


Melt-Spinnable Polyacrylonitrile—An Alternative Carbon Fiber Precursor

 ,
, 
 ,
,  ,
, 

Abstract
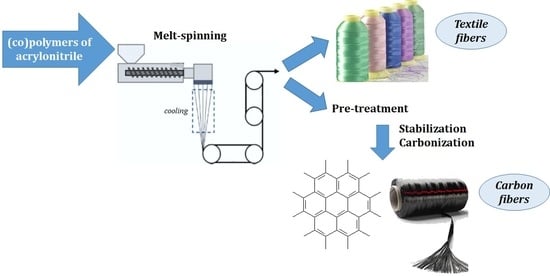
:
1. Introduction
2. Chemical Approach to Reduction of Melting Point: Selection of Copolymer Composition
2.1. Binary and Ternary Copolymers of Acrylonitrile with Methyl Acrylate
2.2. Copolymers of Acrylonitrile with 1-Vinylimidazole
2.3. Copolymers of Acrylonitrile with Other Monomers
3. Physical Approach: Plasticization
3.1. Plasticization by Water (Non-Solvent)
3.2. Plasticization by Water-Organic Additive Mixtures
3.3. Plasticization by Organic Solvents and Non-Solvents
3.4. Eco-Friendly Plasticization
3.4.1. The Use of CO2 as a Plasticizer of AN Copolymers
3.4.2. The Use of Ionic Liquids as Plasticizers of AN Copolymers
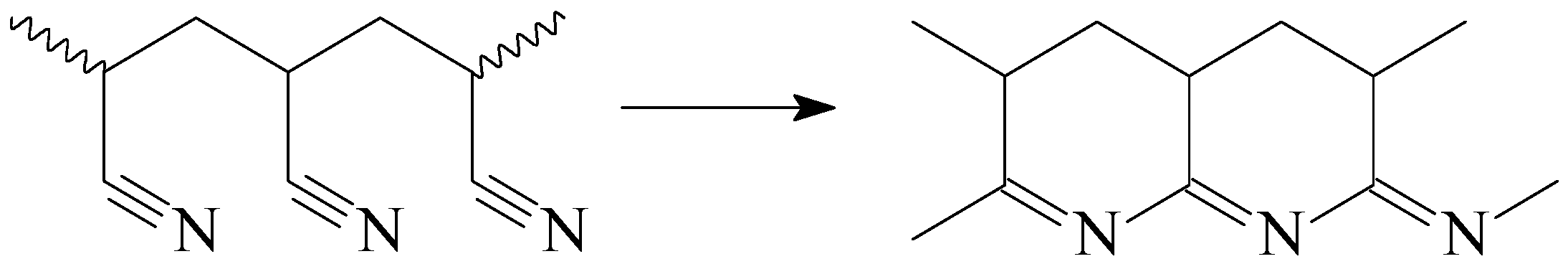
4. How to Conduct Stabilization of the Fiber without Its Melting? Problem and Its Solution
4.1. Chemical Modification at the Synthesis Stage
4.2. Plasticization and Irradiation Approaches
4.2.1. General Notes
4.2.2. UV Irradiation
4.2.3. Electron Beam Irradiation
4.2.4. Plasma Discharge Treatment
4.3. Additional Chemical Modification of Spun Fiber
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Patent Analysis
Appendix A.1. External Plasticizers
Appendix A.2. Internal Plasticizers

References
- Zhang, D. (Ed.) Advances in Filament Yarn Spinning of Textiles and Polymers; Woodhead Publishing: Cambridge, UK, 2009. [Google Scholar]
- Brown, K.R.; Harrell, T.M.; Skrzypczak, L.; Scherschel, A.; Wu, H.F.; Li, X. Carbon fibers derived from commodity polymers: A review. Carbon 2022, 196, 422–439. [Google Scholar] [CrossRef]
- Hufenus, R.; Yan, Y.; Dauner, M.; Kikutani, T. Melt-Spun Fibers for Textile Applications. Materials 2020, 13, 4298. [Google Scholar] [CrossRef]
- Ogale, A.A.; Zhang, M.; Jin, J. Recent advances in carbon fibers derived from biobased precursors. J. Appl. Polym. Sci. 2016, 133, 43794. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Kil, H.-S.; Lee, S. Fabrication of low-cost carbon fibers using economical precursors and advanced processing technologies. Carbon 2018, 142, 610–649. [Google Scholar] [CrossRef]
- Arai, Y. Pitch-Based Carbon Fibers. In High-Performance and Specialty Fibers; Springer: Berlin/Heidelberg, Germany, 2016; pp. 343–354. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Liang, D.; Xie, Q. Development of pitch-based carbon fibers: A review. Energy Sources Part A Recover. Util. Environ. Eff. 2020, 42, 1–21. [Google Scholar] [CrossRef]
- Daulbayev, C.; Kaidar, B.; Sultanov, F.; Bakbolat, B.; Smagulova, G.; Mansurov, Z. The recent progress in pitch derived carbon fibers applications. A Review. S. Afr. J. Chem. Eng. 2021, 38, 9–20. [Google Scholar] [CrossRef]
- Horikiri, A.S.; Iseki, T.J.; Minobe, I.M. Process for Production of Carbon Fiber. U.S. Patent 4070446, 24 January 1978. [Google Scholar]
- Karacan, I.; Benli, H. The effect of sulfonation treatment on the structure and properties of isotactic polypropylene fibers prior to the carbonization stage. J. Appl. Polym. Sci. 2012, 123, 3375–3389. [Google Scholar] [CrossRef]
- Penning, J.P.; Lagcher, R.; Pennings, A.J. The effect of diameter on the mechanical properties of amorphous carbon fibres from linear low density polyethylene. Polym. Bull. 1991, 25, 405–412. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, J.S. Preparation of carbon fibers from linear low density polyethylene. Carbon 2015, 94, 524–530. [Google Scholar] [CrossRef]
- Postema, A.R.; De Groot, H.; Pennings, A.J. Amorphous carbon fibres from linear low density polyethylene. J. Mater. Sci. 1990, 25, 4216–4222. [Google Scholar] [CrossRef]
- Kim, K.-W.; Lee, H.-M.; Kim, B.S.; Hwang, S.-H.; Kwac, L.-K.; An, K.-H.; Kim, B.-J. Preparation and thermal properties of polyethylene-based carbonized fibers. Carbon Lett. 2015, 16, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Wortberg, G.; De Palmenaer, A.; Beckers, M.; Seide, G.; Gries, T. Polyethylene-Based Carbon Fibers by the Use of Sulphonation for Stabilization. Fibers 2015, 3, 373–379. [Google Scholar] [CrossRef] [Green Version]
- De Palmenaer, A.; Wortberg, G.; Drissen, F.; Seide, G.; Gries, T. Production of Polyethylene Based Carbon Fibres. Chem. Eng. Trans. 2015, 43, 1699–1704. [Google Scholar]
- Zhang, N.; Bhat, G.S. Carbon Fibers from Polyethylene-Based Precursors. Mater. Manuf. Process. 1994, 9, 221–235. [Google Scholar] [CrossRef]
- Zhang, D. Carbon Fibers from Oriented Polyethylene Precursors. J. Thermoplast. Compos. Mater. 1993, 6, 38–48. [Google Scholar] [CrossRef]
- Frank, E.; Hermanutz, F.; Buchmeiser, M.R. Carbon Fibers: Precursors, Manufacturing, and Properties. Macromol. Mater. Eng. 2012, 297, 493–501. [Google Scholar] [CrossRef]
- Frank, E.; Steudle, L.M.; Ingildeev, D.; Spörl, D.-C.J.M.; Buchmeiser, M.R. Carbon Fibers: Precursor Systems, Processing, Structure, and Properties. Angew. Chem. Int. Ed. 2014, 53, 5262–5298. [Google Scholar] [CrossRef]
- Huang, X. Fabrication and Properties of Carbon Fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Millington, K.; Smith, S. Producing high-quality precursor polymer and fibers to achieve theoretical strength in carbon fibers: A review. J. Appl. Polym. Sci. 2016, 133, 43963. [Google Scholar] [CrossRef] [Green Version]
- Khayyam, H.; Jazar, R.N.; Nunna, S.; Golkarnarenji, G.; Badii, K.; Fakhrhoseini, S.M.; Kumar, S.; Naebe, M. PAN precursor fabrication, applications and thermal stabilization process in carbon fiber production: Experimental and mathematical modelling. Prog. Mater. Sci. 2019, 107, 100575. [Google Scholar] [CrossRef]
- Newcomb, B.A. Processing, structure, and properties of carbon fibers. Compos. Part A Appl. Sci. Manuf. 2016, 91, 262–282. [Google Scholar] [CrossRef]
- Sumesh, K.R.; Ghanem, Z.; Spatenka, P.; Jenikova, Z. Investigating the influence of plasma treated polyethylene powder, carbon fibers in enhancing the mechanical properties of polymer composites using rotomoulding method. Polym. Compos. 2022. [Google Scholar] [CrossRef]
- Das, T.K.; Ghosh, P.; Das, N.C. Preparation, development, outcomes, and application versatility of carbon fiber-based polymer composites: A review. Adv. Compos. Hybrid Mater. 2019, 2, 214–233. [Google Scholar] [CrossRef]
- Dainton, F.S.; Seaman, P.H. The polymerization of acrylonitrile in aqueous solution. Part I. The reaction catalyzed by Fenton’s reagent at 25 °C. J. Polym. Sci. 1959, 39, 279–297. [Google Scholar] [CrossRef]
- Minato, H.; Furue, H. Polymerization of Acrylonitriie with DMSO—H2O2—Fe3+ System. Polym. J. 1974, 6, 445–447. [Google Scholar] [CrossRef] [Green Version]
- Thomas, W.M.; Gleason, E.H.; Mino, G. Acrylonitrile polymerization in aqueous suspension. J. Polym. Sci. 1957, 24, 43–56. [Google Scholar] [CrossRef]
- Garcia-Rubio, L.H.; Hamielec, A.E. Bulk polymerization of acrylonitrile. I. An experimental investigation of the kinetics of the bulk polymerization of acrylonitrile. J. Appl. Polym. Sci. 1979, 23, 1397–1411. [Google Scholar] [CrossRef]
- Vidotto, G.; Crosato-Amaldi, A.; Talamini, G. Polymerization of acrylonitrile in the presence of different solvents. Die Makromol. Chem. 1969, 122, 91–104. [Google Scholar] [CrossRef]
- Pati, N.C.; Lenka, S.; Nayak, P.L.; Mohanty, T.R. Aqueous polymerization of acrylonitrile initiated by the bromate–thiourea redox system. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 343–351. [Google Scholar] [CrossRef]
- Toms, R.V.; Balashov, M.S.; Gervald, A.Y.; Prokopov, N.I.; Plutalova, A.V.; Chernikova, E.V. Reversible addition–fragmentation chain transfer based copolymers of acrylonitrile and alkyl acrylates as possible precursors for carbon fibers: Synthesis and thermal behavior during stabilization. Polym. Int. 2022, 71, 646–655. [Google Scholar] [CrossRef]
- Polikarpov, V.; Lukhovitskii, V.; Pozdeyeva, R.; Karpov, V. Radiation initiated polymerization of acrylonitrile in emulsion. Polym. Sci. U.S.S.R. 1974, 16, 2559–2566. [Google Scholar] [CrossRef]
- Yoshida, M.; Tanouchi, K. Solution Polymerization of Acrylonitrile in Zinc Chloride Aqueous Solution. Kobunshi Kagaku 1963, 20, 545–550. [Google Scholar] [CrossRef]
- Chand, S. Review Carbon fibers for composites. J. Mater. Sci. 2000, 35, 1303–1313. [Google Scholar] [CrossRef]
- Edie, D.D. The effect of processing on the structure and properties of carbon fibers. Carbon N. Y. 1998, 36, 345–362. [Google Scholar] [CrossRef]
- Bajaj, P.; Paliwal, D.K.; Gupta, A.K. Acrylonitrile–acrylic acids copolymers. I. Synthesis and characterization. J. Appl. Polym. Sci. 1993, 49, 823–833. [Google Scholar] [CrossRef]
- Bansal, R.C.; Donnet, J.B. Pyrolytic Formation of High-performance Carbon Fibres. Compr. Polym. Sci. Suppl. 1989, 6, 501–520. [Google Scholar] [CrossRef]
- Shlyakhtin, A.V.; Lemenovskii, D.A.; Nifant’Ev, I.E. Thermal behaviour of the copolymers of acrylonitrile with methyl acrylate and itaconic acid or its derivatives. Mendeleev Commun. 2013, 23, 277–278. [Google Scholar] [CrossRef]
- Chen, H.; Pan, Y.; Hou, S.; Shao, Z.; Hong, Y.; Ju, A. Poly(acrylonitrile-co-2-methylenesuccinamic acid) as a potential carbon fiber precursor: Preparation and stabilization. RSC Adv. 2017, 7, 54142–54152. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.Y.; McDonnell, J.; Brackley, C.; O’Brien, L.; Church, J.S.; Millington, K.; Smith, S.; Phair-Sorensen, N. Polyacrylonitrile-based precursors and carbon fibers derived from advanced RAFT technology and conventional methods–The 1st comparative study. Mater. Today Commun. 2016, 9, 22–29. [Google Scholar] [CrossRef]
- Moskowitz, J.D.; Abel, B.A.; McCormick, C.L.; Wiggins, J.S. High molecular weight and low dispersity polyacrylonitrile by low temperature RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 54, 553–562. [Google Scholar] [CrossRef]
- Li, J.; Ding, C.; Zhang, Z.; Zhu, J.; Zhu, X. Photo-induced reversible addition-fragmentation chain transfer (RAFT) polymerization of acrylonitrile at ambient temperature: A simple system to obtain high-molecular-weight polyacrylonitrile. React. Funct. Polym. 2017, 113, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chernikova, E.V.; Kostina, Y.V.; Efimov, M.N.; Prokopov, N.I.; Gerval’D, A.Y.; Toms, R.V.; Nikolaev, A.Y.; Shkirev, M.D. Homo- and copolymers of acrylonitrile: Effect of the reaction medium on the thermal behavior in an inert atmosphere. Polym. Sci. Ser. B 2015, 57, 116–131. [Google Scholar] [CrossRef]
- Chiang, R. Crystallization and melting behavior of polyacrylonitrile. J. Polym. Sci. Part A Gen. Pap. 1963, 1, 2765–2775. [Google Scholar] [CrossRef]
- Holland, V.F.; Mitchell, S.B.; Hunter, W.L.; Lindenmeyer, P.H. Crystal structure and morphology of polyacrylonitrile in dilute solution. J. Polym. Sci. 1962, 62, 145–151. [Google Scholar] [CrossRef]
- Bohn, C.R.; Schaefgen, J.R.; Statton, W.O. Laterally ordered polymers: Polyacrylonitrile and poly(vinyl trifluoroacetate). J. Polym. Sci. 1961, 55, 531–549. [Google Scholar] [CrossRef]
- Gohil, R.M.; Patel, K.C.; Patel, R.D. Crystallization of polyacrylonitrile: Growth mechanisms for various growth features. Colloid Polym. Sci. 1976, 254, 859–867. [Google Scholar] [CrossRef]
- Bahrami, S.H.; Bajaj, P.; Sen, K. Thermal behavior of acrylonitrile carboxylic acid copolymers. J. Appl. Polym. Sci. 2003, 88, 685–698. [Google Scholar] [CrossRef]
- Surianarayanan, M.; Rao, S.P.; Vijayaraghavan, R.; Raghavan, K. Thermal behaviour of acrylonitrile polymerization and polyacrylonitrile decomposition. J. Hazard. Mater. 1998, 62, 187–197. [Google Scholar] [CrossRef]
- Dang, W.; Liu, J.; Wang, X.; Yan, K.; Zhang, A.; Yang, J.; Chen, L.; Liang, J. Structural Transformation of Polyacrylonitrile (PAN) Fibers during Rapid Thermal Pretreatment in Nitrogen Atmosphere. Polymers 2020, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Krigbaum, W.R.; Tokita, N. Melting point depression study of polyacrylonitrile. J. Polym. Sci. 1960, 43, 467–488. [Google Scholar] [CrossRef]
- Slade, P. The melting of polyacrylonitrile. Thermochim. Acta 1970, 1, 459–463. [Google Scholar] [CrossRef]
- Wu, Q.-Y.; Chen, X.-N.; Wan, L.-S.; Xu, Z.-K. Interactions between Polyacrylonitrile and Solvents: Density Functional Theory Study and Two-Dimensional Infrared Correlation Analysis. J. Phys. Chem. B 2012, 116, 8321–8330. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, V.G. Untersuchungen zum Schmelzen von Polyacrylnitril. Die Angew. Makromol. Chem. 1971, 20, 121–127. [Google Scholar] [CrossRef]
- Chae, H.G.; Newcomb, B.A.; Gulgunje, P.V.; Liu, Y.; Gupta, K.K.; Kamath, M.G.; Lyons, K.M.; Ghoshal, S.; Pramanik, C.; Giannuzzi, L.; et al. High strength and high modulus carbon fibers. Carbon 2015, 93, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.A.; Weisenberger, M.C.; Bradley, S.B.; Abdallah, M.G.; Mecham, S.J.; Pisipati, P.; McGrath, J.E. Synthesis, spinning, and properties of very high molecular weight poly(acrylonitrile-co-methyl acrylate) for high performance precursors for carbon fiber. Polymer 2014, 55, 6471–6482. [Google Scholar] [CrossRef] [Green Version]
- Naito, K.; Yang, J.-M.; Tanaka, Y.; Kagawa, Y. The effect of gauge length on tensile strength and Weibull modulus of polyacrylonitrile (PAN)- and pitch-based carbon fibers. J. Mater. Sci. 2011, 47, 632–642. [Google Scholar] [CrossRef]
- Matsuhisa, Y.; Kibayashi, M.; Yamasaki, K.; Okuda, A. Carbon Fibers, Acrylic Fibers, and Production Processes Thereof. U.S. Patent US6103211A, 15 August 2000. [Google Scholar]
- Lee, J.H.; Jin, J.-U.; Park, S.; Choi, D.; You, N.-H.; Chung, Y.; Ku, B.-C.; Yeo, H. Melt processable polyacrylonitrile copolymer precursors for carbon fibers: Rheological, thermal, and mechanical properties. J. Ind. Eng. Chem. 2018, 71, 112–118. [Google Scholar] [CrossRef]
- Mukundan, T.; Bhanu, V.; Wiles, K.; Johnson, H.; Bortner, M.; Baird, D.; Naskar, A.; Ogale, A.; Edie, D.; McGrath, J. A photocrosslinkable melt processible acrylonitrile terpolymer as carbon fiber precursor. Polymer 2006, 47, 4163–4171. [Google Scholar] [CrossRef]
- Liu, S.P.; Han, K.Q.; Chen, L.; Zheng, Y.; Yu, M.H. Structure and Performances of Plasticized Melt-Spun Polyacrylonitrile Precursor Fiber. Appl. Mech. Mater. 2015, 723, 652–655. [Google Scholar] [CrossRef]
- Udakhe, J.; Goud, V. Melt Processing of polyacrylonitrile (PAN) polymers. J. Text. Assoc. 2011, 71, 233–241. [Google Scholar]
- Frushour, B. Melting behavior of polyacrylonitrile copolymers. Polym. Bull. 1984, 11, 375–382. [Google Scholar] [CrossRef]
- Han, N.; Zhang, X.-X.; Wang, X.-C.; Wang, N. Fabrication, structures and properties of Acrylonitrile/Vinyl acetate copolymers and copolymers containing microencapsulated phase change materials. Macromol. Res. 2010, 18, 144–152. [Google Scholar] [CrossRef]
- Hao, J.; Liu, Y.; Lu, C. Effect of acrylonitrile sequence distribution on the thermal stabilization reactions and carbon yields of poly(acrylonitrile-co-methyl acrylate). Polym. Degrad. Stab. 2018, 147, 89–96. [Google Scholar] [CrossRef]
- Chang, S.-H. Thermal analysis of acrylonitrile copolymers containing methyl acrylate. J. Appl. Polym. Sci. 1994, 54, 405–407. [Google Scholar] [CrossRef]
- Maksimov, N.M.; Toms, R.V.; Balashov, M.S.; Gerval’D, A.Y.; Prokopov, N.I.; Plutalova, A.V.; Kuzin, M.S.; Skvortsov, I.Y.; Kulichikhin, V.G.; Chernikova, E.V. Novel Potential Precursor of Carbon Fiber Based on Copolymers of Acrylonitrile, Acrylamide, and Alkyl Acrylates. Polym. Sci. Ser. B 2022, 64, 1–18. [Google Scholar] [CrossRef]
- Rangarajan, P.; Yang, J.; Bhanu, V.; Godshall, D.; McGrath, J.; Wilkes, G.; Baird, D. Effect of comonomers on melt processability of polyacrylonitrile. J. Appl. Polym. Sci. 2002, 85, 69–83. [Google Scholar] [CrossRef]
- Bhanu, V.; Rangarajan, P.; Wiles, K.; Bortner, M.; Sankarpandian, M.; Godshall, D.; Glass, T.; Banthia, A.; Yang, J.; Wilkes, G.; et al. Synthesis and characterization of acrylonitrile methyl acrylate statistical copolymers as melt processable carbon fiber precursors. Polymer 2002, 43, 4841–4850. [Google Scholar] [CrossRef]
- Godshall, D.; Rangarajan, P.; Baird, D.; Wilkes, G.; Bhanu, V.; McGrath, J. Incorporation of methyl acrylate in acrylonitrile based copolymers: Effects on melting behavior. Polymer 2003, 44, 4221–4228. [Google Scholar] [CrossRef]
- Wiles, K.B.; Bhanu, V.A.; Pasquale, A.J.; Long, T.E.; McGrath, J.E. Monomer reactivity ratios for acrylonitrile-methyl acrylate free-radical copolymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 2994–3001. [Google Scholar] [CrossRef]
- Peng, W.W.; Han, N.; Tang, X.F.; Liu, H.H.; Zhang, X.X. Preparation and Characterization of Melt-Spun Poly( Acrylonitrile-Methylacrylate) Hollow Fiber. Adv. Mater. Res. 2011, 332, 339–342. [Google Scholar] [CrossRef]
- Han, N.; Gao, X.; Zhang, R.; Zhang, X. Structures and properties of thermoregulated acrylonitrile-methyl acrylate sheet containing microphase change materials. Polym. Compos. 2013, 34, 641–649. [Google Scholar] [CrossRef]
- Han, N.; Chen, S.; Chen, G.; Gao, X.; Zhang, X. Preparation of poly(acrylonitrile-methacrylate) membrane via thermally induced phase separation: Effects of MA with different feeding molar ratios. Desalination Water Treat. 2016, 57, 27531–27547. [Google Scholar] [CrossRef]
- König, S.; Kreis, P.; Herbert, C.; Wego, A.; Steinmann, M.; Wang, D.; Frank, E.; Buchmeiser, M.R. Melt-Spinning of an Intrinsically Flame-Retardant Polyacrylonitrile Copolymer. Materials 2020, 13, 4826. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; Zhang, J.; Horrocks, A.R. The flammability of polyacrylonitrile and its copolymers III. Effect of flame retardants. Fire Mater. 1994, 18, 231–241. [Google Scholar] [CrossRef]
- Rwei, S.-P.; Way, T.-F.; Chiang, W.-Y.; Tseng, J.-C. Thermal analysis and melt spinnability of poly(acrylonitrile-co-methyl acrylate) and poly(acrylonitrile-co-dimethyl itaconate) copolymers. Text. Res. J. 2017, 88, 1479–1490. [Google Scholar] [CrossRef]
- Rangarajan, P.; Bhanu, V.; Godshall, D.; Wilkes, G.; McGrath, J.; Baird, D. Dynamic oscillatory shear properties of potentially melt processable high acrylonitrile terpolymers. Polymer 2002, 43, 2699–2709. [Google Scholar] [CrossRef]
- Way, T.F.; Chen, J.J.; Chen, Y.T.; Hsiao, K.J.; Cheng, S.H.; Chen, J.P. PAN-Based Carbon Fiber and Fabrication Method Therefore and Precursor Raw Material. U.S. Patent 0160369 USA, 30 June 2011. [Google Scholar]
- Rwei, S.-P.; Way, T.-F.; Hsu, Y.-S. Kinetics of cyclization reaction in poly(acrylonitrile/methyl acrylate/dimethyl itaconate) copolymer determined by a thermal analysis. Polym. Degrad. Stab. 2013, 98, 2072–2080. [Google Scholar] [CrossRef]
- Deng, W.; Lobovsky, A.; Iacono, S.T.; Wu, T.; Tomar, N.; Budy, S.M.; Long, T.; Hoffman, W.P.; Dennis, D.W.S., Jr. Poly (acrylonitrile–co -1-vinylimidazole): A new melt processable carbon fiber precursor. Polymer 2011, 52, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, S.F.; Batchelor, B.; Jung, M.; Park, K.; Voit, W.E.; Novak, B.M.; Yang, D. Study of a melt processable polymer precursor for carbon fiber. Carbon Lett. 2019, 29, 605–612. [Google Scholar] [CrossRef]
- Han, N.; Li, H.; Li, C.; Zhang, X.; Li, W.; Wang, D. Synthesis and Properties of Melt Processable Acrylonitrile-N-vinylimidazole Copolymers and Fiber. Chem. J. Chineese Univ. 2015, 36, 2073–2080. [Google Scholar] [CrossRef]
- Toms, R.V.; Gervald, A.Y.; Prokopov, N.I.; Osipova, N.I.; Plutalova, A.V.; Chernikova, E.V. Thermal Behavior of Poly(acrylonitrile-co-1-vinyl imidazole) During Stabilization. Polym. Sci. Ser. B 2022, 64, 294–312. [Google Scholar] [CrossRef]
- Fleming, R.; Candido, T.O.; Granado, N.P.; Pardini, L.C. Melt spinning of poly(acrylonitrile)-co-styrene copolymer. Polym. Degrad. Stab. 2021, 192, 109702. [Google Scholar] [CrossRef]
- Burlyaev, V.V.; Burlyaeva, E.V.; Nikolaev, A.I.; Peshnev, B.V. Functional Modeling of Carbon Sorbents Synthesis Control. Fine Chem. Technol. 2019, 14, 39–46. [Google Scholar] [CrossRef]
- Kressler, J.; Rudolf, B.; Shimomai, K.; Ougizawa, T.; Inoue, T. Cyclization reaction of polyacrylonitrile and poly(styrene-co-acrylonitrile) studied by PVT measurements. Macromol. Rapid Commun. 1995, 16, 631–636. [Google Scholar] [CrossRef]
- Benedict, S.; Curatolo, G.; Li, S. Preparation of Melt-Processable Acrylonitrile/Methacrylonitrile Copolymers. Europa Patent EP0492909B1, 28 February 1996. [Google Scholar]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Melting behavior of acrylonitrile polymers. J. Appl. Polym. Sci. 1998, 70, 2703–2709. [Google Scholar] [CrossRef]
- Frushour, B.G. A new thermal analytical technique for acrylic polymers. Polym. Bull. 1981, 4, 305–314. [Google Scholar] [CrossRef]
- Frushour, B.G. Water as a melting point depressant for acrylic polymers. Polym. Bull. 1982, 7, 1–8. [Google Scholar] [CrossRef]
- Klausner, G.K.; Kreahling, R.P.; Sinha, V.T. Single Phase Extrusion of Acrylic Polymer and Water. U.S. Patent 3,991,153, 9 November 1976. [Google Scholar]
- Grove, D.; Desai, P.; Abhiraman, A. Exploratory experiments in the conversion of plasticized melt spun PAN-based precursors to carbon fibers. Carbon 1988, 26, 403–411. [Google Scholar] [CrossRef]
- Kreahling, R.P.; Pfeiffer, R.E. Melt-Spinning a Plurality of Acrylonitrile Polymer Fibers. U.S. Patent 4,301,107, 17 November 1981. [Google Scholar]
- Coxe, C.D. Preparation of Shaped Articles from Acrylonitrile Polymers. U.S. Patent 2,585,444, 12 February 1952. [Google Scholar]
- George, D.B.; Gerd, R.B.; Byrd, T.T., Jr. Process for Preparing Synthetic Fibers for Paper Products. U.S. Patent 3,402,231, 17 September 1968. [Google Scholar]
- Min, B.G.; Son, T.W.; Jo, W.H.; Choi, S.G. Thermal stability of polyacrylonitrile in the melt formed by hydration. J. Appl. Polym. Sci. 1992, 46, 1793–1798. [Google Scholar] [CrossRef]
- Min, B.G.; Son, T.W.; Kim, B.C.; Jo, W.H. Plasticization Behavior of Polyacrylonitrile and Characterization of Acrylic Fiber Prepared from the Plasticized Melt. Polym. J. 1992, 24, 841–848. [Google Scholar] [CrossRef]
- Min, B.G.; Son, T.W.; Kim, B.C.; Lee, C.J.; Jo, W.H. Effect of solvent or hydrophilic polymer on the hydration melting behavior of polyacrylonitrile. J. Appl. Polym. Sci. 1994, 54, 457–462. [Google Scholar] [CrossRef]
- Min, B.G.; Son, T.W. Effect of Inorganic Fillers on the Properties of Hydrated PAN Melt(I) -Rheological Properties of Hydrated PAN Melt. Korean Soc. Dye. Finish. 2000, 12, 295–300. [Google Scholar]
- Kim, B.C.; Min, B.G.; Son, T.W.; Lee, C.J. A study on the hydrated acrylic polymers. Polym. Int. 1995, 37, 191–195. [Google Scholar] [CrossRef]
- Blickenstaff, R.A. Acrylonitrile Polymer Filaments. U.S. Patent 3,984,601, 5 October 1976. [Google Scholar]
- Goodman, A.; Mark, A.S. Melt-Extrusion of Acrylonitrile Polymers into Filaments. U.S. Patent 3,896,204, 22 July 1975. [Google Scholar]
- Beebe, E.V. Production of Plexifilament Strands. U.S. Patent 4,166,091, 28 August 1979. [Google Scholar]
- Cramer, F.B. Process for Preparing Acrylic Polymer Plexifilaments. U.S. Patent 4,238,441, 9 December 1980. [Google Scholar]
- Cline, E.T.; Francis, B.C. Process for Melt Spinning Acrylonitrile Polymer Hydrates. U.S. Patent 4,238,442, 9 December 1980. [Google Scholar]
- American Cyanamid Company. Melt-Spinning Process for the Production of Shaped Articles from Acrylic Nitrile Polymers. Germany Patent 2,403,947, 5 February 1974.
- Porosoff, H. Melt-Spinning Acrylonitrile Polymer Fibers. U.S. Patent 4,163,770, 7 August 1979. [Google Scholar]
- DeMaria, F.; Young, C.C. Process for Melt Spinning Acrylonitrile Polymer Fiber Using Hot Water as Stretching Aid. U.S. Patent 4,303,607, 1 December 1981. [Google Scholar]
- Coleman, D.; Koroscil, A. Hydrophilic Acrylonitrile Polymers for Melt-Spinning. U.S. Patent 4,271,056, 2 June 1981. [Google Scholar]
- Siegman, E.J. Purging for Spinning Hydrated Acrylic Polymer Melt. U.S. Patent 4,226,817, 7 October 1980. [Google Scholar]
- Young, C.C.; DeMaria, F. Process for Melt-Spinning Acrylonitrile Polymer Fiber Using Vertically Disposed Compression Zone. U.S. Patent 4,283,365, 11 August 1981. [Google Scholar]
- Young, C.C.; DeMaria, F. Melt Spinning Process for Acrylonitrile Polymer Fiber-Three or More Stretch Stages. U.S. Patent 4,379,113, 5 April 1983. [Google Scholar]
- Young, C.C.; DeMaria, F. Continuous Liquid Phase Process for Melt Spinning Acrylonitrile Polymer. U.S. Patent 4,461,739, 24 July 1983. [Google Scholar]
- Miller, G.; Yu, J.; Joseph, R.; Choudhury, S.; Mecham, S.; Baird, D.; Bortner, M.; Norris, R.; Paulauskas, F.; Riffle, J. Melt-spinnable polyacrylonitrile copolymer precursors for carbon fibers. Polymer 2017, 126, 87–95. [Google Scholar] [CrossRef]
- Yu, J.; Miller, G.C.; Riffle, J.S.; Baird, D.G. Identifying Melt Processing Conditions for a Polyacrylonitrile Copolymer Plasticized with Water, Acetonitrile and their Mixtures. Int. Polym. Process. 2019, 34, 307–313. [Google Scholar] [CrossRef]
- Yu, J.; Baird, D.G. Study of Melt Spinning Processing Conditions for a Polyacrylonitrile Copolymer with a Water/Ethanol Mixture as a Plasticizer. Int. Polym. Process. 2019, 34, 557–563. [Google Scholar] [CrossRef]
- Yu, J. Establishing the Conditions for Stable Extrusion of Melt Spun Polyacrylonitrile with Water Based Plasticizers. Ph.D. Thesis, Dissertation in Chemical Engineering, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 18 June 2019. [Google Scholar]
- Júnior, C.A.R.B.; Fleming, R.R.; Pardini, L.C.; Alves, N.P. Análise térmica da poliacrilonitrila plastificada com glicerol em extrusora. Polímeros 2012, 22, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.F.; Pardini, L.C.; Alves, N.P.; Júnior, C.A.R.B. Thermal Stabilization study of polyacrylonitrile fiber obtained by extrusion. Polímeros 2015, 25, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Alves, N.P. Acrylic and modacrylic polymer fusion process derivated from acrylonitrile and molded articles made from the same. WO Patent 147224 A2, 27 December 2007. [Google Scholar]
- Im, Y.M.; Choi, H.M.; Nathanael, A.J.; Jeong, M.H.; Lee, S.O.; Yun, S.N.; Oh, T.H. Effects of Glycerol on Melt Spinning of Polyacrylonitrile Copolymer and Tetrapolymer. Fibers Polym. 2020, 21, 376–383. [Google Scholar] [CrossRef]
- Atureliya, S.K.; Bashir, Z. Continuous plasticized melt-extrusion of polyacrylonitrile homopolymer. Polymer 1993, 34, 5116–5122. [Google Scholar] [CrossRef]
- König, S.; Kreis, P.; Reinders, L.; Beyer, R.; Wego, A.; Herbert, C.; Steinmann, M.; Frank, E.; Buchmeiser, M.R. Melt spinning of propylene carbonate-plasticized poly(acrylonitrile)-co-poly(methyl acrylate). Polym. Adv. Technol. 2020, 31, 1827–1835. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Su, J.H.; Manivannan, G.; Lee, P.H.C.; Sawan, S.P.; Spall, W.D. Interaction of supercritical carbon dioxide with polymers. II. Amorphous polymers. J. Appl. Polym. Sci. 1996, 59, 707–717. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Su, J.H.; Manivannan, G.; Lee, P.H.C.; Sawan, S.P.; Spall, W.D. Interaction of supercritical carbon dioxide with polymers. I. Crystalline polymers. J. Appl. Polym. Sci. 1996, 59, 695–705. [Google Scholar] [CrossRef]
- Bae, Y.C.; Gulari, E. Viscosity reduction of polymeric liquid by dissolved carbon dioxide. J. Appl. Polym. Sci. 1997, 63, 459–466. [Google Scholar] [CrossRef]
- Gerhardt, L.J.; Manke, C.W.; Gulari, E. Rheology of polydimethylsiloxane swollen with supercritical carbon dioxide. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 523–534. [Google Scholar] [CrossRef]
- Royer, J.R.; Gay, Y.J.; DeSimone, J.M.; Khan, S.A. High-pressure rheology of polystyrene melts plasticized with CO2: Experimental measurement and predictive scaling relationships. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 3168–3180. [Google Scholar] [CrossRef]
- Royer, J.R.; DeSimone, J.M.; Khan, S.A. High-pressure rheology and viscoelastic scaling predictions of polymer melts containing liquid and supercritical carbon dioxide. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 3055–3066. [Google Scholar] [CrossRef]
- Bortner, M.J.; Baird, D.G. Absorption of CO2 and subsequent viscosity reduction of an acrylonitrile copolymer. Polymer 2004, 45, 3399–3412. [Google Scholar] [CrossRef]
- Bortner, M.J.; Bhanu, V.A.; McGrath, J.E.; Baird, D.G. Absorption of CO2 in high acrylonitrile content copolymers: Dependence on acrylonitrile content. Polymer 2004, 45, 3413–3422. [Google Scholar] [CrossRef]
- Wilding, M.D.; Baird, D.G. Melt processing and rheology of an acrylonitrile copolymer with absorbed carbon dioxide. Polym. Eng. Sci. 2009, 49, 1990–2004. [Google Scholar] [CrossRef]
- Lee, J.S.; Wang, X.; Luo, H.; Baker, G.A.; Dai, S. Facile Ionothermal Synthesis of Microporous and Mesoporous Carbons from Task Specific Ionic Liquids. J. Am. Chem. Soc. 2009, 131, 4596–4597. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dai, S. Ionic Liquids as Versatile Precursors for Functionalized Porous Carbon and Carbon-Oxide Composite Materials by Confined Carbonization. Angew. Chem. Int. Ed. 2010, 49, 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cheng, L.; Zhang, H.; Zhang, Y.; Wang, H.; Yu, M. Rheological Behaviors of Polyacrylonitrile/1-Butyl-3- Methylimidazolium Chloride Concentrated Solutions. Int. J. Mol. Sci. 2007, 8, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Saba, H.; Zhang, Y.; Wang, H. The viscoelastic behavior of concentrated polyacrylonitrile/1-butyl-3-methylimidazolium chloride from solution to gel. Polym. Eng. Sci. 2013, 54, 598–606. [Google Scholar] [CrossRef]
- Yang, T.; Yao, Y.; Lin, Y.; Wang, B.; Niu, A.; Wu, D. Rheological Behaviour of Polyacrylonitrile in an Ionic Liquid Solution. Iran. Polym. J. 2010, 19, 843–852. [Google Scholar]
- Li, X.; Qin, A.; Zhao, X.; Ma, B.; He, C. The plasticization mechanism of polyacrylonitrile/1-butyl-3-methylimidazolium chloride system. Polymer 2014, 55, 5773–5780. [Google Scholar] [CrossRef]
- Tian, Y.C.; Han, K.Q.; Zhang, W.H.; Zhang, J.J.; Zhang, R.; Liu, S.P.; Yu, M.H. Influence of Melt Temperature on Structure of Polyacrylonitrile in Ionic Liquids during Plasticized Melt Spinning Process. Appl. Mech. Mater. 2012, 268, 483–486. [Google Scholar] [CrossRef]
- Tian, Y.; Han, K.; Zhang, W.; Zhang, J.; Rong, H.; Wang, D.; Yan, B.; Liu, S.; Yu, M. Influence of residence time on the structure of polyacrylonitrile in ionic liquids during melt spinning process. Mater. Lett. 2013, 92, 119–121. [Google Scholar] [CrossRef]
- Liu, S.; Han, K.; Chen, L.; Zheng, Y.; Yu, M. Structure and properties of partially cyclized polyacrylonitrile-based carbon fiber-precursor fiber prepared by melt-spun with ionic liquid as the medium of processing. Polym. Eng. Sci. 2015, 55, 2722–2728. [Google Scholar] [CrossRef]
- Martin, H.J.; Luo, H.; Chen, H.; Do-Thanh, C.-L.; Kearney, L.T.; Mayes, R.T.; Naskar, A.K.; Dai, S. Effect of the Ionic Liquid Structure on the Melt Processability of Polyacrylonitrile Fibers. ACS Appl. Mater. Interfaces 2020, 12, 8663–8673. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Luo, H.; Martin, H.J.; Wang, T.; Sun, Y.; Arnould, M.A.; Thapaliya, B.P.; Dai, S. Controlling the elasticity of polyacrylonitrile fibers via ionic liquids containing cyano-based anions. RSC Adv. 2022, 12, 8656–8660. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhang, Y.; Wang, H. Acrylic fibers processing with ionic liquid as solvent. Polym. Adv. Technol. 2008, 20, 857–862. [Google Scholar] [CrossRef]
- Jiang, J.; Srinivas, K.; Kiziltas, A.; Geda, A.; Ahring, B.K. Rheology of Polyacrylonitrile/Lignin Blends in Ionic Liquids under Melt Spinning Conditions. Molecules 2019, 24, 2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oroumei, A.; Fox, B.; Naebe, M. Thermal and Rheological Characteristics of Biobased Carbon Fiber Precursor Derived from Low Molecular Weight Organosolv Lignin. ACS Sustain. Chem. Eng. 2015, 3, 758–769. [Google Scholar] [CrossRef]
- König, S.; Clauss, M.M.; Giebel, E.; Buchmeiser, M.R. N,N’-Substituted acryloamidines–novel comonomers for melt-processible poly(acrylonitrile)-based carbon fiber precursors. Polym. Chem. 2019, 10, 4469–4476. [Google Scholar] [CrossRef]
- Morgan, P. (Ed.) Carbon Fibers and Their Composites; Taylor and Francis: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Badawy, A.S.M.; Dessouki, A.M. Cross-Linked Polyacrylonitrile Prepared by Radiation-Induced Polymerization Technique. J. Phys. Chem. B 2003, 107, 11273–11279. [Google Scholar] [CrossRef]
- Bullock, R. Radiation-enhanced oxidation of PAN-based carbon fibres. Fibre Sci. Technol. 1974, 7, 157–160. [Google Scholar] [CrossRef]
- Stephenson, C.V.; Lacey, J.C.; Wilcox, W.S. Ultraviolet irradiation of plastics III. Decomposition products and mechanisms. J. Polym. Sci. 1961, 55, 477–488. [Google Scholar] [CrossRef]
- Jellinek, H.H.G.; Bastien, I.J. Photolysis of polyacrylonitrile. Can. J. Chem. 1961, 39, 2056–2068. [Google Scholar] [CrossRef]
- Ranby, B.; Rabek, J.F. (Eds.) Photodegradation, Photo-Oxidation and Photostabilization of Polymers; Wiley: London, UK, 1975. [Google Scholar] [CrossRef]
- Paiva, M.; Kotasthane, P.; Edie, D.; Ogale, A. UV stabilization route for melt-processible PAN-based carbon fibers. Carbon 2003, 41, 1399–1409. [Google Scholar] [CrossRef]
- Jo, A.Y.; Yoo, S.H.; Chung, Y.-S.; Lee, S. Effects of ultraviolet irradiation on stabilization of textile-grade polyacrylonitrile fibers without photo-initiator for preparing carbon fibers. Carbon 2018, 144, 440–448. [Google Scholar] [CrossRef]
- Zhao, W.; Lu, Y.; Jiang, J.; Hu, L.; Zhou, L. The effect of γ-ray irradiation on the microstructure and thermal properties of polyacrylonitrile fibers. RSC Adv. 2015, 5, 23508–23518. [Google Scholar] [CrossRef]
- Liu, Y.; Kumar, S. Recent Progress in Fabrication, Structure, and Properties of Carbon Fibers. Polym. Rev. 2012, 52, 234–258. [Google Scholar] [CrossRef]
- Dietrich, J.; Hirt, P.; Herlinger, H. Electron-beam-induced cyclisation to obtain C-fibre precursors from polyacrylonitrile homopolymers. Eur. Polym. J. 1996, 32, 617–623. [Google Scholar] [CrossRef]
- Park, S.; Yoo, S.H.; Kang, H.R.; Jo, S.M.; Joh, H.-I.; Lee, S. Comprehensive stabilization mechanism of electron-beam irradiated polyacrylonitrile fibers to shorten the conventional thermal treatment. Sci. Rep. 2016, 6, 27330. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.H.; Park, S.; Park, Y.; Lee, D.; Joh, H.-I.; Shin, I.; Lee, S. Facile method to fabricate carbon fibers from textile-grade polyacrylonitrile fibers based on electron-beam irradiation and its effect on the subsequent thermal stabilization process. Carbon 2017, 118, 106–113. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Liu, W.; Yang, C.; Wu, G. Higher dose rate effect of 500-keV EB irradiation favoring free radical annealing and pre-oxidation of polyacrylonitrile fibers. Polym. Degrad. Stab. 2019, 167, 201–209. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, X.; Liu, J.; Liang, J.; Wang, X. Structure and tensile properties of carbon fibers based on electron-beam irradiated polyacrylonitrile fibers. J. Mater. Sci. 2020, 55, 4962–4969. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, Y.; Liu, P.; Yu, H.; Ge, B.; Mei, Y. Effect of electron beam irradiation on polyacrylonitrile precursor fibers and stabilization process. J. Appl. Polym. Sci. 2011, 122, 90–96. [Google Scholar] [CrossRef]
- Yu, H.W.; Yuan, H.W.; Wang, Y.S.; Wei, Z. The Characterization of Different Doses of Electron Beam Irradiation on the Structure and Properties of PAN Precursor Fibers. Adv. Mater. Res. 2011, 328, 1594–1597. [Google Scholar] [CrossRef]
- Sedghi, A.; Farsani, R.E.; Shokuhfar, A. The effect of commercial polyacrylonitrile fibers characterizations on the produced carbon fibers properties. J. Mater. Process. Technol. 2008, 198, 60–67. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y.; Liu, J.; Shen, Z.; Liang, J.; Wang, X. Rapid and Continuous Preparation of Polyacrylonitrile-Based Carbon Fibers with Electron-Beam Irradiation Pretreatment. Materials 2018, 11, 1270. [Google Scholar] [CrossRef] [Green Version]
- König, S.; Bauch, V.; Herbert, C.; Wego, A.; Steinmann, M.; Frank, E.; Buchmeiser, M.R. High-Performance Carbon Fibers Prepared by Continuous Stabilization and Carbonization of Electron Beam-Irradiated Textile Grade Polyacrylonitrile Fibers. Macromol. Mater. Eng. 2021, 306, 2100484. [Google Scholar] [CrossRef]
- Shin, H.K.; Park, M.; Kang, P.H.; Choi, H.-S.; Park, S.-J. Preparation and characterization of polyacrylonitrile-based carbon fibers produced by electron beam irradiation pretreatment. J. Ind. Eng. Chem. 2014, 20, 3789–3792. [Google Scholar] [CrossRef]
- Shin, H.K.; Park, M.; Kim, H.-Y.; Park, S.-J. Influence of orientation on ordered microstructure of PAN-based fibers during electron beam irradiation stabilization. J. Ind. Eng. Chem. 2015, 32, 120–122. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Shin, H.-K.; Jeun, J.-P.; Kim, H.-B.; Oh, S.-H.; Kang, P.-H. Characterization of polyacrylonitrile based carbon nanofiber mats via electron beam processing. J. Nanosci. Nanotechnol. 2012, 12, 6120–6124. [Google Scholar] [CrossRef]
- Paulauskas, F.L.; White, T.L.; Sherman, D.M. Apparatus and Method for Oxidation and Stabilization of Polymeric Materials. U.S. Patent 0263295 A1, 22 October 2009. [Google Scholar]
- Lee, S.-W.; Lee, H.-Y.; Jang, S.-Y.; Jo, S.; Lee, H.-S.; Choe, W.-H.; Lee, S. Efficient preparation of carbon fibers using plasma assisted stabilization. Carbon 2013, 55, 361–365. [Google Scholar] [CrossRef]
- Bortner, M.J.; Bhanu, V.; McGrath, J.E.; Baird, D.G. Shear rheological properties of acrylic copolymers and terpolymers suitable for potentially melt processable carbon fiber precursors. J. Appl. Polym. Sci. 2004, 93, 2856–2865. [Google Scholar] [CrossRef]
- Naskar, A.K.; Walker, R.A.; Proulx, S.; Edie, D.D.; Ogale, A.A. UV assisted stabilization routes for carbon fiber precursors produced from melt-processible polyacrylonitrile terpolymer. Carbon 2005, 43, 1065–1072. [Google Scholar] [CrossRef]
- Walker, R.A.; Naskar, A.K.; Ogale, A.A. Carbon mats from melt spun polyacrylonitrile based precursors for automotive composites. Plast. Rubber Compos. 2006, 35, 242–246. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Wang, S.G.; Liu, J.L. Polyacrylonitrile Based Carbon Fibers Obtained from a Melt Spun Route. Key Eng. Mater. 2013, 575, 151–155. [Google Scholar] [CrossRef]
- Bhanu, V.A.; Wiles, K.B.; Bortner, M.; Glass, T.E.; Godshall, D.; Baird, D.G.; Wilkes, G.L.; McGrath, J.E. Designing the molecular composition of the melt processable acrylonitrile copolymer carbon fiber precursors. Polymer Prep. 2002, 43, 1017. [Google Scholar]
- Mukundan, T.; Bhanu, V.A.; Wiles, K.B.; Bortner, M.; Baird, D.G.; McGrath, J.E. Photocrosslinkable acrylonitrile terpolymers as carbon fiber precursors. Polymer Prepr. 2003, 44, 651. [Google Scholar]
- Czech, Z. New copolymerizable photoinitiators for radiation curing of acrylic PSA. Int. J. Adhes. Adhes. 2007, 27, 195–199. [Google Scholar] [CrossRef]
- Chen, J.; Han, N.; Wu, C.; Sun, Z.; Wu, Y.; Sun, Z.; Wang, L.; Zhang, X. Preparation and properties of pitch plasticized melt spinning polyacrylonitrile-based carbon fibers precursor. Acta Mater. Compos. Sin. 2022, 39, 4540–4550. [Google Scholar]
- Han, S.Y.; Tae, W.S.; Byung, C.K.; Byung, G.M.; Jae, W.C.; Chul, J.L. Heat And Chemical Resistant Acrylic Short Fibers Without Spinning. U.S. Patent 5,401,576, 28 March 1995. [Google Scholar]
- Shiota, H.; Nishida, R.; Kida, T.; Kohara, N.; Watanabe, Y.; Kasahara, R. Process For Producing Homogeneous Phase Melt of Polyacrylonitrile. U.S. Patent 5,681,512 A, 28 October 1997. [Google Scholar]
- Hirotaka, S.; Ryosuke, N.; Takahisa, K.; Noriyuki, K.; Yoshihiro, W.; Ryuichi, K. Acrylonitrile Polymer Compositions, Method For Producing The Compositions, And Method For Producing Shaped Articles From The Compositions. U.S. Patent 5,973,106, 26 October 1999. [Google Scholar]
- Serad, G.A. Melt Extrusion Of Acrylonitrile Polymers. U.S. Patent 3,634,575, 11 January 1972. [Google Scholar]
- Daumit, G.P.; Ko, Y.S.; Slater, C.R.; Venner, J.G.; Young, C.C. Formation of Melt-Spun Acrylic Fibers Which Are Particularly Suited For Thermal Conversion to High Strength Carbon Fibers. U.S. Patent 4,921,656, 1 May 1990. [Google Scholar]
- John, F. Production of Melt Fused Synthetic Fibers Using A Spinneret. U.S. Patent 2003/ 0,020,190 A1, 30 January 2003. [Google Scholar]
- Norman, F. Acrylonitrile Polymer Compositions And Articles And Methods For Their Preparation. U.S. Patent 5,434,205, 18 July 1995. [Google Scholar]
- Edmund, H.; Merz, R.A.; White, J.P.; Fouser, N.F. Acrylonitrile Polymer Compositions And Articles And Methods For Their Preparation. U.S. Patent 5,861,442, 18 January 1999. [Google Scholar]
- Alves, N.P. Thermoplastic Polyacrylonitrile Production Process. U.S. Patent 2011/0,024,939 A1, 3 February 2011. [Google Scholar]
- Alves, N.P. Composition of Polyacrylonitrile/Lignin Blend And Use Thereof In Melt Spinning Carbon Fibre Precursors. U.S. Patent 2018/0,282,535 A1, 4 October 2018. [Google Scholar]
- Chang, H.C.; Chang, S.W.; Kap, S.Y.; Chang, H.L.; Sun, S.C.; Hong, M.K.; Kyung, A.O.; Eun, J.K. Method Of Producing Carbon Fiber. U.S. Patent 1,1268,215 B2, 8 March 2022. [Google Scholar]
- George, P.S. Method of Processing Polyacrylonitrile. U.S. Patent 6,593,451 B1, 15 July 2003. [Google Scholar]
- Yu, M.; Rong, H.; Han, K.; Wang, Z.; Tian, Y.; Zhang, H. Process of Melt-Spinning Polyacrylonitrile Fiber. U.S. Patent 9,644,290 B2, 9 May 2017. [Google Scholar]
- Lutzmann, H.H.; Miller, J.D.I.G.W. Thermoplastic Polyacrylonitrile Compositions. U.S. Patent 7,541,400 B2, 2 June 2009. [Google Scholar]
- Kühne, A.; Möller, M.; Hoffmann, A.; Usselmann, M. Melt-Processable Acrylonitrile-Based Copolymers And Their Acidic Prestabilization For Conversion Into Carbon Fibers And Workpieces. E.P. Patent 3,872,103 A1, 1 September 2021. [Google Scholar]
- Herfurth, C.; Lieske, A.; Hahn, M. Melt Spinnable Copolymers From Polyacrylonitrile, Method For Producing Fibers Or Fiber Precursors By Means Of Melt Spinning, And Fibers Produced Accordingly. W.O. Patent 2017/1,67,355 A1, 5 October 2017. [Google Scholar]
- Benedict, S.; Curatolo, G.S.L. Preparation Of Melt-Processable Acrylonitrile/Methacrylonitrile Copolymers. E.P. Patent 0,492,909 A1, 1 July 1992. [Google Scholar]
- Hahn, M.; Lieske, A.; Knoop, M. Melt Spinnable Copolymers Of Polyacrylonitrile, Method For Producing Fibers Or Threir Precursors By Means Of Melt Spinning And Correspondingly Produced Fibres. E.P. Patent 3,201,248 B1, 7 November 2018. [Google Scholar]
- Knoop, M.T.; Lieske, A. Melt Spinnable Copolymers From Polyacrylonitrile, Method For Producing Fibers Or Fiber Precursors By Means Of Melt Spinning, And Fibers Produced Accordingly. U.S. Patent 2019/0,100,856 A1, 4 April 2019. [Google Scholar]
- Erdmann, J.; Ganster, J. Continuous Method For Producing A Thermally Stabilized Multifilament Thread, Multifilament Thread, And Fiber. U.S. Patent 1,1242,623 B2, 8 February 2022. [Google Scholar]
- Percec, E.S.; Ii Jorkasky, L.E.B.R.J. Carbon Fibers Or Sheets Made From Copolymers Of Acrylonitrile. W.O. Patent 2000/0,506,75 A1, 31 August 2000. [Google Scholar]
- Dugan, J.S. Dissociable Multicomponent Fibers Containing A Polyacrylonitrile Polymer Component. U.S. Patent 6,583,075 B1, 24 June 2003. [Google Scholar]
- Dugan, J.S. Splittable Multicomponent Fibers Containing A Polyacrylonitrile Polymer Component. U.S. Patent 6,444,312 B1, 3 September 2002. [Google Scholar]
- Tsotsis, T.K. Continuous Carbon-Nanotube-Reinforced Polymer Precursors And Carbon Fibers. U.S. Patent 8,642,167 B2, 4 February 2014. [Google Scholar]
- Ball, L.E.; Wu, M.; Wardlow, E. Stabilizers For High Nitrile Multipolymers. U.S. Patent 5,714,535, 3 February 1998. [Google Scholar]
- Ball, L.E.; Richard, J.; Jorkasky, C.E.; Uebele, M.; Wu, M. Melt Spun Acrylonitrile Olefinically Unsaturated Fibers And A Process To Make Fibers. E.P. Patent 0,780,498 B1, 3 December 2016. [Google Scholar]
- Yang, K.-S.; Lee, S.-H.; Chae, H.-H.; Lee, D.-H. Method For Preparing Polyacrylonitrile-Based Polymer For Preparation Of Carbon Fiber Using Microwave And Method For Preparing Carbon Fiber Using The Same. U.S. Patent 8,685,361 B2, 1 April 2014. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernikova, E.V.; Osipova, N.I.; Plutalova, A.V.; Toms, R.V.; Gervald, A.Y.; Prokopov, N.I.; Kulichikhin, V.G. Melt-Spinnable Polyacrylonitrile—An Alternative Carbon Fiber Precursor. Polymers 2022, 14, 5222. https://doi.org/10.3390/polym14235222
Chernikova EV, Osipova NI, Plutalova AV, Toms RV, Gervald AY, Prokopov NI, Kulichikhin VG. Melt-Spinnable Polyacrylonitrile—An Alternative Carbon Fiber Precursor. Polymers. 2022; 14(23):5222. https://doi.org/10.3390/polym14235222
Chicago/Turabian StyleChernikova, Elena V., Natalia I. Osipova, Anna V. Plutalova, Roman V. Toms, Alexander Y. Gervald, Nickolay I. Prokopov, and Valery G. Kulichikhin. 2022. "Melt-Spinnable Polyacrylonitrile—An Alternative Carbon Fiber Precursor" Polymers 14, no. 23: 5222. https://doi.org/10.3390/polym14235222
APA StyleChernikova, E. V., Osipova, N. I., Plutalova, A. V., Toms, R. V., Gervald, A. Y., Prokopov, N. I., & Kulichikhin, V. G. (2022). Melt-Spinnable Polyacrylonitrile—An Alternative Carbon Fiber Precursor. Polymers, 14(23), 5222. https://doi.org/10.3390/polym14235222