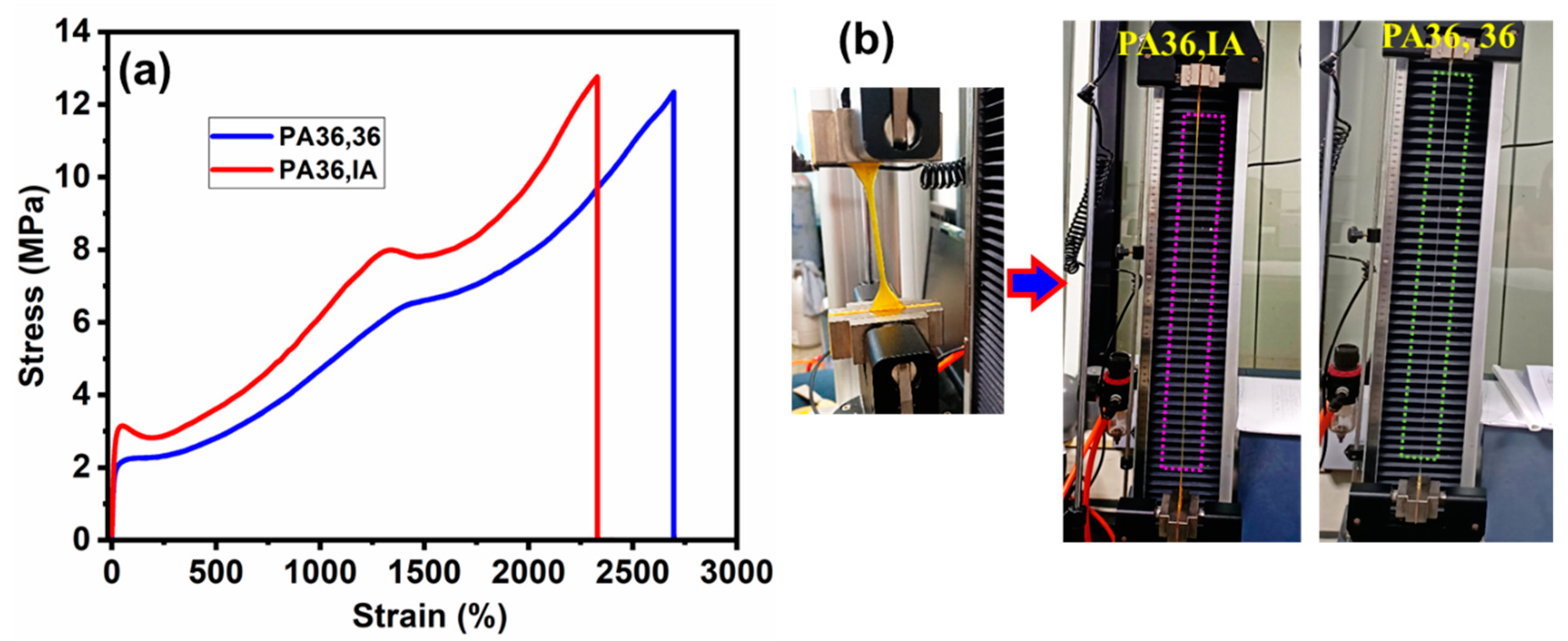
3.3. Mechanical Properties
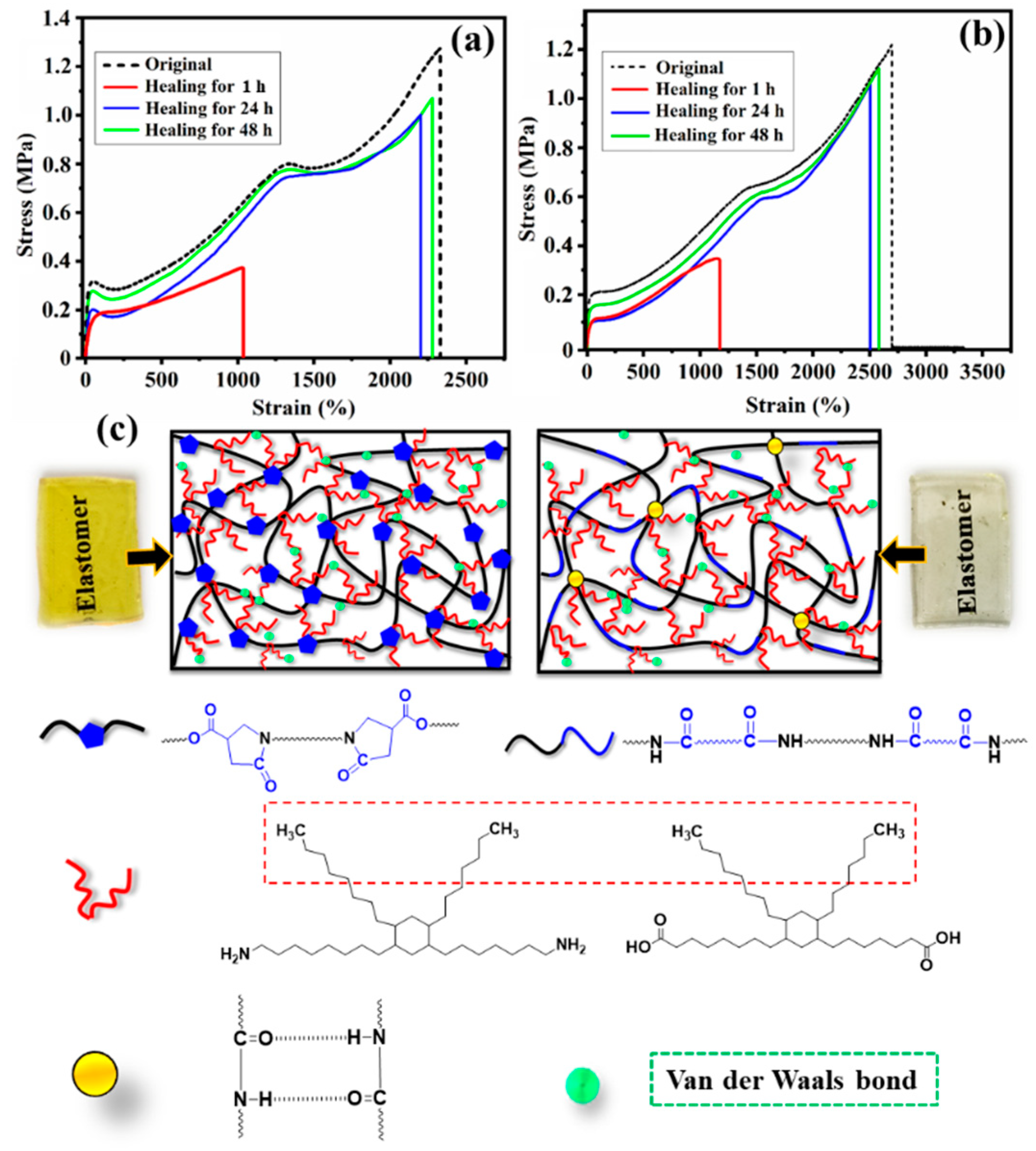
3.4. Self-Healing Properties of the Bio-Based PA Elastomers
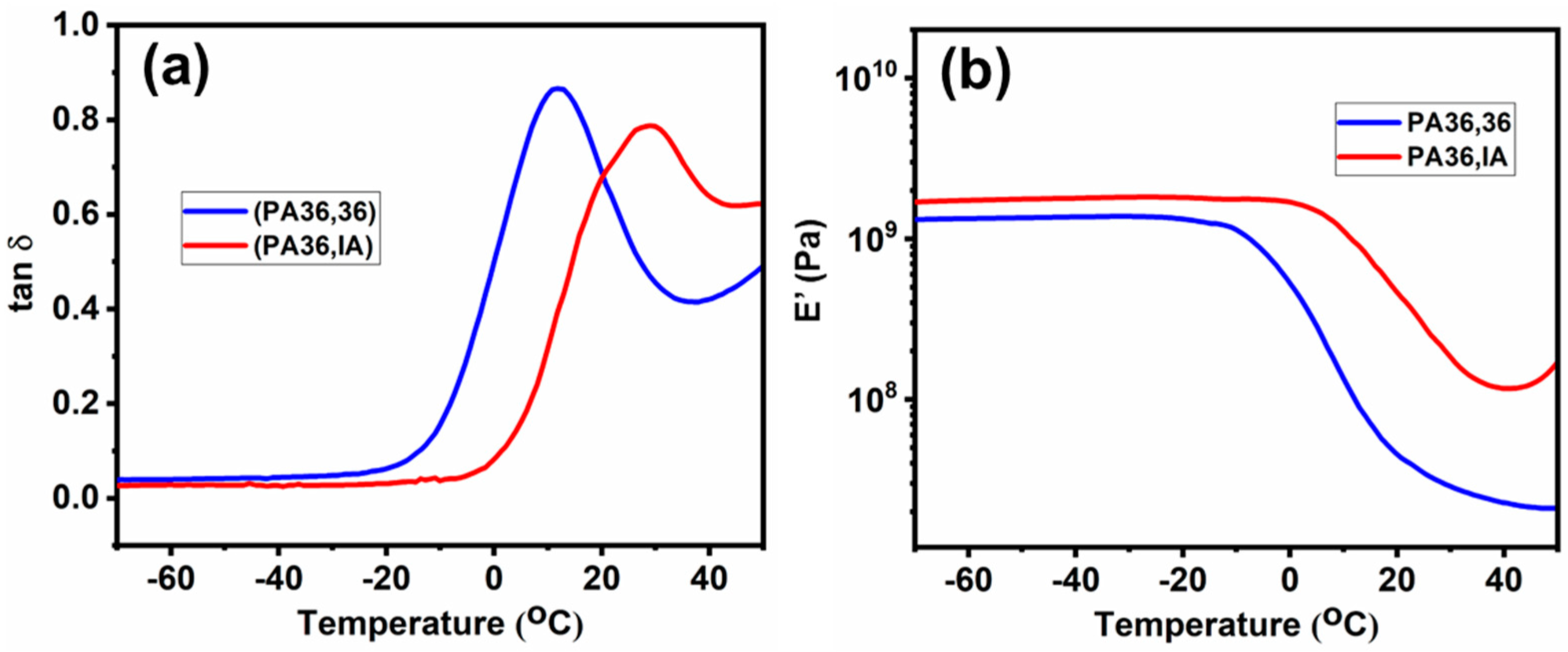
3.5. Viscoelastic Properties
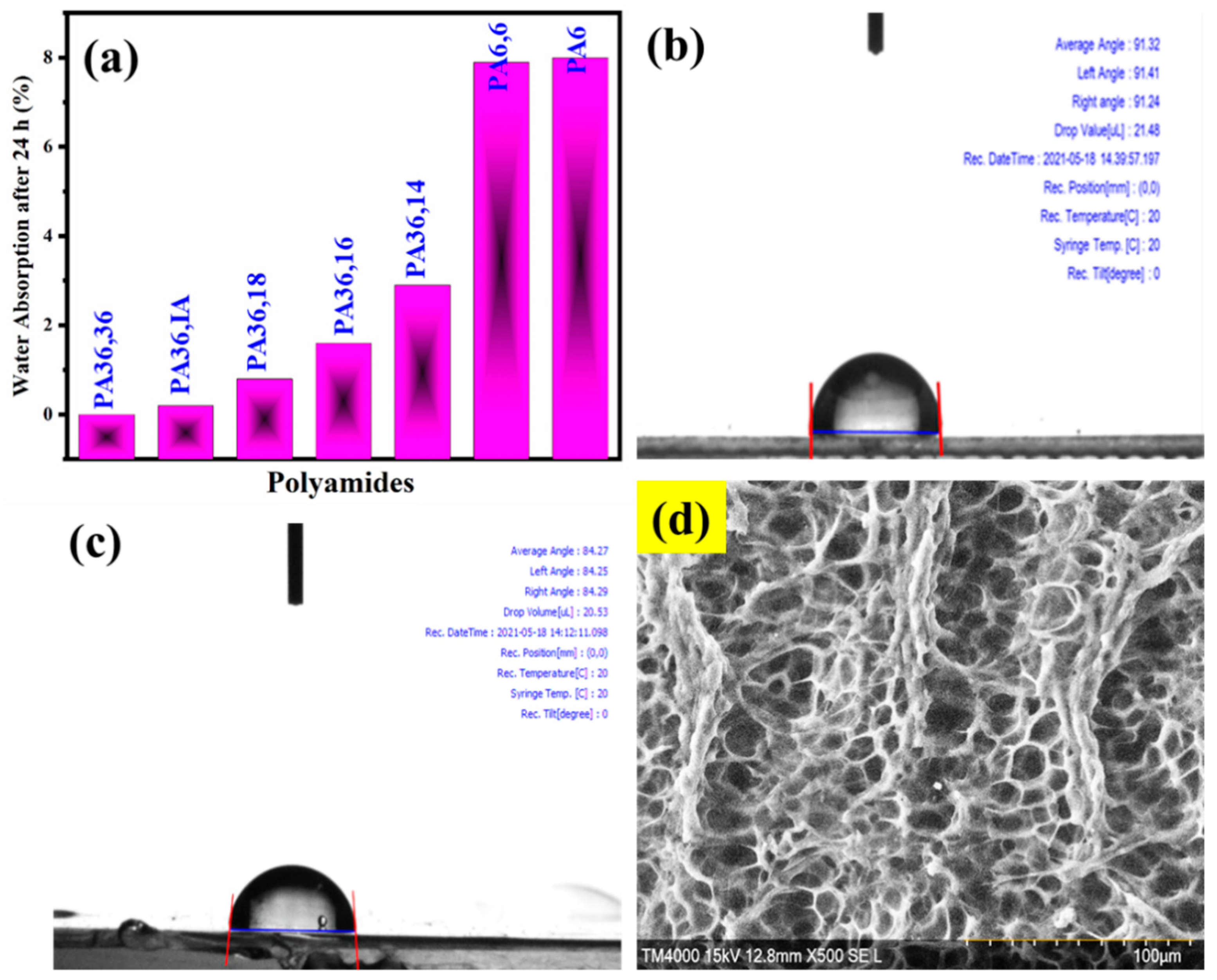
3.6. Water Uptake Study
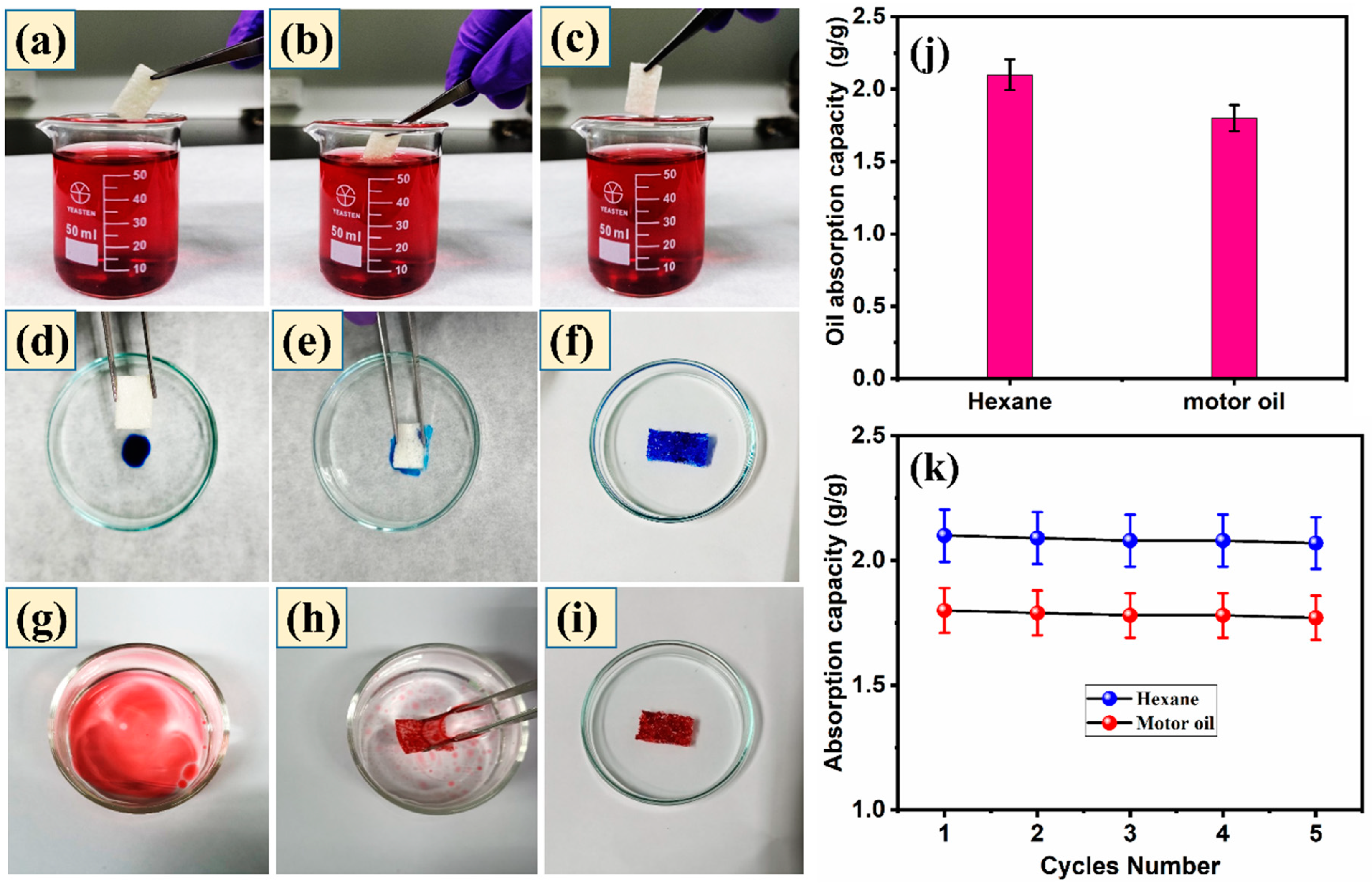
3.7. Absorption Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Tondi, G.; Schnabel, T. Bio-Based Polymers for Engineered Green Materials. Polymers 2020, 12, 775. [Google Scholar] [CrossRef] [Green Version]
- Blondiaux, E.; Bomon, J.; Smolen, M.; Kaval, N.; Lemiere, F.; Sergeyev, S.; Diels, L.; Sels, B.F.; Maes, B.U.W. Bio-based Aromatic Amines from Lignin-Derived Monomers. ACS Sustain. Chem. Eng. 2019, 7, 6906–6916. [Google Scholar] [CrossRef]
- Nakajima, H.; Dijkstra, P.; Loos, K. The Recent Developments in Biobased Polymers Toward General and Engineering Ap-plications: Polymers that are Upgraded from Biodegradable Polymers, Analogous to Petroleum-Derived Polymers, and Newly Developed. Polymers 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A. Sustainable polymers: Replacing polymers derived from fossil fuels. Polym. Chem. 2014, 5, 3117–3118. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, J.; Zhang, L.; Yue, D. Bio-Based Self-Healing Eucommia Ulmoides Ester Elastomer with Damping and Oil Re-sistance. J. Mater. Sci. 2020, 55, 4940–4951. [Google Scholar] [CrossRef]
- Raut, S.K.; Mondal, P.; Parameswaran, B.; Sarkar, S.; Dey, P.; Gilbert, R.; Bhadra, S.; Naskar, K.; Nair, S.; Singha, N.K. Self-healable ultrahydrophobic modified bio-based elastomer using Diels-Alder ‘click chemistry’. Eur. Polym. J. 2021, 146, 110204. [Google Scholar] [CrossRef]
- Lei, W.; Russell, T.P.; Hu, L.; Zhou, X.; Qiao, H.; Wang, W.; Wang, R.; Zhang, L. Pendant Chain Effect on the Synthesis, Characterization, and Structure–Property Relations of Poly(di-n-alkyl itaconate-co-isoprene) Biobased Elastomers. ACS Sustain. Chem. Eng. 2017, 5, 5214–5223. [Google Scholar] [CrossRef]
- Yoda, R. Elastomers for biomedical applications. J. Biomater. Sci. Polym. Ed. 1998, 9, 561–626. [Google Scholar] [CrossRef]
- Wei, T.; Lei, L.; Kang, H.; Qiao, B.; Wang, Z.; Zhang, L.; Coates, P.; Hua, K.C.; Kulig, J. Tough Bio-Based Elastomer Nano-composites with High Performance for Engineering Applications. Adv. Eng. Mater. 2012, 14, 112–118. [Google Scholar] [CrossRef]
- Koenig, J.L. The Chemical Reactions of Network Structures in Elastomers. Accounts Chem. Res. 1999, 32, 1–8. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nat. Cell Biol. 2008, 451, 977–980. [Google Scholar] [CrossRef]
- Amaral, A.J.R.; Pasparakis, G. Stimuli responsive self-healing polymers: Gels, elastomers and membranes. Polym. Chem. 2017, 8, 6464–6484. [Google Scholar] [CrossRef] [Green Version]
- Gerratt, A.P.; Michaud, H.O.; Lacour, S.P. Elastomeric Electronic Skin for Prosthetic Tactile Sensation. Adv. Funct. Mater. 2015, 25, 2287–2295. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.; Wang, G.-J.N.; Zhu, C.; Luo, S.; Jin, L.; Gu, X.; Chen, S.; Feig, V.R.; To, J.W.F.; et al. Highly stretchable polymer semiconductor films through the nanoconfinement effect. Science 2017, 355, 59–64. [Google Scholar] [CrossRef]
- Kim, Y.; Yuk, H.; Zhao, R.; Chester, S.A.; Zhao, X. Printing ferromagnetic domains for untethered fast-transforming soft materials. Nat. Cell Biol. 2018, 558, 274–279. [Google Scholar] [CrossRef]
- Huang, S.; Liu, Y.; Zhao, Y.; Ren, Z.; Guo, C.F. Flexible Electronics: Stretchable Electrodes and Their Future. Adv. Funct. Mater. 2019, 29, 1805924. [Google Scholar] [CrossRef]
- Wu, T.; Chen, B. Synthesis of Multiwalled Carbon Nanotube-Reinforced Polyborosiloxane Nanocomposites with Mechani-cally Adaptive and Self-Healing Capabilities for Flexible Conductors. ACS Appl. Mater. Interfaces 2016, 8, 24071–24078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, M.W.; Davydovich, D.; Yang, Y.; Demir, T.; Zhang, Y.; Casabianca, L. Key-and-Lock Commodity Self-Healing Co-polymers. Science 2018, 362, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hager, M.D. Self-Healing Materials. In Handbook of Solid State Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 201–225. [Google Scholar]
- Liu, J.; Tan, C.S.; Yu, Z.; Li, N.; Abell, C.; Scherman, O.A. Tough Supramolecular Polymer Networks with Extreme Stretch-ability and Fast Room-Temperature Self-Healing. Adv. Mater. 2017, 29, 1605325. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Son, D.; Wang, G.J.; Liu, Y.; Lopez, J.; Kim, Y.; Oh, J.Y.; Katsumata, T.; Mun, J.; Lee, Y.; et al. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Mater. 2018, 30, 1706846. [Google Scholar] [CrossRef]
- Mozhdehi, D.; Ayala, S.; Cromwell, O.R.; Guan, Z. Self-Healing Multiphase Polymers via Dynamic Metal–Ligand Interactions. J. Am. Chem. Soc. 2014, 136, 16128–16131. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, L.H.; Weitz, D.A. Tough Self-Healing Elastomers by Molecular Enforced Integration of Covalent and Reversible Networks. Adv. Mater. 2017, 29, 1702616. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, R.; He, Y.; Zhang, Y.; Yang, X.; Zhao, X.; Huang, W. Self-Healing Polyurethane Elastomers Based on a Disulfide Bond by Digital Light Processing 3D Printing. ACS Macro Lett. 2019, 8, 1511–1516. [Google Scholar] [CrossRef]
- Nasresfahani, A.; Zelisko, P.M. Synthesis of a self-healing siloxane-based elastomer cross-linked via a furan-modified polyhedral oligomeric silsesquioxane: Investigation of a thermally reversible silicon-based cross-link. Polym. Chem. 2017, 8, 2942–2952. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Ranganathan, P.; Chen, C.W.; Tasi, M.C.; Rwei, S.P.; Lee, Y.H. Biomass Thermoplastic (Co) polyamide Elastomers Synthesized from a Fatty Dimer Acid: A Sustainable Route toward a New Era of Uniform and Bimodal Foams. Ind. Eng. Chem. Res. 2021, 6, 12139–12154. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, H.; Li, Y.; Meng, J.; Yao, Y.; Yao, C. Biomass polyamide elastomers based on hydrogen bonds with rapid self-healing properties. Eur. Polym. J. 2020, 133, 109802. [Google Scholar] [CrossRef]
- Zhang, R.; Yan, T.; Lechner, B.D.; Schröter, K.; Liang, Y.; Li, B.; Furtado, F.; Sun, P.; Saalwächter, K. Heterogeneity, Segmental and Hydrogen Bond Dynamics, and Aging of Supramolecular Self-Healing Rubber. Macromolecules 2013, 46, 1841–1850. [Google Scholar] [CrossRef]
- Guo, W.; Wang, X.; Lu, X.; Li, X.; Li, Y.; Sun, J. Plant oil and amino acid-derived elastomers with rapid room temperature self-healing ability. J. Mater. Chem. A 2019, 7, 21927–21933. [Google Scholar] [CrossRef]
- Pathan, J.R.; Sureshan, K.M. Solvent-Free and Catalyst-Free Synthesis of Cross-Linkable Polyfumaramides via Topochemical Azide-Alkyne Cycloaddition Polymerization. ACS Sustain. Chem. Eng. 2021, 9, 9871–9878. [Google Scholar] [CrossRef]
- Anantharaj, S.; Jayakannan, M. Polymers from Amino Acids: Development of Dual Ester-Urethane Melt Condensation Ap-proach and Mechanistic Aspects. Biomacromolecules 2012, 13, 2446–2455. [Google Scholar] [CrossRef]
- Whinfield, J.R. Chemistry of ‘Terylene’. Nature 1946, 158, 930–941. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, F.; Weng, D.; Li, X.; Li, Y.; Sun, J. Superhydrophobic Foams with Chemical- and Mechanical-Damage-Healing Abilities Enabled by Self-Healing Polymers. ACS Appl. Mater. Interfaces 2019, 11, 37285–37294. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Shen, C.; Liu, C.; Liu, X. Facile Thermally Impacted Water-Induced Phase Separation Approach for the Fabrication of Skin-Free Thermoplastic Polyurethane Foam and its Recyclable Counterpart for Oil–Water Separation. Macro-mol. Rapid Commun. 2018, 39, 1800635. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Liu, X.; Liu, H.; Li, N.; Liu, C.; Schubert, D.W.; Shen, C. Facile Fabrication of Superhydrophobic and Eco-Friendly Poly(lactic acid) Foam for Oil–Water Separation via Skin Peeling. ACS Appl. Mater. Interfaces 2019, 11, 14362–14367. [Google Scholar] [CrossRef]
- Gao, J.; Song, X.; Huang, X.; Wang, L.; Li, B.; Xue, H. Facile preparation of polymer microspheres and fibers with a hollow core and porous shell for oil adsorption and oil/water separation. Appl. Surf. Sci. 2018, 439, 394–404. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhu, Y.J.; Xiong, Z.C.; Wu, J.; Chen, F. Bioinspired Ultralight Inorganic Aerogel for Highly Efficient Air Filtration and Oil–Water Separation. ACS Appl. Mater. Interfaces 2018, 10, 13019–13027. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Samal, S.K.; Liu, Z.; Xiong, R.; De Smedt, S.C.; Bhushan, B.; Zhang, Q.; Huang, C. Dual pH-and Ammonia-Vapor-Responsive Electrospun Nanofibrous Membranes for Oil-Water Separations. J. Membr. Sci. 2017, 1, 128–139. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Wang, J.; Ren, Y.; Xuan, C.; Liu, C.; Shen, C. Superhydrophobic and superoleophilic porous reduced graphene oxide/polycarbonate monoliths for high-efficiency oil/water separation. J. Hazard. Mater. 2018, 344, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, C.; Yang, C.; Yi, C. Flexible preparation of polyamide-6 based thermoplastic elastomers via amide exchange. J. Mater. Sci. 2021, 56, 12018–12029. [Google Scholar] [CrossRef]
- Błażek, K.; Kasprzyk, P.; Datta, J. Diamine Derivatives of Dimerized Fatty Acids and Bio-Based Polyether Polyol as Sustaina-ble Platforms for the Synthesis of Non-Isocyanate Polyurethanes. Polymer 2020, 28, 122768. [Google Scholar] [CrossRef]
- Hablot, E.; Donnio, B.; Bouquey, M.; Avérous, L. Dimer acid-based thermoplastic bio-polyamides: Reaction kinetics, properties and structure. Polymer 2010, 51, 5895–5902. [Google Scholar] [CrossRef]
- Van Velthoven, J.L.; Gootjes, L.; Noordover, B.A.; Meuldijk, J. Bio-based, amorphous polyamides with tunable thermal properties. Eur. Polym. J. 2015, 66, 57–66. [Google Scholar] [CrossRef]
- Martino, L.; Basilissi, L.; Farina, H.; Ortenzi, M.A.; Zini, E.; Di Silvestro, G.; Scandola, M. Bio-based polyamide 11: Synthesis, rheology and solid-state properties of star structures. Eur. Polym. J. 2014, 59, 69–77. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D.; Lewin, M. Thermal Decomposition of Aliphatic Nylons. Polym. Int. 1999, 48, 532–557. [Google Scholar] [CrossRef]
- Herrera, M.; Matuschek, G.; Kettrup, A. Main products and kinetics of the thermal degradation of polyamides. Chemosphere 2001, 42, 601–607. [Google Scholar] [CrossRef]
- Ali, M.A.; Tateyama, S.; Oka, Y.; Kaneko, D.; Okajima, M.K.; Kaneko, T. Syntheses of High-Performance Biopolyamides De-rived from Itaconic Acid and their Environmental Corrosion. Macromolecules 2013, 46, 3719–3725. [Google Scholar] [CrossRef]
- Khatri, B.S.; Byrne, K.; Kawakami, M.; Brockwell, D.J.; Smith, D.A.; Radford, S.E.; McLeish, T.C.B. Internal friction of single polypeptide chains at high stretch. Faraday Discuss. 2008, 139, 35–51. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Spoljaric, S.; Seppälä, J. Redefining Polyamide Property Profiles via Renewable Long-Chain Aliphatic Segments: Towards Impact Resistance and Low Water Absorption. Eur. Polym. J. 2018, 109, 16–25. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, X.; Urban, M.W. Chemical and physical aspects of self-healing materials. Prog. Polym. Sci. 2015, 49–50, 34–59. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-H.; Wang, C.; Keplinger, C.; Zuo, J.-L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C.; et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Lips, P.; Broos, R.; van Heeringen, M.; Dijkstra, P.; Feijen, J. Synthesis and characterization of poly(ester amide)s containing crystallizable amide segments. Polymer 2005, 46, 7823–7833. [Google Scholar] [CrossRef]
- Haque, A.; Kurokawa, T.; Gong, J.P. Super tough double network hydrogels and their application as biomaterials. Polymer 2012, 53, 1805–1822. [Google Scholar] [CrossRef]
- Sheth, J.P.; Xu, J.; Wilkes, G.L. Solid state structure–property behavior of semicrystalline poly(ether-block-amide) PEBAX® thermoplastic elastomers. Polymer 2003, 44, 743–756. [Google Scholar] [CrossRef]
- Zeng, C.; Seino, H.; Ren, J.; Hatanaka, K.; Yoshie, N. Bio-Based Furan Polymers with Self-Healing Ability. Macromolecules 2013, 46, 1794–1802. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Oh, K.W.; Kim, S.H. Synthesis and characterization of a furan-based self-healing polymer. Macromol. Res. 2016, 24, 874–880. [Google Scholar] [CrossRef]
- Susa, A.; Bose, R.K.; Grande, A.M.; Van Der Zwaag, S.; Garcia, S.J. Effect of the Dianhydride/Branched Diamine Ratio on the Architecture and Room Temperature Healing Behavior of Polyetherimides. ACS Appl. Mater. Interfaces 2016, 8, 34068–34079. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef]
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- Wang, X.; Liu, W.; Li, H.; Du, Z.; Zhang, C. Role of maleic- anhydride-grafted- polypropylene in supercritical CO2 foaming of poly (lactic acid) and its effect on cellular morphology. J. Cell. Plast. 2014, 52, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Extrand, C. Water Contact Angles and Hysteresis of Polyamide Surfaces. J. Colloid Interface Sci. 2002, 248, 136–142. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Mw (g mol−1) | Mn (g mol−1) | Ð |
|---|---|---|---|
| PA36,IA | 30,889 | 19,262 | 1.60 |
| PA36,36 | 32,938 | 21,045 | 1.57 |
| Samples | Tdmax (°C) | Tg (°C) |
|---|---|---|
| IA | 217.0 | - |
| Priamine®1075, | 356.6 | - |
| PripolTM1009 | 385.3 | - |
| PA36,IA | 471.0 | 24.0 |
| PA36,36 | 470.7 | 11.7 |
| Sample | Strength (MPa) | Strain (%) |
|---|---|---|
| PA36,IA (original) | 1.29 ± 0.04 | 2330 ± 112 |
| PA36,36 (original) | 1.22 ± 0.02 | 2698 ± 108 |
| PA36,IA (healing for 1 h) | 0.39 ± 0.05 | 1029 ± 217 |
| PA36,IA (healing for 24 h) | 1.00 ± 0.11 | 2206 ± 170 |
| PA36,IA (healing for 48 h) | 1.08 ± 0.06 | 2265 ± 156 |
| PA36,36 (healing for 1 h) | 0.35 ± 0.09 | 1196 ± 195 |
| PA36,36 (healing for 24 h) | 1.07 ± 0.13 | 2502 ± 147 |
| PA36,36 (healing for 48 h) | 1.11 ± 0.15 | 2648 ± 125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranganathan, P.; Chen, C.-W.; Rwei, S.-P. Highly Stretchable Fully Biomass Autonomic Self-Healing Polyamide Elastomers and Their Foam for Selective Oil Absorption. Polymers 2021, 13, 3089. https://doi.org/10.3390/polym13183089
Ranganathan P, Chen C-W, Rwei S-P. Highly Stretchable Fully Biomass Autonomic Self-Healing Polyamide Elastomers and Their Foam for Selective Oil Absorption. Polymers. 2021; 13(18):3089. https://doi.org/10.3390/polym13183089
Chicago/Turabian StyleRanganathan, Palraj, Chin-Wen Chen, and Syang-Peng Rwei. 2021. "Highly Stretchable Fully Biomass Autonomic Self-Healing Polyamide Elastomers and Their Foam for Selective Oil Absorption" Polymers 13, no. 18: 3089. https://doi.org/10.3390/polym13183089