
Study on GAP Adhesive-Based Polymer Films, Energetic Polymer Composites and Application


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
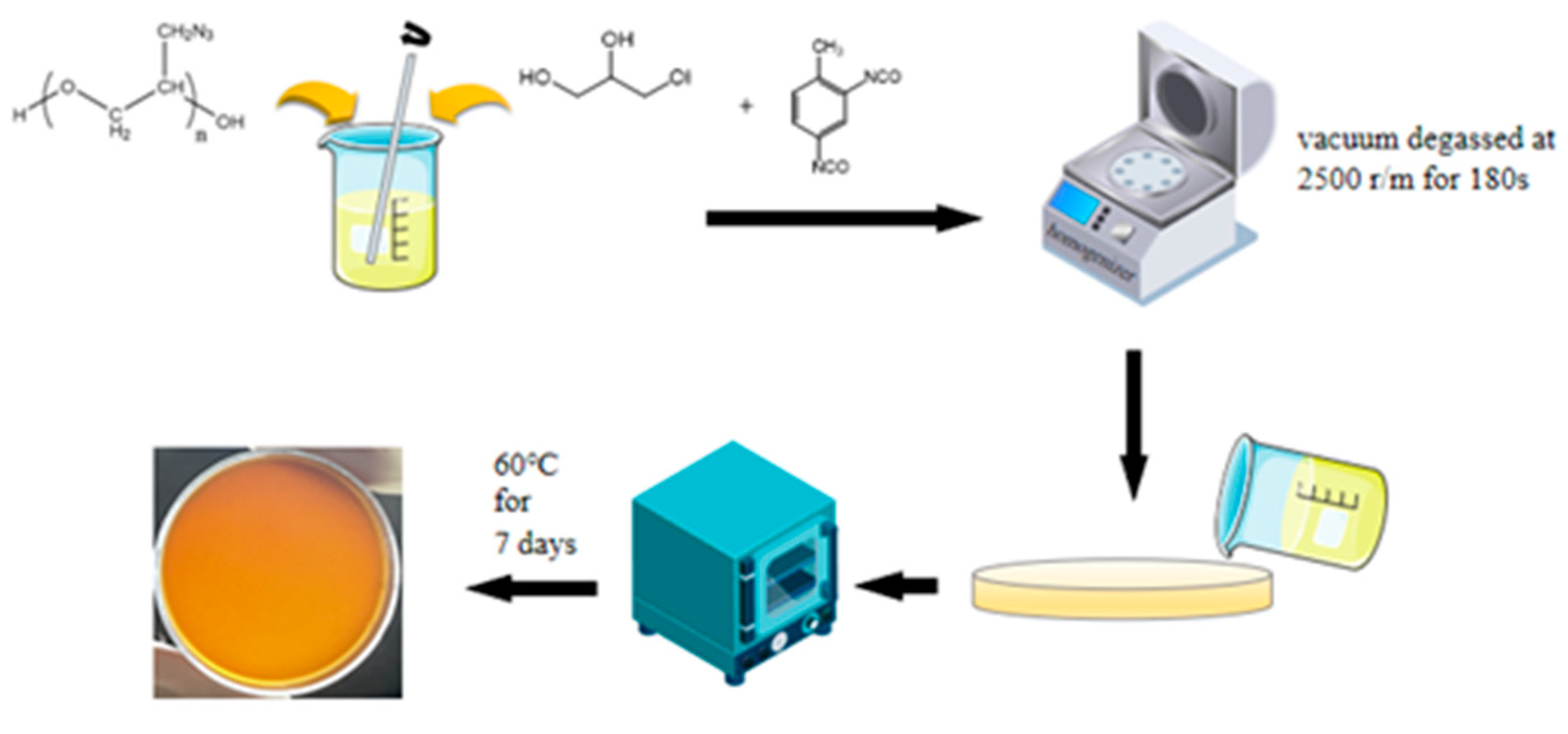
2.2. Preparation of Polymer Films
2.3. Preparation of Energetic Polymer Composites
2.4. Characterization
3. Results and Discussion
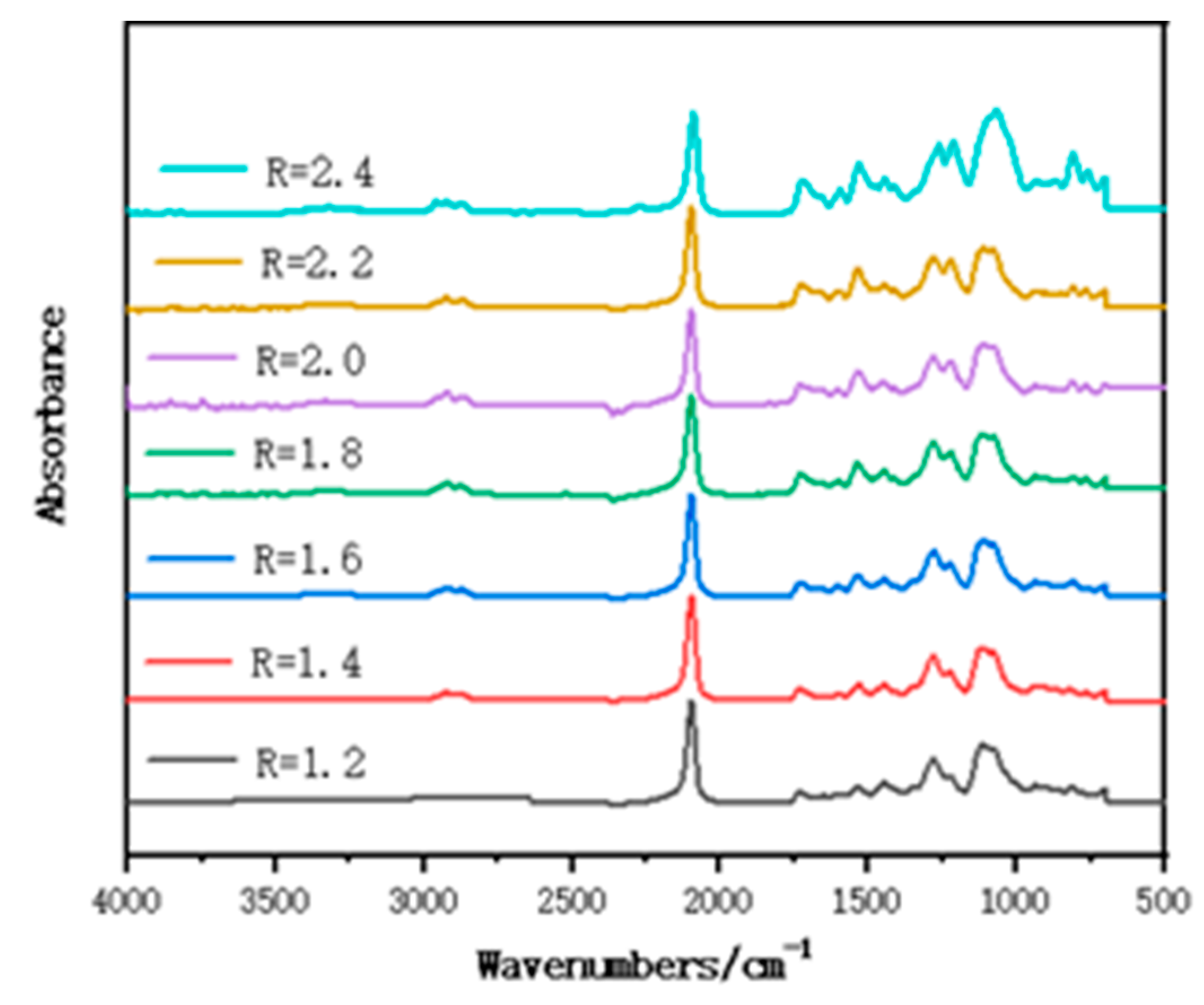
3.1. FT-IR of Polymer Films with Different R
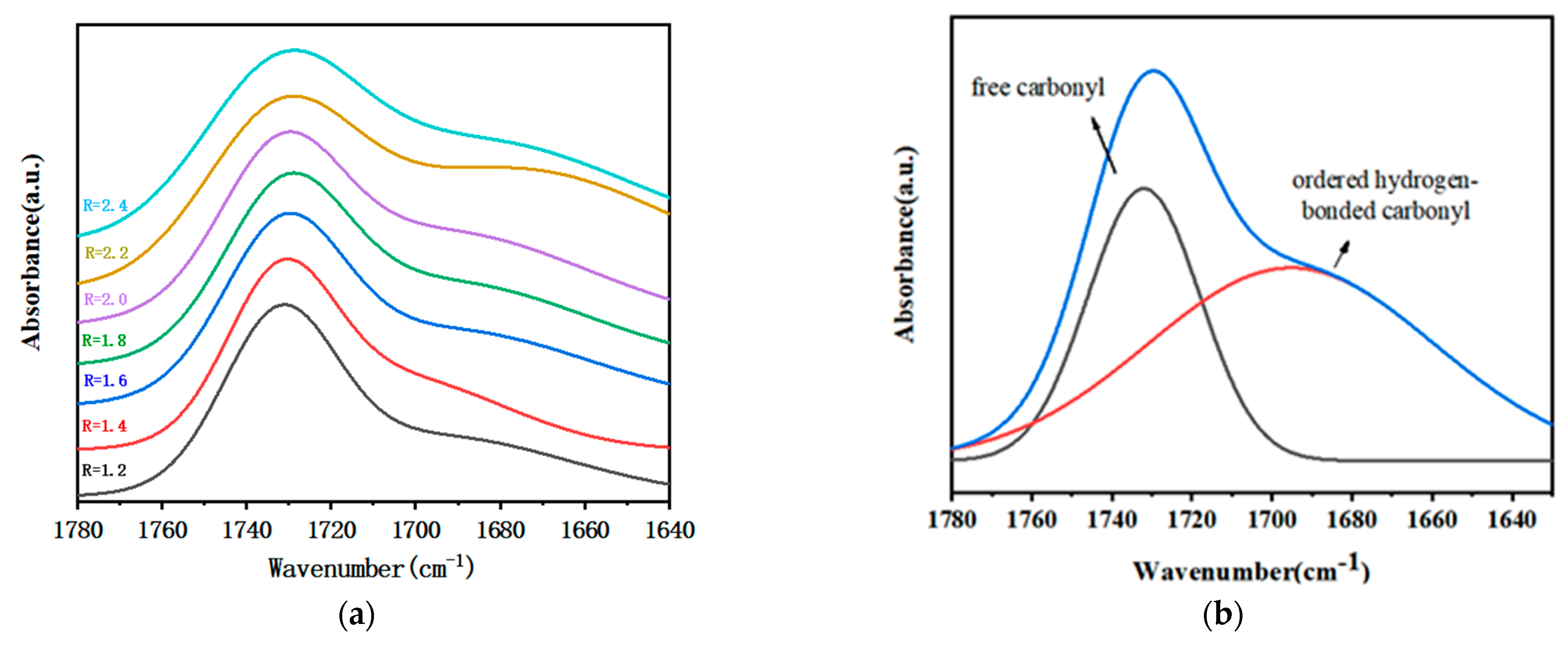
3.2. Crosslink Density of Polymer Films with Different R
3.3. Mechanical Property of Polymer Films with Different R
3.4. The SEM of Tensile Cross-Section of Energetic Polymer Composites
3.5. Mechanical Property of Energetic Polymer Composites with Different RDX Contents
3.6. The Heat of Explosive and Residual Carbon Rate of Energetic Polymer Composites with Different RDX Contents
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boshra, I.K.; Elbeih, A.; Mostafa, H.E. Composite Solid Rocket Propellant Based on GAP Polyurethane Matrix with Different Binder Contents. Chin. J. Explos. Propellants 2020, 43, 6. [Google Scholar]
- Frankel, M.B.; Grant, L.R.; Flanagan, J.E. Historical development of glycidyl azide polymer. J. Propuls. Power 1992, 8, 560–563. [Google Scholar] [CrossRef]
- Dey, A.; Sikder, A.K.; Talawar, M.B.; Chottopadhyay, S. Towards new directions in oxidizers/energetic fillers for composite propellants: An overview. Cent. Eur. J. Energetic Mater. 2015, 12, 377–399. [Google Scholar]
- Selim, K.; Yilmaz, L. Thermal characterization of glycidyl azide polymer (GAP) and GAP-based binders for composite propellants. J. Appl. Polym. Sci. 2000, 77, 538–546. [Google Scholar] [CrossRef]
- Li, M.-M.; Hu, R.; Xu, M.-H.; Wang, Q.-L.; Yang, W.-T. Burning characteristics of high density foamed GAP/CL-20 propellants. Def. Technol. 2021, 18, 1914–1921. [Google Scholar] [CrossRef]
- Xu, M.; Ge, Z.; Lu, X.; Mo, H.; Ji, Y.; Hu, H. Fluorinated glycidyl azide polymers as potential energetic binders. RSC Adv. 2017, 7, 47271–47278. [Google Scholar] [CrossRef] [Green Version]
- Shedge, M.; Tadkod, S.; Murthy, G.; Patel, C. Polyvinyl Acetate Resin as a Binder Effecting Mechanical and CombustionProperties of Combustible Cartridge Case Formulations. Def. Sci. J. 2008, 58, 390–397. [Google Scholar] [CrossRef] [Green Version]
- Robbins, F.; Colburn, J.; Zoltani, C. Combustible Cartridge Cases: Current Status and Future Prospects; Army Ballistic Research Lab.: Aberdeen Proving Ground, MD, USA, 1992. [Google Scholar]
- DeLuca, P.L.; Williams, J.C. Fibrillated polyacrylic fiber in combustible cartridge cases. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 438–441. [Google Scholar] [CrossRef]
- Stacer, R.G.; Husband, D.M. Molecular structure of the ideal solid propellant binder. Propellants Explos. Pyrotech. 1991, 16, 167–176. [Google Scholar] [CrossRef]
- Xu, M.; Ge, Z.; Lu, X.; Mo, H.; Ji, Y.; Hu, H. Structure and mechanical properties of fluorine-containing glycidyl azide polymer-based energetic binders. Polym. Int. 2017, 66, 9. [Google Scholar]
- Fataliev, R.V.; Eneikina, T.A.; Kozlova, L.A.; Gatina, R.F.; Mikhailov, Y.M. Study of an effect of promising cellulose components on the physico-mechanical properties and a burning rate of a combustible material for combustible cartridge cases produced according to the filtration casting technology. J. Phys. Conf. Ser. 2020, 1666, 012054. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Zhang, W.; Fan, X.; Xie, W.; Xu, H.; Liu, Y.; Du, J. GAP Factors affecting the mechanical properties of gap binder films. Chin. J. Explos. Propellants 2016, 39, 79–83. [Google Scholar]
- Ma, S.; Li, Y.; Li, Y.; Li, G.; Luo, Y. Research on the Mechanical Properties and Curing Networks of Energetic GAP/TDI Binders. Cent. Eur. J. Energetic Mater. 2017, 14, 708–725. [Google Scholar] [CrossRef]
- Chen, S.; Shi, H.; Tian, S.; Li, Z.; Xiao, Z. Design and fabrication of a composite coating to improve the environmental adaptability of combustible cartridges. Polym. Compos. 2018, 40, 685–694. [Google Scholar] [CrossRef]
- Yang, Z.; Long, X.-P.; Zeng, Q.-X. Simulation study of the morphologies of energetic block copolymers based on glycidyl azide polymer. J. Appl. Polym. Sci. 2012, 129, 480–486. [Google Scholar] [CrossRef]
- Yuan, S.; Jiang, S.; Luo, Y. Cross-linking network structures and mechanical properties of novel HTPE/PCL binder for solid propellant. Polym. Bull. 2020, 78, 313–334. [Google Scholar] [CrossRef]
- Haska, S.B.; Bayramli, E.; Pekel, F. Adhesion of an HTPB-IPDI-based liner elastomer to composite matrix and metal case. J. Appl. Polym. Sci. 1997, 64, 2355–2362. [Google Scholar] [CrossRef]
- Kakade, S.D.; Navale, S.B.; Narsimhan, V.L. Studies on Interface Properties of Propellant Liner for Case-Bonded Composite Propellants. J. Energetic Mater. 2003, 21, 73–85. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | GAP/% | TDI/% | GLY/% |
|---|---|---|---|
| 1.2 | 89.71 | 8.74 | 1.03 |
| 1.4 | 87.03 | 11.16 | 1.31 |
| 1.6 | 83.81 | 14.04 | 1.65 |
| 1.8 | 79.88 | 17.56 | 2.06 |
| 2.0 | 74.95 | 21.97 | 2.58 |
| 2.2 | 68.60 | 27.65 | 3.25 |
| 2.4 | 60.12 | 35.24 | 4.14 |
| R | Free-Carbonyl | Bonded-Carbonyl | Total-Carbonyl | Bonded-Carbonyl-Ratio (%) |
|---|---|---|---|---|
| 1.2 | 2.0771 | 2.0006 | 4.0778 | 49.06 |
| 1.4 | 3.0331 | 3.1961 | 6.2291 | 51.31 |
| 1.6 | 9.9886 | 3.9343 | 6.9228 | 56.83 |
| 1.8 | 1.5847 | 2.1949 | 3.7796 | 58.07 |
| 2.0 | 0.6030 | 1.0923 | 1.6953 | 64.43 |
| 2.2 | 1.2901 | 2.3794 | 3.6695 | 64.84 |
| 2.4 | 8.8463 | 9.7253 | 16.572 | 58.69 |
| R | νe × 10−4 (mol/cm−3) | ρ (g/cm−3) | E (MPa) | Mc (kg/cm−3) | A |
|---|---|---|---|---|---|
| 1.2 | 1.115 | 1.299 | 0.055 | 1.165 | −0.235 |
| 1.4 | 1.231 | 1.297 | 0.126 | 1.054 | −0.237 |
| 1.6 | 1.302 | 1.295 | 0.226 | 0.995 | −0.240 |
| 1.8 | 4.823 | 1.291 | 2.196 | 0.268 | −0.361 |
| 2.0 | 8.033 | 1.288 | 6.709 | 0.160 | 0.409 |
| 2.2 | 11.06 | 1.283 | 17.001 | 0.116 | 3.157 |
| 2.4 | 12.84 | 1.277 | 71.228 | 0.099 | 20.815 |
| RDX Content (%) | The Heat of Explosive (MJ/kg) | Residue Carbon Rate (%) |
|---|---|---|
| 10 | 2.25 | 6.75 |
| 20 | 2.41 | 5.28 |
| 30 | 2.51 | 4.09 |
| 40 | 2.87 | 2.47 |
| 50 | 3.09 | 2.20 |
| 60 | 3.29 | 1.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Li, X.; Ge, Z.; Luo, Y. Study on GAP Adhesive-Based Polymer Films, Energetic Polymer Composites and Application. Polymers 2023, 15, 1538. https://doi.org/10.3390/polym15061538
Wu S, Li X, Ge Z, Luo Y. Study on GAP Adhesive-Based Polymer Films, Energetic Polymer Composites and Application. Polymers. 2023; 15(6):1538. https://doi.org/10.3390/polym15061538
Chicago/Turabian StyleWu, Siyuan, Xiaomeng Li, Zhen Ge, and Yunjun Luo. 2023. "Study on GAP Adhesive-Based Polymer Films, Energetic Polymer Composites and Application" Polymers 15, no. 6: 1538. https://doi.org/10.3390/polym15061538
APA StyleWu, S., Li, X., Ge, Z., & Luo, Y. (2023). Study on GAP Adhesive-Based Polymer Films, Energetic Polymer Composites and Application. Polymers, 15(6), 1538. https://doi.org/10.3390/polym15061538