

Preparation of Molecularly Imprinted Polymer Microspheres for Selective Solid-Phase Extraction of Capecitabine in Urine Samples


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. UV Determination
2.4. Synthesis of MIP Microspheres
2.5. Preparation of MISPE
2.6. Pretreatment of Urine Samples
3. Results and Discussion
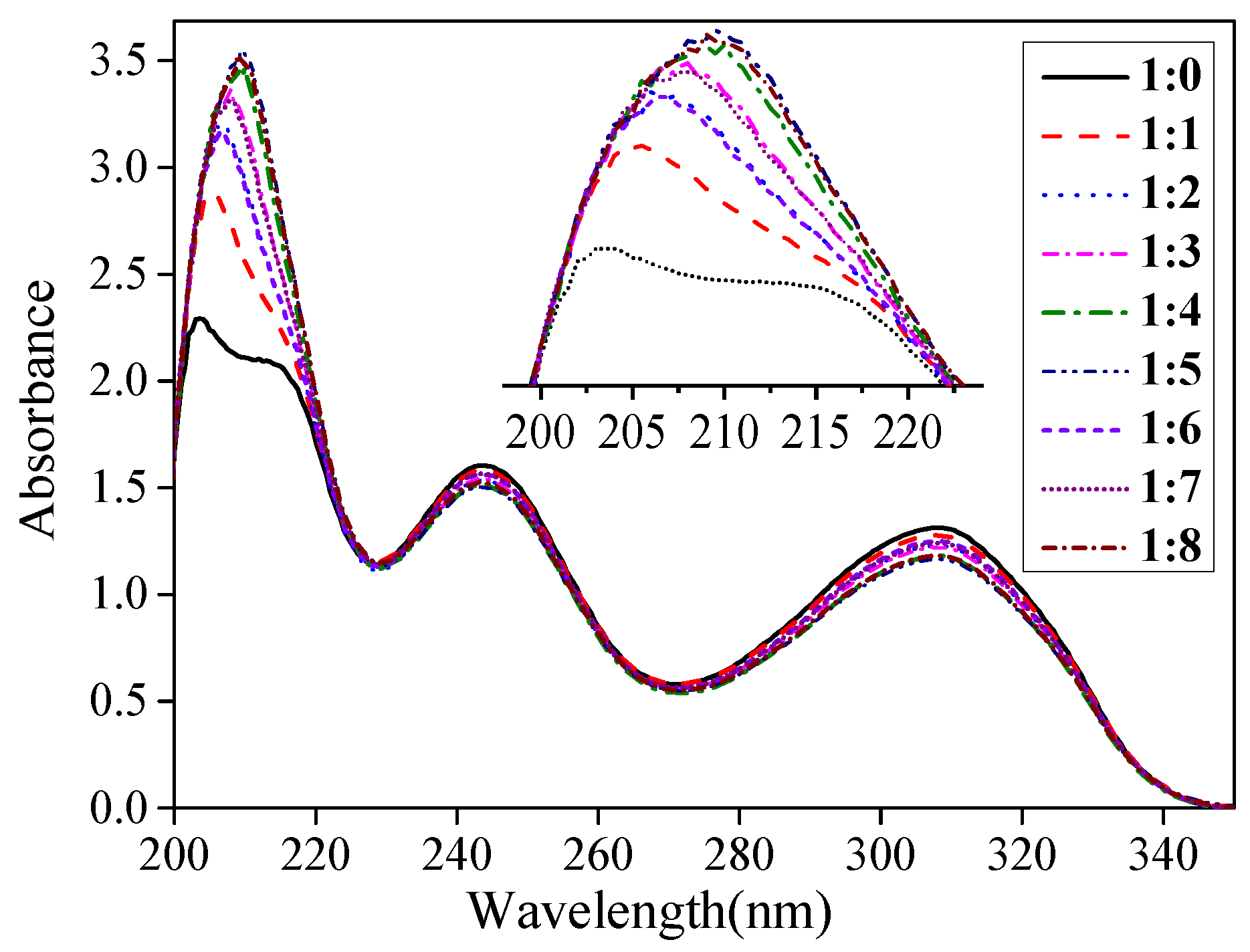
3.1. Interaction between the Template Molecule and Functional Monomer
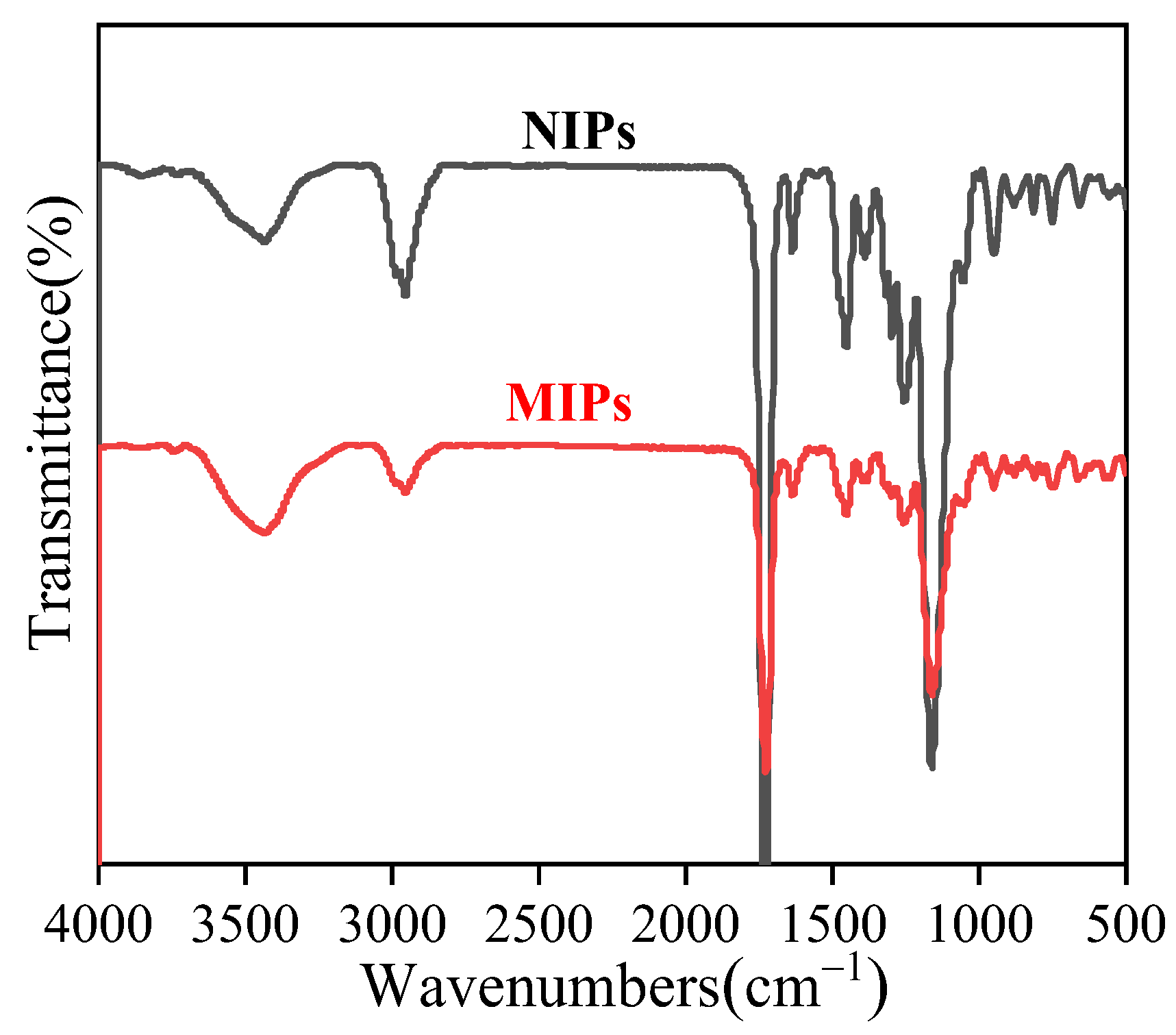
3.2. FT-IR Analysis of MIP Microspheres
3.3. SEM Analysis of the MIP Microspheres
3.4. N2 Adsorption Analysis of the MIP Microspheres
3.5. MISPE Analysis of CAP in Urine Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schreiber, V.; Kitzmueller, M.; Poxhofer, M.; Gintersdorfer, S.; Neudorfer, C.; Lichtneckert, M.; Dittrich, C.; Czejka, M. Impact of co-administered drugs on drug monitoring of capecitabine in patients with advanced colorectal cancer. Anticancer Res. 2014, 34, 3371–3376. [Google Scholar] [PubMed]
- Niazi Saei, J.; Mokhtari, A.; Karimian, H. Stopped-flow chemiluminescence determination of the anticancer drug capecitabine: Application in pharmaceutical analysis and drug-delivery systems. Luminescence 2020, 35, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Dhananjeyan, M.R.; Liu, J.; Bykowski, C.; Trendel, J.A.; Sarver, J.G.; Ando, H.; Erhardt, P.W. Rapid and simultaneous determination of capecitabine and its metabolites in mouse plasma, mouse serum, and in rabbit bile by high-performance liquid chromatography. J. Chromatogr. A 2007, 1138, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, S.; Farhadi, K.; Molaei, R.; Forough, M. Silver nanoparticles-tragacanth gel as a green membrane for effective extraction and determination of capecitabine. J. Sep. Sci. 2020, 43, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, H.; Basan, H. Development of a molecularly imprinted solid-phase extraction sorbent for the selective extraction of telmisartan from human urine. J. Sep. Sci. 2015, 38, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Tulipani, S.; Llorach, R.; Urpi-Sarda, M.; Andres-Lacueva, C. Comparative analysis of sample preparation methods to handle the complexity of the blood fluid metabolome: When less is more. Anal. Chem. 2013, 85, 341–348. [Google Scholar] [CrossRef]
- Jalili, V.; Barkhordari, A.; Ghiasvand, A. Solid-phase microextraction technique for sampling and preconcentration of polycyclic aromatic hydrocarbons: A review. Rev. Anal. Chem. 2020, 39, 1–19. [Google Scholar] [CrossRef]
- Wang, D.A.; Zeng, J.B.; Xiang, W.A.; Yin, M.A.; Zhong, G.B.; Xia, Z.A. Online coupling of the using chamber, solid-phase extraction and high-performance liquid chromatography for screening and analysis of active constituents of traditional Chinese medicines. J. Chromatogr. A 2020, 1609, 460480. [Google Scholar] [CrossRef]
- García-Atienza, P.; Esteve-Turrillas, F.A.; Armenta, S.; Herrero-Martínez, J. Ethylphenidate determination in oral fluids by molecularly imprinted polymer extraction and ion mobility spectrometry. Microchem. J. 2022, 178, 107423. [Google Scholar] [CrossRef]
- Kliangsuwan, A.; Phonchai, A.; Bunkoed, O. A magnetic molecularly imprinted polymer hierarchical composite adsorbent embedded with a zinc oxide carbon foam nanocomposite for the extraction of sulfonamides. Microchem. J. 2022, 179, 107443. [Google Scholar] [CrossRef]
- Ulusoy, M.; Aslyüce, S.; Keskin, N.; Denizli, A. Beauvericin purification from fungal strain using molecularly imprinted cryogels. Process Biochem. 2022, 113, 185–193. [Google Scholar] [CrossRef]
- Fu, J.; Zhou, S.; Zhao, P.; Wu, X.; Tang, S.; Chen, S.; Yang, Z.; Zhang, Z. A dual-response ratiometric fluorescence imprinted sensor based on metal-organic frameworks for ultrasensitive visual detection of 4-nitrophenol in environments. Biosens. Bioelectron. 2022, 198, 113848. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Zhou, D.B.; Song, W.; Hu, Y.Y.; Deng, X.J. Computational design and synthesis of molecular imprinted polymers for selective solid phase extraction of sulfonylurea herbicides. J. Chromatogr. A 2021, 1651, 462321. [Google Scholar] [CrossRef]
- At, A.; Npk, A.; Ak, B.; Kgf, B.; Vfs, A. Bisphenol a migration to alcoholic and non-alcoholic beverages-an improved molecular imprinted solid phase extraction method prior to detection with HPLC-DAD. Microchem. J. 2021, 162, 105846. [Google Scholar]
- Li, Z.; Chen, X.; Zhang, X.; Wang, Y.; Li, D.; Gao, H.; Duan, X. Selective solid-phase extraction of four phenylarsonic compounds from feeds, edible chicken and pork with tailoring imprinted polymer. Food Chem. 2021, 347, 129054. [Google Scholar] [CrossRef]
- Cheng, G.; Li, X.; Li, X.; Chen, J.; Liu, Y.; Zhao, G.; Zhu, G. Surface imprinted polymer on a metal-organic framework for rapid and highly selective adsorption of sulfamethoxazole in environmental samples. J. Hazard. Mater. 2022, 423, 127087. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Diniz, M.; Pianetti, G.A.; César, I.C.; Scarpelli, R.; Fernando, D.; Sousa, R.D.; Fernandes, C. Molecularly imprinted polymer for determination of lumefantrine in human plasma through chemometric-assisted solid-phase extraction and liquid cromatography. Talanta 2018, 184, 173–183. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, W.; Tan, L.; Wang, J.; Li, H.; Wang, J. Separation and detection of trace atrazine from seawater using dummy-template molecularly imprinted solid-phase extraction followed by high-performance liquid chromatography. Mar. Pollut. Bull. 2019, 149, 110502. [Google Scholar] [CrossRef]
- Ji, L.; Xiaoling, H.; Ping, G.; Xiaoyan, Z.; Liwei, Q.; Nan, Z.; Chunbao, D.; Renyuan, S. Preparation of l-phenylalanine-imprinted solid-phase extraction sorbent by pickering emulsion polymerization and the selective enrichment of l-phenylalanine from human urine. J. Sep. Sci. 2016, 39, 1863–1872. [Google Scholar]
- Sun, X.; Wang, M.; Peng, J.; Yang, L.; Wang, X.; Wang, F.; Zhang, X.; Wu, Q.; Chen, R.; Chen, J. Dummy molecularly imprinted solid phase extraction of climbazole from environmental water samples. Talanta 2019, 196, 47–53. [Google Scholar] [CrossRef]
- Song, R.; Hu, X.; Guan, P.; Li, J.; Wang, Q.; Zhao, N. Molecularly imprinted solid-phase extraction of glutathione from urine samples. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 44, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Otsu, T.; Matsunaga, T.; Doi, T.; Matsumoto, A. Features of living radical polymerization of vinyl monomers in homogeneous system using N,N-diethyldithiocarbamate derivatives as photoiniferters. Eur. Polym. J. 1995, 31, 67–78. [Google Scholar] [CrossRef]
- Hwang, C.C.; Lee, W.C. Chromatographic characteristics of cholesterol-imprinted polymers prepared by covalent and non-covalent imprinting methods. J. Chromatogr. A 2002, 962, 69–78. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kusunoki, T.; Zhang, Q.; Takeda, K. Bile Acid. Imprinting polymers prepared by covalent-ester monomer-template technique: Synthesis, characterization and fluorescence application for BA recognition. J. Chem. Eng. Jpn. 2007, 40, 516–522. [Google Scholar] [CrossRef]
- Karim, K.; Breton, F.; Rouillon, R.; Piletska, E.V.; Piletsky, S.A. How to find effective functional monomers for effective molecularly imprinted polymers? Adv. Drug Deliv. Rev. 2005, 57, 1795–1808. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.B.; Tang, S.-S.; Jin, R.F. Preparation of melamine molecularly imprinted polymer by computer-aided design. J. Sep. Sci. 2015, 38, 2647–2654. [Google Scholar] [CrossRef]
- Gueltekin, A.; Ersoz, A.; Denizli, A.; Say, R. Preparation of new molecularly imprinted nanosensor for cholic acid determination. Sens. Actuators 2012, 162, 153–158. [Google Scholar] [CrossRef]
- Wang, X.Y.; Sun, F.Q.; Zeng, G.P.; Liu, X.X.; Ji-Wen, Y.U.; Lai, J.P. Preparation and application of molecularly imprinted polymers for chloramphenicol based on ionic liquids assistance. J. Instrum. Anal. 2017, 36, 18–24. [Google Scholar]
- Liu, H.; Wu, D.; Zhou, K.; Wang, J.; Sun, B. Development and applications of molecularly imprinted polymers based on hydrophobic CdSe/ZnS quantum dots for optosensing of Nε-carboxymethyllysine in foods. Food Chem. 2016, 211, 34–40. [Google Scholar] [CrossRef]
- O’Mahony, J.; Molinelli, A.; Nolan, K.; Smyth, M.R.; Mizaikoff, B. Towards the rational development of molecularly imprinted polymers: 1H NMR studies on hydrophobicity and ion-pair interactions as driving forces for selectivity. Biosens. Bioelectron. 2005, 20, 1884–1893. [Google Scholar] [CrossRef]
- Mansouri, M.; Pirouzi, M.; Saberi, M.R.; Ghaderabad, M.; Chamani, J. Investigation on the interaction between cyclophosphamide and lysozyme in the presence of three different kind of cyclodextrins: Determination of the binding mechanism by spectroscopic and molecular modeling techniques. Molecules 2013, 18, 789–813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Hu, X.; Guan, P.; Song, R.; Zhang, X. Preparation of erythromycin-imprinted polymeric microspheres by emulsion polymerization and their adsorption properties. Acta Phys. Chim. Sin. 2014, 30, 121–128. [Google Scholar]
- Li, P.; Rong, F.; Xie, Y.; Hu, V.; Yuan, C. Study on the binding characteristic of S-naproxen imprinted polymer and the interactions between templates and monomers. J. Anal. Chem. 2004, 59, 939–944. [Google Scholar] [CrossRef]
- Giussi, J.M.; Blaszczyk-Lezak, I.; Sanz, B.; Allegretti, P.E.; Mijangos, C.; Cortizo, M.S. Tautomeric acetoacetate monomers as building units of functional copolymers. Eur. Polym. J. 2014, 59, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Attaran, A.M.; Mohammadi, N.; Javanbakht, M.; Akbari-Adergani, B. Molecularly imprinted solid-phase extraction for selective trace analysis of trifluoperazine. J. Chromatogr. A 2014, 52, 730–738. [Google Scholar] [CrossRef]
- Quaglia, M.; De Lorenzi, E.; Sulitzky, C.; Caccialanza, G.; Sellergren, B. Molecularly imprinted polymer films grafted from porous or nonporous silica: Novel affinity stationary phases in capillary electrochromatography. Electrophoresis 2003, 24, 952–957. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | dp (nm) | Vp (cm3·g−1) | S (m2·g−1) |
|---|---|---|---|
| NIPs | 13.17 | 0.0462 | 69.85 |
| MIPs | 25.25 | 0.0846 | 117.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, R.; Xie, J.; Yu, X.; Ge, J.; Liu, M.; Guo, L. Preparation of Molecularly Imprinted Polymer Microspheres for Selective Solid-Phase Extraction of Capecitabine in Urine Samples. Polymers 2022, 14, 3968. https://doi.org/10.3390/polym14193968
Song R, Xie J, Yu X, Ge J, Liu M, Guo L. Preparation of Molecularly Imprinted Polymer Microspheres for Selective Solid-Phase Extraction of Capecitabine in Urine Samples. Polymers. 2022; 14(19):3968. https://doi.org/10.3390/polym14193968
Chicago/Turabian StyleSong, Renyuan, Jiawei Xie, Xiaofeng Yu, Jinlong Ge, Muxin Liu, and Liping Guo. 2022. "Preparation of Molecularly Imprinted Polymer Microspheres for Selective Solid-Phase Extraction of Capecitabine in Urine Samples" Polymers 14, no. 19: 3968. https://doi.org/10.3390/polym14193968
APA StyleSong, R., Xie, J., Yu, X., Ge, J., Liu, M., & Guo, L. (2022). Preparation of Molecularly Imprinted Polymer Microspheres for Selective Solid-Phase Extraction of Capecitabine in Urine Samples. Polymers, 14(19), 3968. https://doi.org/10.3390/polym14193968