
Phenolation to Improve Hardwood Kraft Lignin for Wood Adhesive Application

Abstract
:1. Introduction
2. Materials and Methods
2.1. Kraft Lignin Preparation
2.2. Lignin Content and Neutral Sugar Analyses
2.3. Uronic Acid, Element, and Ash Content Determination
2.4. Phenolation of Kraft Lignin
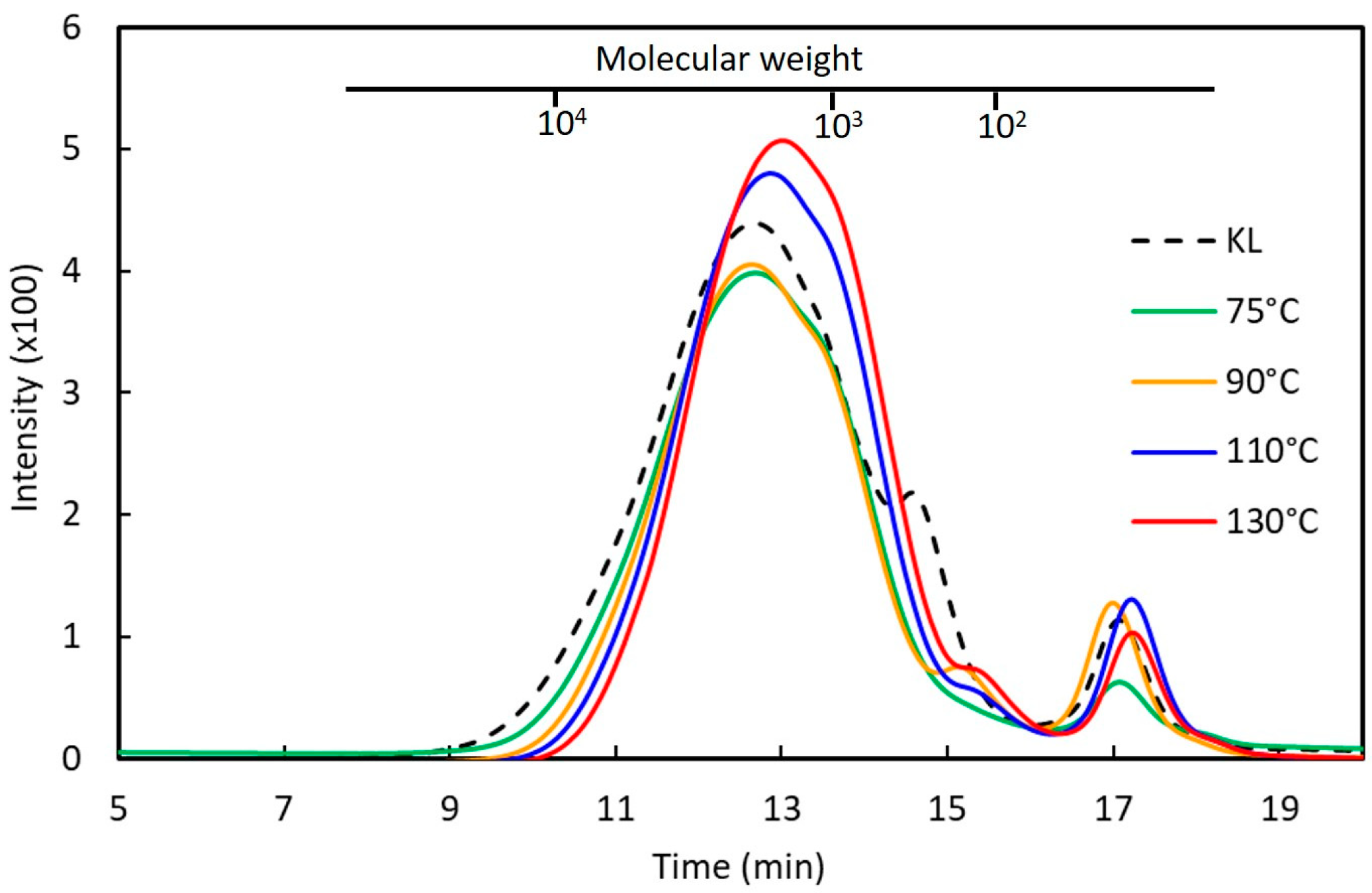
2.5. Molecular Weight Determination
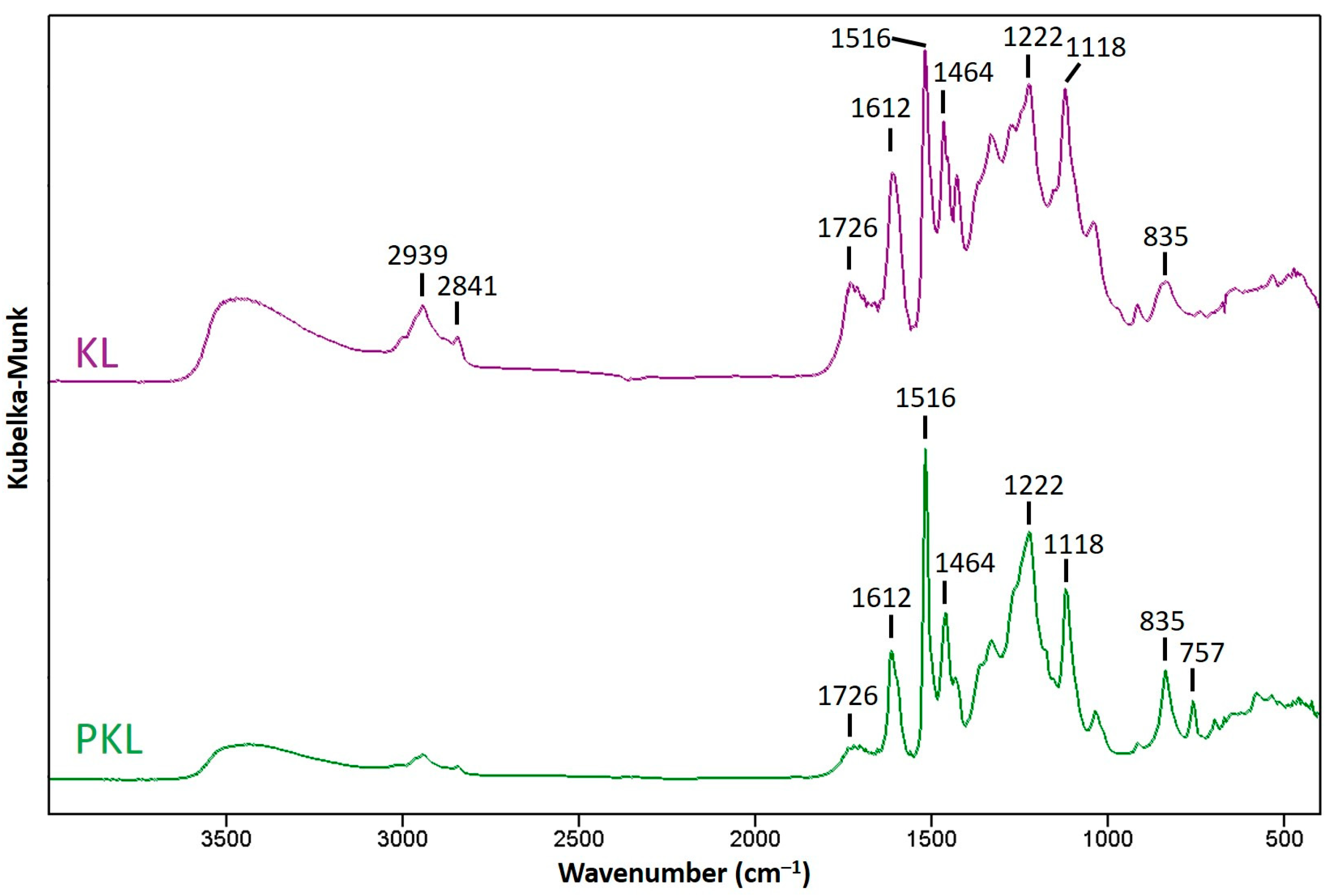
2.6. UV–Vis and Fourier Transform Infrared Spectrometry
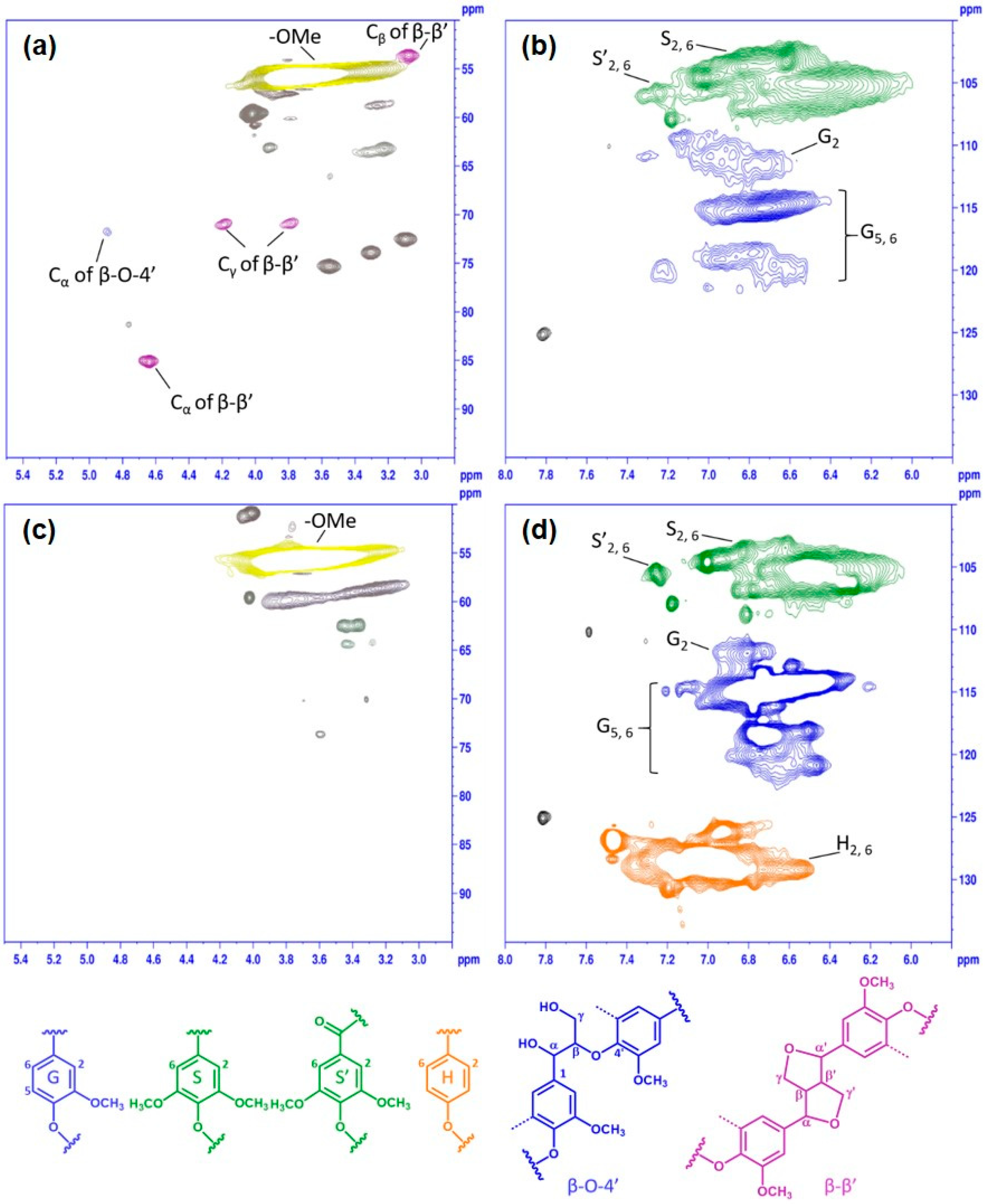
2.7. NMR Analyses of Lignin Samples
2.8. Adhesives Synthesis
2.9. Plywood Bonding Strength and Formaldehyde Emission Evaluation
2.10. Statistical Analyses
3. Results and Discussion
3.1. Basic Properties of the Extractive-Free Kraft Lignin
3.2. Effects of Phenolation Conditions on the Extractive-Free Kraft Lignin
3.3. Chemical Structure Variation in the Phenolated Kraft Lignin
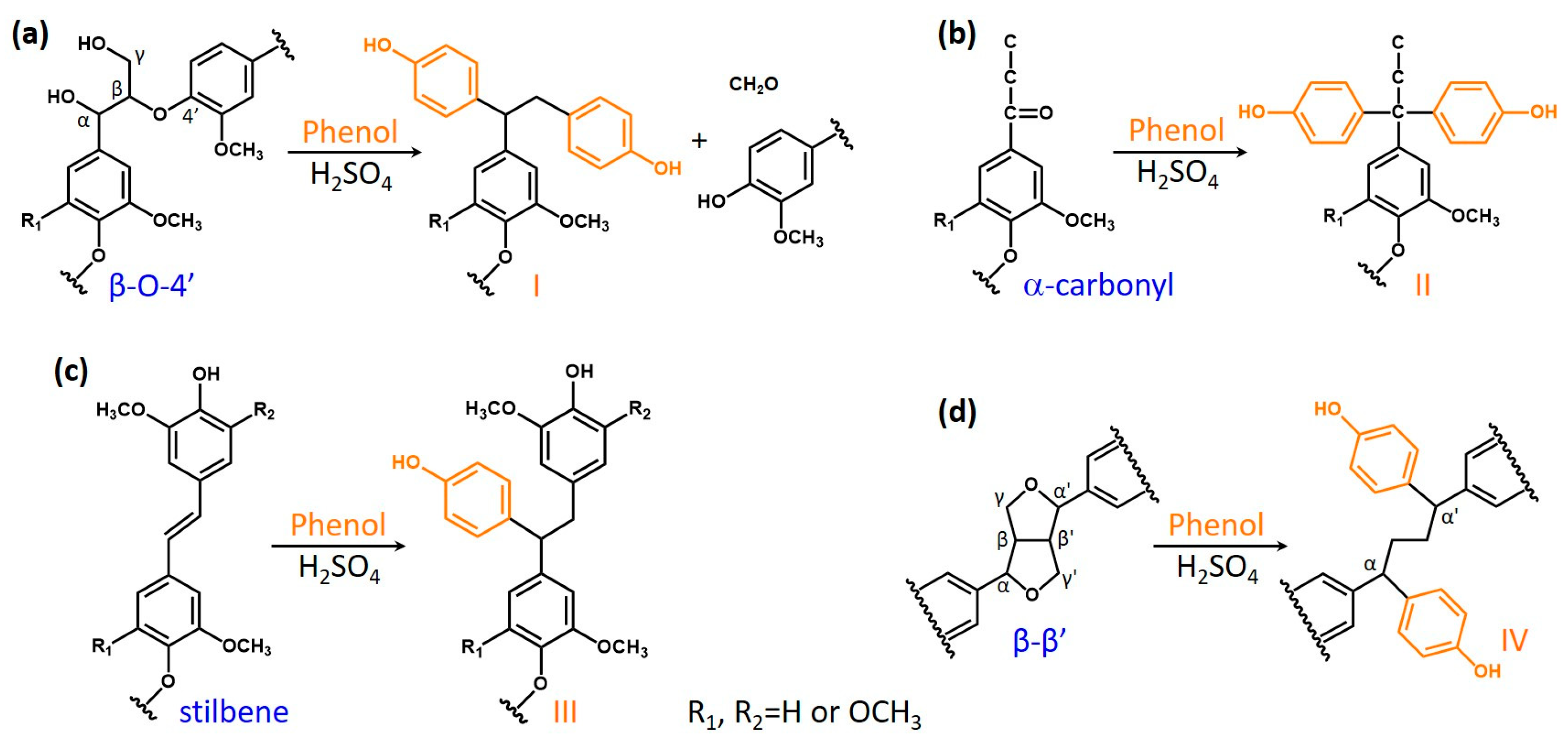
3.4. Plausible Reactions in Acid-Catalyzed Phenolation on Hardwood Kraft Lignin
3.5. Adhesive and Plywood Property Evaluation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuck, C.O.; Perez, E.; Horvath, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Funaoka, M. Sequential transformation and utilization of natural network polymer “LIGNIN”. React. Funct. Polym. 2013, 73, 396–404. [Google Scholar] [CrossRef]
- Berlin, A.; Balakshin, M. Industrial lignin: Analysis, properties, and applications. In Bioenergy Research: Advances and Applications; Gupta, V.K., Kubicek, C.P., Saddler, J., Xu, F., Tuohy, M.G., Eds.; Elsevier: Waltham, MA, USA, 2014; pp. 315–336. [Google Scholar]
- Balakshin, M.Y.; Capanema, E.A.; Sulaeva, I.; Schlee, P.; Huang, Z.E.; Feng, M.; Borghei, M.; Rojas, O.J.; Potthast, A.; Rosenau, T. New opportunities in the valorization of technical lignins. ChemSusChem 2021, 14, 1016–1036. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, A.R.; Wuzella, G.; Kandelbauer, A. Thermal characterization of kraft ligin phenol-formaldehyde resin for paper impregnation. In Wood Adhesives; Pizzi, A., Mittal, K.L., Eds.; CRC Press: New York, NY, USA, 2011; pp. 291–303. [Google Scholar]
- Pilato, L. Phenolic resins: 100 years and still going strong. React. Funct. Polym. 2013, 73, 270–277. [Google Scholar] [CrossRef]
- Xu, C.; Ferdosian, F. Lignin-based phenol-formaldehyde (LPF) resins/adhesives. In Conversion of Lignin into Bio-Based Chemicals and Materials; Springer: Heidelberg, Germany, 2017; pp. 91–109. [Google Scholar]
- Pizzi, A.; Cameron, F.A.; van der Klashorst, G.H. Soda bagasse lignin adhesive for particleboard. In Adhesives from Renewable Resources; ACS Symposium Series 385; American Chemical Society: Washington, DC, USA, 1989; pp. 82–95. [Google Scholar]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Kalami, S.; Chen, N.S.; Borazjani, H.; Nejad, M. Comparative analysis of different lignins as phenol replacement in phenolic adhesive formulations. Ind. Crops Prod. 2018, 125, 520–528. [Google Scholar] [CrossRef]
- Funaoka, M.; Abe, I.; Chiang, V.L. Nucleus exchange reaction. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: New York, NY, USA, 1992; pp. 369–386. [Google Scholar]
- Bertella, S.; Luterbacher, J.S. Simultaneous extraction and controlled chemical functionalization of hardwood lignin for improved phenolation. Green. Chem. 2021, 23, 3459–3467. [Google Scholar] [CrossRef]
- Podschun, J.; Stucker, A.; Saake, B.; Lehnen, R. Structure-function relationships in the phenolation of lignins from different sources. ACS Sustain. Chem. Eng. 2015, 3, 2526–2532. [Google Scholar] [CrossRef]
- Vazquez, G.; Gonzalez, J.; Freire, S.; Antorrena, G. Effect of chemical modification of lignin on the gluebond performance of lignin-phenolic resins. Bioresour. Technol. 1997, 60, 191–198. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, J.; Du, X.Y.; Hu, Z.J.; Chang, H.M.; Jameel, H. Phenolation to improve lignin reactivity toward thermosets application. ACS Sustain. Chem. Eng. 2018, 6, 5504–5512. [Google Scholar] [CrossRef]
- Ou, J.F.; Li, S.X.; Li, W.Y.; Liu, C.G.; Ren, J.L.; Yue, F.X. Revealing the structural influence on lignin phenolation and its nanoparticle fabrication with tunable sizes. ACS Sustain. Chem. Eng. 2022, 10, 14845–14854. [Google Scholar] [CrossRef]
- Tran, N.T.; Ko, Y.; Kim, S.; Moon, J.; Choi, J.W.; Kim, K.H.; Kim, C.S.; Ha, J.M.; Kim, H.; Jeong, K.; et al. Microwave-assisted phenolation of acid-insoluble Klason lignin and its application in adhesion. Green Chem. 2022, 24, 2051–2061. [Google Scholar] [CrossRef]
- Maree, C.; Gorgens, J.F.; Tyhoda, L. Lignin phenol formaldehyde resins synthesised using south African spent pulping liquor. Waste Biomass Valorization 2022, 13, 3489–3507. [Google Scholar] [CrossRef]
- Falireas, P.G.; Gracia-Vitoria, J.; Hensen, A.; Vanbroekhoven, K.; Vendamme, R. Incorporating phenolated lignin into polyurethane materials: Impact on mechanical, thermal, and adhesion performance. Ind. Eng. Chem. Res. 2024, 63, 3921–3935. [Google Scholar] [CrossRef]
- Wang, L.Y.; Lagerquist, L.; Zhang, Y.C.; Koppolu, R.; Tirri, T.; Sulaeva, I.; von Schoultz, S.; Vahasalo, L.; Pranovich, A.; Rosenau, T.; et al. Tailored thermosetting wood adhesive based on well-defined hardwood lignin fractions. ACS Sustain. Chem. Eng. 2020, 8, 13517–13526. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, X.; Lin, J.; Zhao, G.; Chang, H.-m.; Jameel, H. Reactivity improvement by phenolation of wheat straw lignin isolated from a biorefinery process. N. J. Chem. 2019, 43, 2238–2246. [Google Scholar] [CrossRef]
- Jardim, J.M.; Hart, P.W.; Lucia, L.; Jameel, H. Insights into the potential of hardwood kraft lignin to be a green platform material for emergence of the biorefinery. Polymers 2020, 12, 1795. [Google Scholar] [CrossRef]
- Saulnier, B.K.; Siahkamari, M.; Singh, S.K.; Nejad, M.; Hodge, D.B. Effect of dilute acid pretreatment and lignin extraction conditions on lignin properties and suitability as a phenol replacement in phenol-formaldehyde wood adhesives. J. Agric. Food Chem. 2023, 71, 592–602. [Google Scholar] [CrossRef]
- Dence, C.W. The determination of lignin. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: New York, NY, USA, 1992; pp. 34–40. [Google Scholar]
- Blakeney, A.B.; Harris, P.J.; Henry, R.J.; Stone, B.A. A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydr. Res. 1983, 113, 291–299. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Liu, J.; Du, J.; Hu, Z.; Du, X.; Jameel, H.; Chang, H.m. A novel phenolation process of softwood kraft lignin for adhesive application. In Proceedings of the 18th International Symposium on Wood, Fiber and Pulp Chemistry (ISWFPC), Vienna, Austria, 9–11 September 2015; pp. 177–180. [Google Scholar]
- Gellerstedt, G. Gellerstedt, G. Gel permeation chromatography. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: New York, NY, USA, 1992; pp. 487–497. [Google Scholar]
- Hu, Z.J.; Du, X.Y.; Liu, J.; Chang, H.M.; Jameel, H. Structural characterization of pine kraft lignin: BioChoice lignin vs Indulin AT. J. Wood Chem. Technol. 2016, 36, 432–446. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A. Comprehensive structural analysis of biorefinery lignins with a quantitative 13C NMR approach. RSC Adv. 2015, 5, 87187–87199. [Google Scholar] [CrossRef]
- CNS 5808; Methods of Test for Strength Properties of Adhesives for Wood in Shear by Tension Loading. Bureau of Standards, Metrology and Inspection: Taipei, Taiwan, 1980.
- CNS 1349; Plywood. Bureau of Standards, Metrology and Inspection: Taipei, Taiwan, 2014.
- CNS 12001; Phenol Resin Adhesives for Wood. Bureau of Standards, Metrology and Inspection: Taipei, Taiwan, 1987.
- Alen, R.; Patja, P.; Sjostrom, E. Carbon dioxide precipitation of lignin from pine kraft black liquor. Tappi J. 1979, 62, 108–110. [Google Scholar]
- Alen, R.; Sjostrom, E.; Vaskikari, P. Carbon dioxide precipitation of lignin from alkaline pulping liquors. Cellul. Chem. Technol. 1985, 19, 537–541. [Google Scholar]
- Evdokimov, A.N.; Kurzin, A.V.; Fedorova, O.V.; Lukanin, P.V.; Kazakov, V.G.; Trifonova, A.D. Desulfurization of kraft lignin. Wood Sci. Technol. 2018, 52, 1165–1174. [Google Scholar] [CrossRef]
- Yang, S.; Wen, J.L.; Yuan, T.Q.; Sun, R.C. Characterization and phenolation of biorefinery technical lignins for lignin-phenol-formaldehyde resin adhesive synthesis. RSC Adv. 2014, 4, 57996–58004. [Google Scholar] [CrossRef]
- Gan, L.H.; Pan, X.J. Phenol-enhanced depolymerization and activation of kraft lignin in alkaline medium. Ind. Eng. Chem. Res. 2019, 58, 7794–7800. [Google Scholar] [CrossRef]
- Faix, O. Fourier transform infrared spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer-Verlag: New York, NY, USA, 1992; pp. 83–109. [Google Scholar]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds; John Wiley & Sons, Inc.: New York, NY, USA, 1991; p. 419. [Google Scholar]
- Alonso, M.V.; Oliet, M.; Rodriguez, F.; Garcia, J.; Gilarranz, M.A.; Rodriguez, J.J. Modification of ammonium lignosulfonate by phenolation for use in phenolic resins. Bioresour. Technol. 2005, 96, 1013–1018. [Google Scholar] [CrossRef]
- Balakshin, M.Y.; Capanema, E.A.; Santos, R.B.; Chang, H.M.; Jameel, H. Structural analysis of hardwood native lignins by quantitative 13C NMR spectroscopy. Holzforschung 2016, 70, 95–108. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, R.K.; Kollman, M.; Chang, H.m.; Jameel, H. Structure and property variations of mixed hardwood kraft lignins. J. Wood Chem. Technol. 2023, 43, 320–336. [Google Scholar] [CrossRef]
- Luo, B.; Jia, Z.; Jiang, H.R.; Wang, S.F.; Min, D.Y. Improving the reactivity of sugarcane bagasse kraft lignin by a combination of fractionation and phenolation for phenol-formaldehyde adhesive applications. Polymers 2020, 12, 1825. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T. Revisiting the mechanism of β-O-4 bond cleavage during acidolysis of lignin: Part 6. A review. J. Wood Chem. Technol. 2015, 35, 27–42. [Google Scholar] [CrossRef]
- Lin, L.Z.; Nakagame, S.; Yao, Y.G.; Yoshioka, M.; Shiraishi, N. Liquefaction mechanism of β-O-4 lignin model compound in the presence of phenol under acid catalysis: Part 2. Reaction behavior and pathways. Holzforschung 2001, 55, 625–630. [Google Scholar] [CrossRef]
- Funaoka, M.; Abe, I. Phenyl nucleus-exchange method for the degradation of lignin. Wood Sci. Technol. 1987, 21, 261–279. [Google Scholar] [CrossRef]
- Li, S.X.; Shi, L.L.; Wang, C.; Yue, F.X.; Lu, F.C. Naphthalene structures derived from lignins during phenolation. ChemSusChem 2020, 13, 5549–5555. [Google Scholar] [CrossRef]
- Santos, J.; Delgado, N.; Fuentes, J.; Fuentealba, C.; Vega-Lara, J.; Garcia, D.E. Exterior grade plywood adhesives based on pine bark polyphenols and hexamine. Ind. Crops Prod. 2018, 122, 340–348. [Google Scholar] [CrossRef]
- Pilato, L. Resin chemistry. In Phenolic Resins: A Century of Progress; Pilato, L., Ed.; Springer: Berlin, Germany, 2010; pp. 41–91. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Weight Percentage 2 |
|---|---|
| Total lignin | 87.5 ± 0.3 |
| Acid-soluble | 4.9 ± 0.2 |
| Acid-insoluble | 82.5 ± 0.2 |
| Residual polysaccharides 1 | 6.6 ± 0.1 |
| Rhamnan | 0.4 ± 0.1 |
| Arabinan | 0.1 ± 0.1 |
| Xylan | 3.9 ± 0.0 |
| Mannan | 0.1 ± 0.0 |
| Galactan | 0.0 ± 0.1 |
| Glucan | 2.0 ± 0.1 |
| Uronic acid | 0.6 ± 0.3 |
| Sample | Temperature (°C) | Mw (g mol−1) | Polydispersity (Mw/Mn) | PKL Yield 2 (%) |
|---|---|---|---|---|
| KL | 2181 | 2.60 | ||
| PKLs 1 | 75 | 1940 | 2.07 | 115 |
| 90 | 1760 | 2.02 | 127 | |
| 110 | 1539 | 1.94 | 127 | |
| 130 | 1347 | 1.91 | 126 |
| Sample | Time (h) | H2SO4 (%) | Mw (g mol−1) | Polydispersity (Mw/Mn) | PKL Yield 2 (%) |
|---|---|---|---|---|---|
| KL | 2181 | 2.60 | |||
| PKLs 1 | 1 | 5 | 1799 | 2.39 | 110 |
| 2 | 5 | 1760 | 2.02 | 127 | |
| 4 | 5 | 1703 | 1.95 | 128 | |
| 2 | 1 | 1775 | 2.41 | 103 | |
| 2 | 10 | 1676 | 1.61 | 121 |
| Adhesive 1 | Viscosity (cP) | Dry Strength 2 (MPa) | Wet Strength 2 (MPa) | Formaldehyde Emission 2 (μg L−1) |
|---|---|---|---|---|
| PF | 75 | 2.07 ± 0.04 * | 1.46 ± 0.03 * | 3.9 ± 0.9 * |
| KLPF | 225 | 1.12 ± 0.04 | 0.75 ± 0.02 | 3.8 ± 0.1 * |
| PKLPF-90 | 725 | 1.71 ± 0.04 * | 1.10 ± 0.02 * | 1.1 ± 0.3 * |
| PKLPF-130 | 3625 | 1.64 ± 0.06 * | 1.06 ± 0.01 * | 0.8 ± 0.1 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.-Y.; Chiang, W.-S.; Chang, H.-m.; Yeh, T.-F. Phenolation to Improve Hardwood Kraft Lignin for Wood Adhesive Application. Polymers 2024, 16, 1923. https://doi.org/10.3390/polym16131923
Liu L-Y, Chiang W-S, Chang H-m, Yeh T-F. Phenolation to Improve Hardwood Kraft Lignin for Wood Adhesive Application. Polymers. 2024; 16(13):1923. https://doi.org/10.3390/polym16131923
Chicago/Turabian StyleLiu, Li-Yuan, Wan-Shuan Chiang, Hou-min Chang, and Ting-Feng Yeh. 2024. "Phenolation to Improve Hardwood Kraft Lignin for Wood Adhesive Application" Polymers 16, no. 13: 1923. https://doi.org/10.3390/polym16131923