


Synthesis of Poly(L–lactide)–poly(ε–caprolactone)–poly(ethylene glycol) Terpolymer Grafted onto Partially Oxidized Carbon Nanotube Nanocomposites for Drug Delivery

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation, Purification, and Chemical Modification of the Prepared CNTs
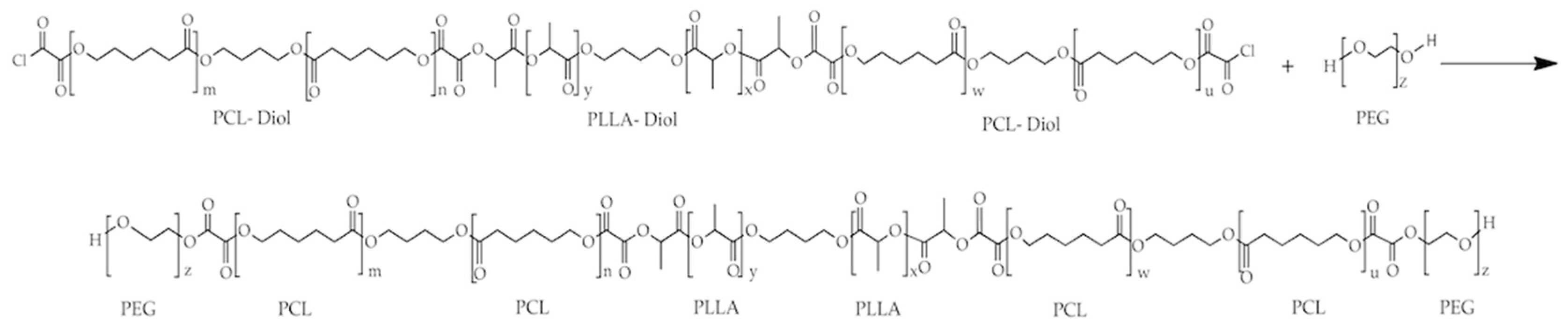
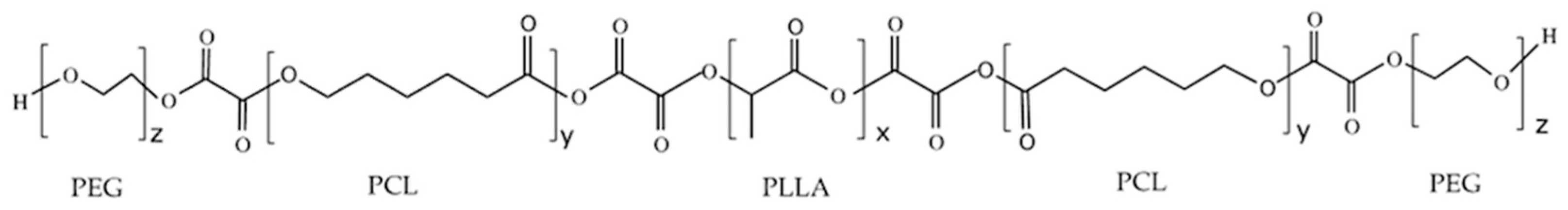
2.3. Synthesis of PLLA–PCL–PEG Terpolymers
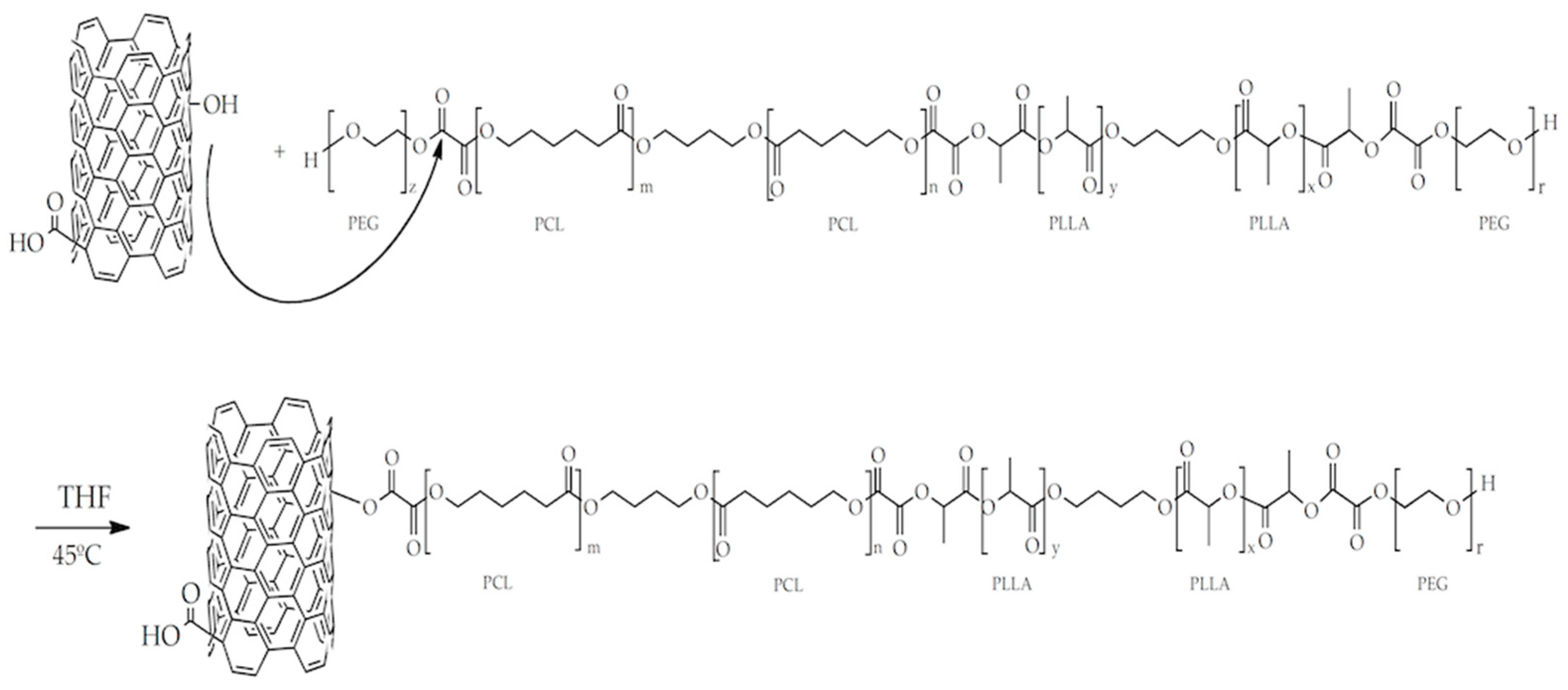
2.4. Synthesis of PLLA–PCL–PEG/CNTpo Nanocomposites
2.5. Characterization of CNTspo, PLLA–PCL–PEG Terpolymers, and PLLA–PCL–PEG/CNTpo Nanocomposites
2.5.1. Characterization of the Functionalized CNTs
2.5.2. GPC
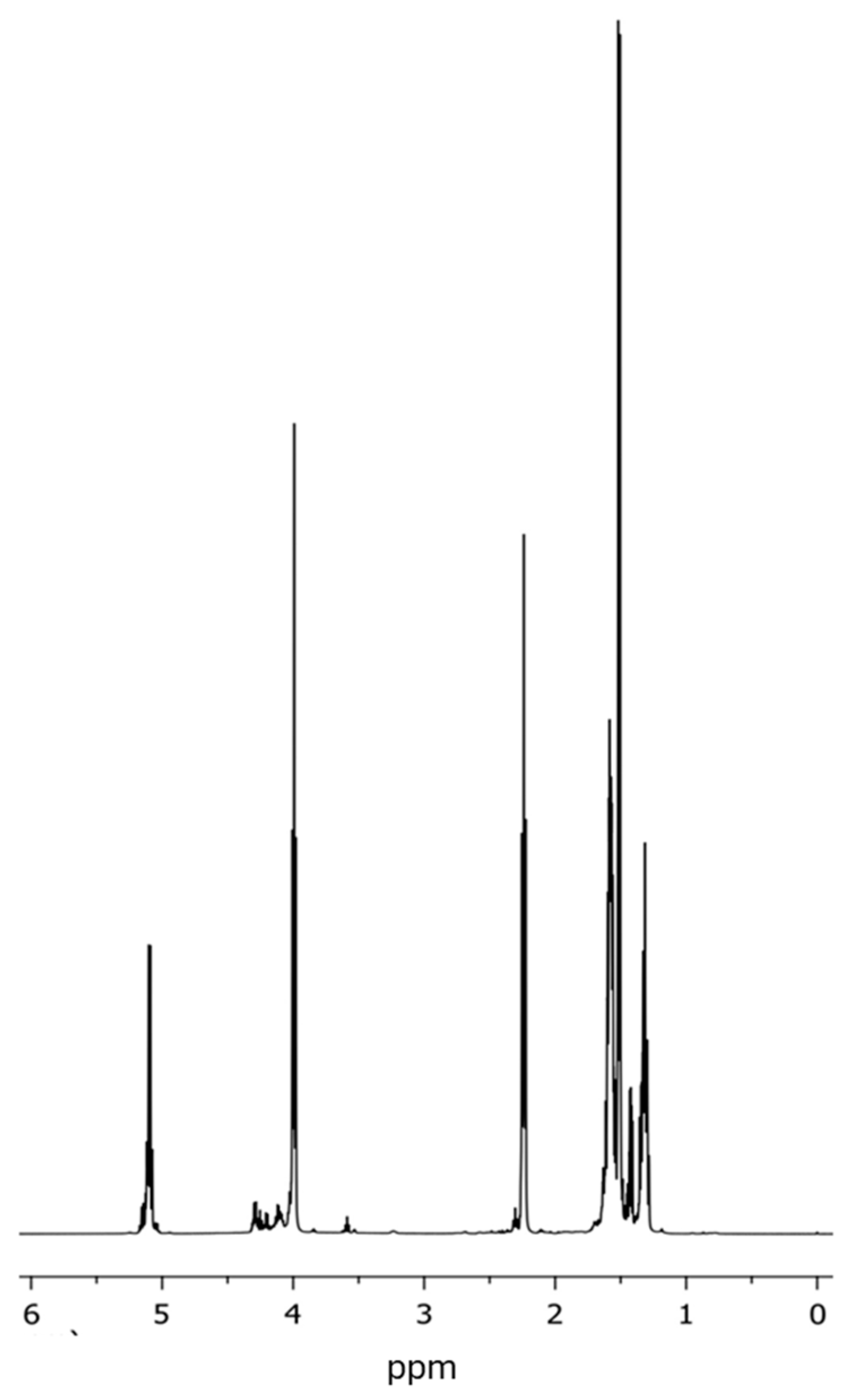
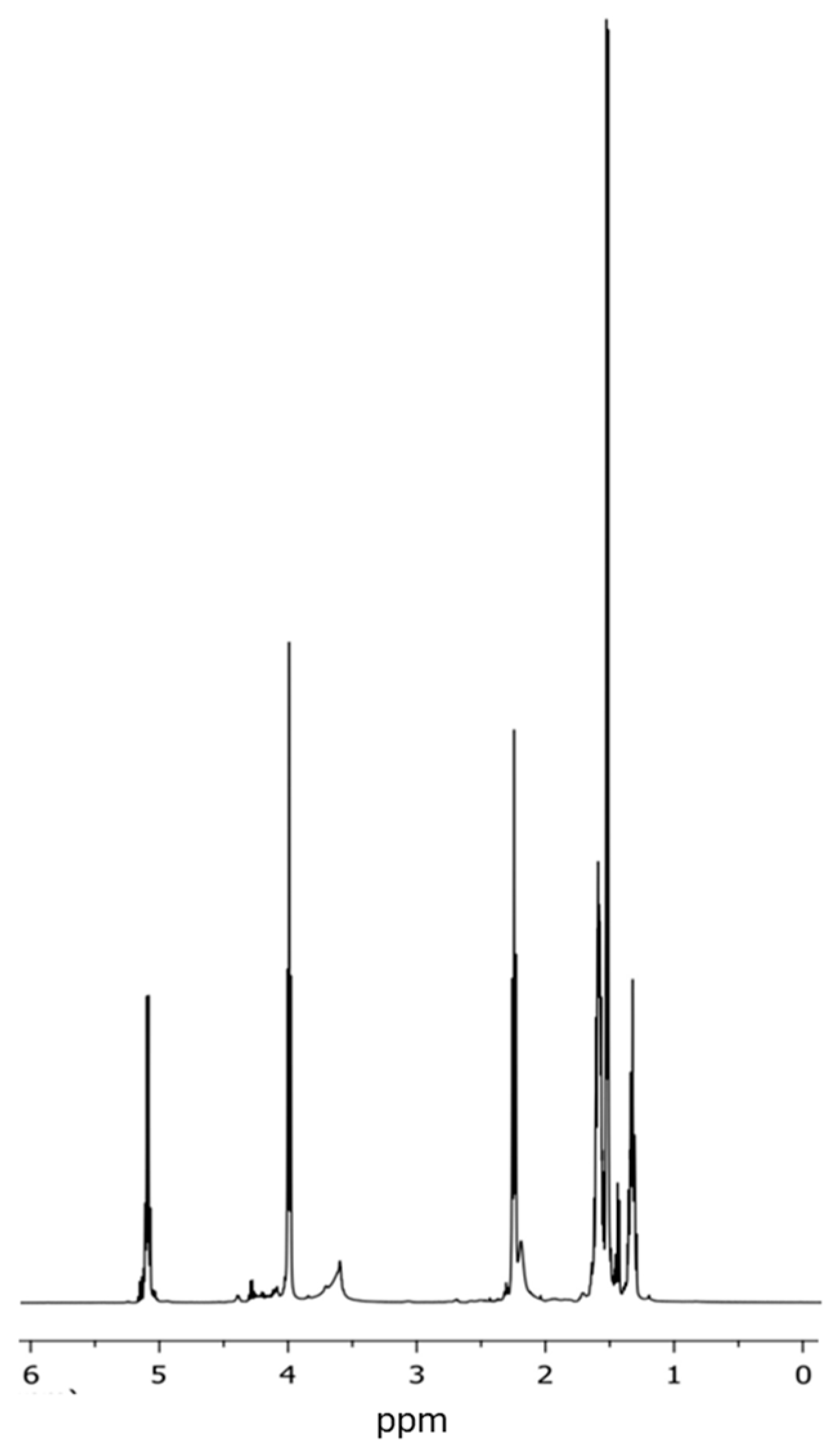
2.5.3. Proton and Carbon–13 Nuclear Magnetic Resonance (1H– and 13C–NMR)
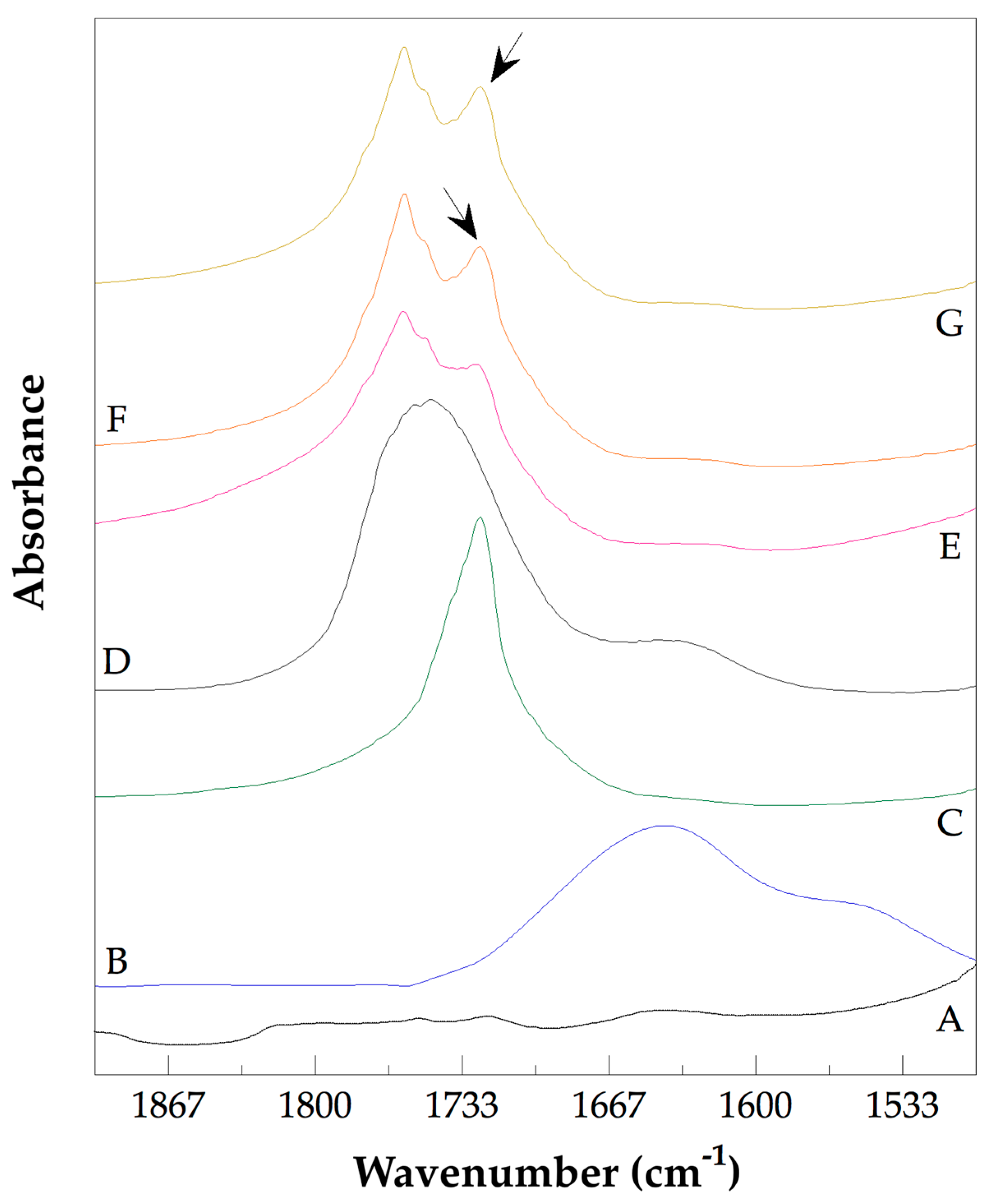
2.5.4. Fourier-Transform Infrared Spectroscopy (FTIR)
2.5.5. XPS
2.5.6. DSC
2.5.7. Ultraviolet–Visible (UV–vis) Spectroscopy
3. Results and Discussion
3.1. Characterization of the Prepared and Functionalized CNTs
3.2. Characterization via GPC of the Homopolymers of PLLA and PCL
3.3. Analysis of the Chemical Structure of the PLLA–PCL Copolymers, PLLA–PCL–PEG Terpolymers, and PLLA–PCL–PEG/CNTpo Nanocomposites
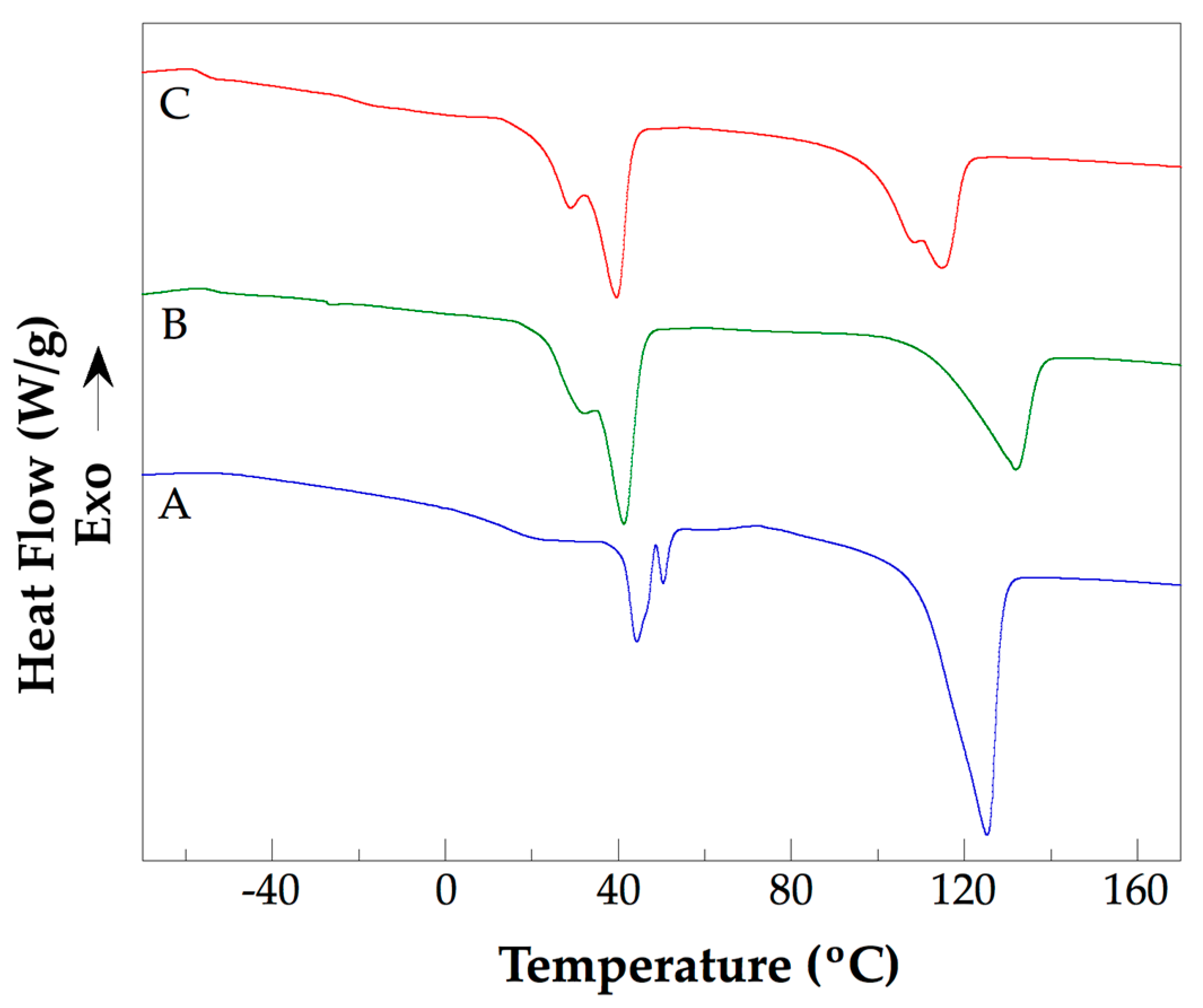
3.4. Thermal Analysis of the PLLA–PCL–PEG Terpolymers and PLLA–PCL–PEG/CNTpo Nanocomposites
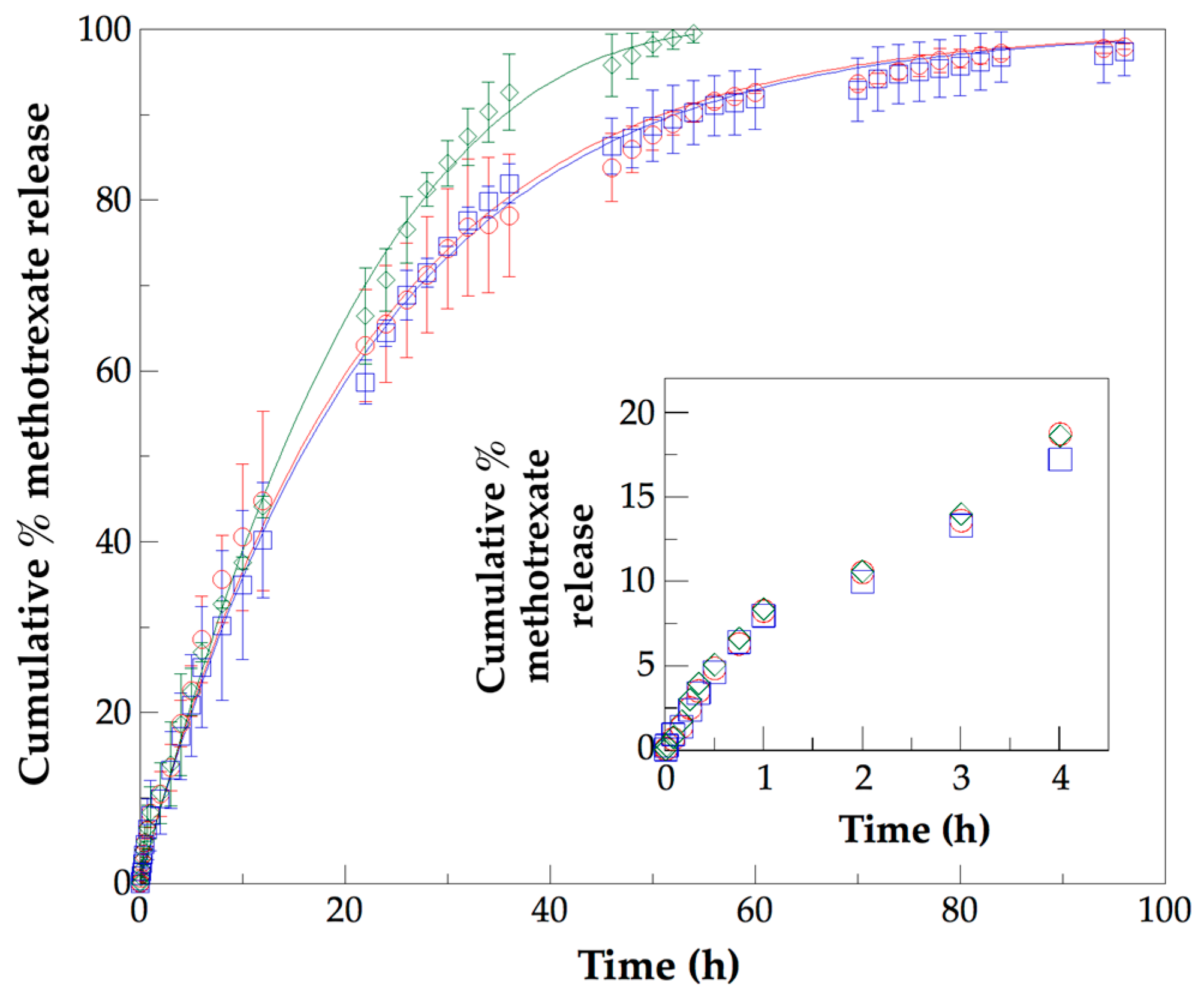
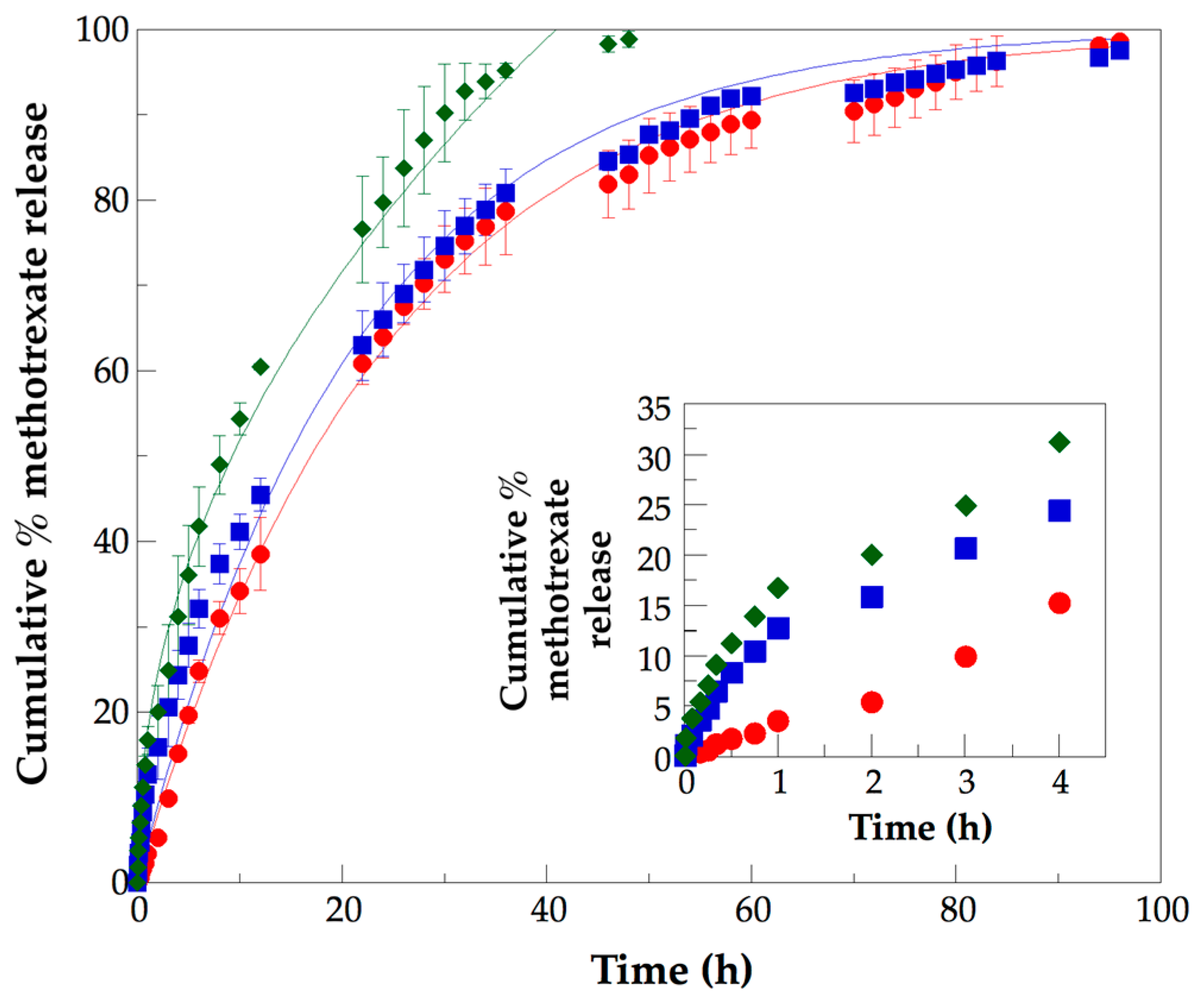
3.5. Analysis of In Vitro Methotrexate Release Profiles from PLLA–PCL–PEG Terpolymers and PLLA–PCL–PEG/CNTpo Nanocomposites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopes, J.R.; Santos, G.; Barata, P.; Oliveira, R.; Lopes, C.M. Physical and chemical stimuli–responsive drug delivery systems: Targeted delivery and main routes of administration. Curr. Pharm. Des. 2013, 19, 7169–7184. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.F.; Ferreira, P.C.; Alves, P.; Cordeiro, R.; Fonseca, A.C.; Góis, J.R.; Gil, M.H. Drug delivery systems: Advances technologies potentially applicable in personalized treatments. EPMA J. 2010, 1, 164–209. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Holá, K.; Ŝubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted drug delivery with polymers and magnetic nanoparticles: Covalent and noncovalent approaches, release control, and clinical studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef]
- Oladipo, A.O.; Lebelo, S.L.; Msagati, T.A.M. Nanocarrier design–function relationship: The prodigious role of properties in regulation biocompatibility for drug delivery applications. Chem. Biol. Interact. 2023, 377, 110466. [Google Scholar] [CrossRef] [PubMed]
- Raquez, J.-M.; Habibi, Y.; Murariu, M.; Dubois, P. Polylactide (PLA)–based nanocomposites. Prog. Polym. Sci. 2013, 38, 1504–1542. [Google Scholar] [CrossRef]
- Saska, S.; Pilatti, L.; Blay, A.; Shibli, J.A. Bioresorbable polymers: Advanced materials and 4D printing for tissue engineering. Polymers 2021, 13, 563. [Google Scholar] [CrossRef]
- Tokiwa, Y.; Jarerat, A. Biodegradation of poly(L–lactide). Biotechnol. Lett. 2004, 26, 771–777. [Google Scholar] [CrossRef]
- Sosnowski, S.; Gadzinowski, M.; Slomkowski, S. Poly(L,L–lactide) microspheres by ring–opening polymerization. Macromol. 1996, 29, 4556–4564. [Google Scholar] [CrossRef]
- Brzeziński, M.; Biela, T. Polylactide nanocomposites with functionalized carbon nanotubes and their stereocomplexes: A focused review. Mater. Lett. 2014, 121, 244–250. [Google Scholar] [CrossRef]
- El Yousfi, R.; Brahmi, R.; Dalli, M.; Achalhi, N.; Azougagh, O.; Tahani, A.; Touzani, R.; El Idrissi, A. Recent advances in nanoparticle development for drug delivery: A comprehensive review of polycaprolactone–based multi–arm architerctures. Polymers 2023, 15, 1835. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer–Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Tambralli, A.; Blakeney, B.; Anderson, J.; Kushwaha, M.; Andukuri, A.; Dean, D.; Jun, H.-W. A hybrid biomimetic scaffold composed of electrospun polycaprolactone nanofibers and self–assembled peptide amphiphile nanofibers. Biofabrication 2009, 1, 025001. [Google Scholar] [CrossRef] [PubMed]
- Aghdam, R.M.; Najarian, S.; Shakhesi, S.; Khanlari, S.; Shaabani, K.; Sharifi, S. Investigating the effect of PGA on physical and mechanical properties of electrospun PCL/PGA blend nanofibers. J. Appl. Polym. Sci. 2012, 124, 123–131. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Wei, K.; Zhao, N.; Zhang, S.; Chen, J. Drug distribution within poly(ε–caprolactone) microspheres and in vitro release. J. Mater. Process. Technol. 2009, 209, 348–354. [Google Scholar] [CrossRef]
- Constantino, V.R.L.; Figueiredo, M.P.; Magri, V.R.; Eulálio, D.; Cunha, V.R.R.; Alcântara, A.C.S.; Perotti, G.F. Biomaterials based on organic polymers and layered double hydroxides nanocomposites: Drug delivery and tissue engineering. Pharmaceutics 2023, 15, 413. [Google Scholar] [CrossRef]
- Yadav, D.; Dewangan, H.K. PEGYLATION: An important approach for novel drug delivery system. J. Biomater. Sci. Polym. Ed. 2021, 32, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Tian, H.; Tang, Z.; Zhuang, X.; Chen, X.; Jing, X. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012, 37, 237–280. [Google Scholar] [CrossRef]
- Tamboli, V.; Mishra, G.P.; Mitra, A.K. Novel pentablock copolymer (PLA–PCL–PEG–PCL–PLA) based nanoparticles for controlled drug delivery: Effect of copolymer compositions on the crystallinity of copolymers and in vitro drug release profile from nanoparticles. Colloid Polym. Sci. 2013, 291, 1235–1245. [Google Scholar] [CrossRef]
- Ramasamy, T.; Ruttala, H.B.; Gupta, B.; Poudel, B.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Smart chemistry–based nanosized drug delivery systems for systemic applications: A comprehensive review. J. Control. Release 2017, 258, 226–253. [Google Scholar] [CrossRef]
- Celentano, W.; Pizzocri, M.; Moncalvo, F.; Pessina, F.; Matteoli, M.; Cellesi, F.; Passoni, L. Functional poly(ε–caprolactone)/poly(ethylene glycol) copolymers with complex topologies for doxorubicin delivery to a proteinase–rich tumor environment. ACS Appl. Polym. Mater. 2022, 4, 8043–8056. [Google Scholar] [CrossRef]
- Rad, A.H.; Asiaee, F.; Jafari, S.; Shayanfar, A.; Lavasanifar, A.; Molavi, O. Poly(ethylene glycol)–poly(ε–caprolactone)–based micelles for solubilization and tumor–targeted delivery of silibinin. Bioimpacts 2020, 10, 87–95. [Google Scholar]
- Chu, B.; Zhang, L.; Qu, Y.; Chen, X.; Peng, J.; Huang, Y.; Qian, Z. Synthesis, characterization and drug loading property of monomethoxy–poly(ethylene glycol)–poly(ε–caprolactone)–poly(D,L–lactide) (MPEG–PCLA) copolymers. Sci. Rep. 2016, 6, 34069. [Google Scholar] [CrossRef]
- Jikei, M.; Takeyama, Y.; Yamadoi, Y.; Shinbo, N.; Matsumoto, K.; Motokawa, M.; Ishibashi, K.; Yamamoto, F. Synthesis and properties of poly(L–lactide)–poly(ε–caprolactone) multiblock copolymers by self–polycondensation of diblock macromonomers. Polym. J. 2015, 47, 657–665. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, C.; Duan, J.; Liang, X.; Li, X.; Sun, H.; Kong, D.; Yang, J. Synthesis of three–arm block copolymer poly(lactic–co–glycolic acid)–poly(ethylene glycol) with oxalyl chloride and its application in hydrophobic drug delivery. Int. J. Nanomed. 2016, 11, 6065–6077. [Google Scholar] [CrossRef]
- Jia, W.; Gu, Y.; Gou, M.; Dai, M.; Li, X.; Kan, B.; Yang, J.; Song, Q.; Wei, Y.; Qian, Z. Preparation of biodegradable polycaprolactone/poly(ethylene glycol)/polycaprolactone (PCEC) nanoparticles. Drug Deliv. 2008, 15, 409–416. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix–controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Qnouch, A.; Solarczyk, V.; Verin, J.; Tourrel, G.; Stahl, P.; Danede, F.; Willart, J.F.; Lemesre, P.E.; Vincent, C.; Siepmann, J.; et al. Dexamethasone–loaded cochlear implants: How to provide a desired “burst release”. Int. J. Pharm. X 2021, 3, 100088. [Google Scholar] [CrossRef]
- Bakhrushina, E.O.; Sakharova, P.S.; Konogorova, P.D.; Pyzhov, V.S.; Kosenkova, S.I.; Bardakov, A.I.; Zubareva, I.M.; Krasnyuk, I.I.; Krasnyuk, I.I., Jr. Burst release from in situ forming PLGA–based implants: 12 effectors and ways of correction. Pharmaceutics 2024, 16, 115. [Google Scholar] [CrossRef]
- Li, J.; Yap, S.Q.; Yoong, S.L.; Nayak, T.R.; Chandra, G.W.; Ang, W.H.; Panczyk, T.; Ramaprabhu, S.; Vashist, S.K.; Sheu, F.-S.; et al. Carbon nanotube bottles for incorporation, release and enhanced cytotoxic effect of cisplatin. Carbon 2012, 50, 1625–1634. [Google Scholar] [CrossRef]
- Heister, E.; Neves, V.; Lamprecht, C.; Silva, S.R.P.; Coley, H.M.; McFadden, J. Drug loading, dispersion stability, and therapeutic efficacy in targeted drug delivery with carbon nanotubes. Carbon 2012, 50, 622–632. [Google Scholar] [CrossRef]
- Ye, L.; Chen, W.; Chen, Y.; Qiu, Y.; Yi, J.; Li, X.; Lin, Q.; Guo, B. Functionalized multiwalled carbon nanotube–ethosomes for transdermal delivery of ketoprofen: Ex vivo and in vivo evaluation. J. Drug Deliv. Sci. Technol. 2022, 69, 103098. [Google Scholar] [CrossRef]
- Liu, Z.; Tabakman, S.; Welsher, K.; Dai, H. Carbon nanotubes in biology and medicine: In vitro and in vivo detection, imaging and drug delivery. Nano Res. 2009, 2, 85–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Clancy, A.J.; Fernández–Toribio, J.C.; Anthony, D.B.; White, E.R.; Solano, E.; Leese, H.S.; Vilatela, J.J.; Shaffer, M.S.P. Interfacially–grafted single–walled carbon nanotube/poly(vinyl alcohol) composite fibers. Carbon 2019, 146, 162–171. [Google Scholar] [CrossRef]
- Kalay, S.; Stetsyshyn, Y.; Donchak, V.; Harhay, K.; Lishchynskyi, O.; Ohar, H.; Panchenko, Y.; Voronov, S.; Çulha, M. pH–Controlled fluorescence switching in water–dispersed polymer brushes grafted to modified boron nitride nanotubes for cellular imaging. Beilstein J. Nanotechnol. 2019, 10, 2428–2439. [Google Scholar] [CrossRef]
- Silva–Jara, J.M.; Manríquez–González, R.; López–Dellamary, F.A.; Puig, J.E.; Nuño–Donlucas, S.M. Semi–continuous heterophase polymerization to synthesize nanocomposites of poly(acrylic acid)–functionalized carbon nanotubes. J. Macromol. Sci. Part A 2015, 52, 732–744. [Google Scholar] [CrossRef]
- Sandoval–García, K.; Alvarado–Mendoza, A.G.; Jiménez–Avalos, J.A.; García–Carvajal, Z.Y.; Olea–Rodríguez, M.A.; Cajero–Zul, L.R.; Nuño–Donlucas, S.M. Synthesis, characterization and evaluation of the toxicity, drug release ability and antibacterial capacity of nanocomposites of polyethylene glycol and functionalized carbon nanotubes. J. Macromol. Sci. Part A 2022, 59, 889–903. [Google Scholar] [CrossRef]
- Xia, R.; Li, M.; Zhang, Y.; Qian, J.; Yuan, X. Surface modification of MWNTs with BA–MMA–GMA terpolymer by single–step grafting technique. J. Appl. Polym. Sci. 2011, 119, 282–289. [Google Scholar] [CrossRef]
- Villar–Alvarez, E.; Figueroa–Ochoa, E.; Barbosa, S.; Soltero, J.F.A.; Taboada, P.; Mosquera, V. Reverse poly(butylene oxide)–poly(ethylene oxide)–poly(butylene oxide) block copolymers with lengthy hydrophilic blocks as efficient single and dual drug–loaded nanocarriers with synergistic toxic effects on cancer cells. RSC Adv. 2015, 5, 52105–52120. [Google Scholar] [CrossRef]
- Rúan–Esparza, L.; Soto, V.; Gómez–Salazar, S.; Rabelero, M.; Ávalos–Borja, M.; Luna–Bárcenas, G.; Prokhorov, E.; Nuño–Donlucas, S.M. Poly[ethylene–co–(acrylic acid)]–based nanocomposites: Thermal and mechanical properties and their structural characteristics studied by Raman spectroscopy. Polym. Compos. 2011, 32, 1181–1189. [Google Scholar] [CrossRef]
- Storey, R.F.; Taylor, A.E. End–group analysis of poly(e–caprolactone) initiated with water, ethylene glycol, and 1,4–butanediol. J. Macromol. Sci. Part A 1996, 33, 77–89. [Google Scholar] [CrossRef]
- Abdolmaleki, A.; Mallakpour, S.; Rostami, M. Development of carboxylated multi–walled carbon nanotubes reinforced potentially biodegradable poly(amide–imide) based on N–trimellitylimido–S–valine matrixes: Preparation, processing, and thermal properties. Prog. Org. Coat. 2015, 80, 71–76. [Google Scholar] [CrossRef]
- Alghunaim, N.S. Optimization and spectroscopic studies on carbon nanotubes/PVA nanocomposites. Results Phys. 2016, 6, 456–460. [Google Scholar] [CrossRef]
- Omran, S.M.; Abdullah, E.T.; Al–Zuhairi, O.A. Structural and infrared spectroscopy of polyvinylpyrrolidone/multi–walled carbon nanotubes nanocomposite. Iraqi J. Phys. 2021, 19, 1–6. [Google Scholar] [CrossRef]
- Parada, L.G.; Cesteros, L.C.; Meurio, E.; Katime, I. Miscibility and specific interactions in blends of poly(vinyl acetate–co–vinyl alcohol) with poly(ethyloxazoline). Macromol. Chem. Phys. 1997, 198, 2505–2517. [Google Scholar]
- Antolín–Cerón, V.H.; Gómez–Salazar, S.; Soto, V.; Ávalos–Borja, M.; Nuño–Donlucas, S.M. Polymer nanocomposites containing carbon nanotubes and miscible polymer blends based on poly[ethylene–co–(acrylic acid)]. J. Appl. Polym. Sci. 2008, 108, 1462–1472. [Google Scholar] [CrossRef]
- AAnalyzer®: A Peak–Fitting Program for Photoemission Data. Available online: https://xpsoasis.org/ (accessed on 8 January 2024).
- Ratner, B.D.; Castner, D.G. Electron spectroscopy for chemical analysis. In Surface Analysis: The Principal Techniques, 2nd ed.; Vickerman, J.C., Gilmore, I.S., Eds.; Wiley: Chichester, UK, 2009; pp. 53–79. [Google Scholar]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, 1st ed.; Wiley: Chichester, UK, 1992; pp. 84–85, 136–137, 142–143. [Google Scholar]
- Wingrove, A.S.; Caret, R.L. Organic Chemistry, 1st ed.; Harla: Mexico City, Mexico, 1984; pp. 1103–1104. [Google Scholar]
- Bogdanov, B.; Vidts, A.; Van Den Buicke, A.; Verbeeck, R.; Schacht, E. Synthesis and thermal properties of poly(ethylene glycol)–poly(ε–caprolactone) copolymers. Polymer 1998, 39, 1631–1636. [Google Scholar] [CrossRef]
- Bogdanov, B.; Vidts, A.; Schacht, E.; Berghmans, H. Isothermal crystallization of poly(ε–caprolactone–ethylene glycol) block copolymers. Macromolecules 1999, 32, 726–731. [Google Scholar] [CrossRef]
- Takeshita, H.; Fukumoto, K.; Ohnishi, T.; Ohkubo, T.; Miya, M.; Takenaka, K.; Shiomi, T. Formation of lamellar structure by competition in crystalization of both components for crystalline–crystallines block copolymers. Polymer 2006, 47, 8210–8218. [Google Scholar] [CrossRef]
- Xu, Y.; He, Y.; Wei, J.; Fan, Z.; Li, S. Morphology and melt crystallization of PCL–PEG diblocj copolymers. Macromol. Chem. Phys. 2008, 209, 1836–1844. [Google Scholar] [CrossRef]
- Sun, J.; He, C.; Zhuang, X.; Jing, X.; Chen, X. The crystallization behavior of poly(ethylene glycol)–poly(ε–caprolactone) diblock copolymers with asymmetric block compositions. J. Polym. Res. 2011, 18, 2161–2168. [Google Scholar] [CrossRef]
- Shokrolahi, F.; Yeganeh, H. Soft segment composition and its influence on phase–separated morphology of PCL/PEG–based poly(urethane urea)s. Iran Polym. J. 2014, 23, 505–512. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Fan, Z.; Li, S. Melt crystallization and morphology of poly(ε–caprolactone)–poly(ethylene glycol) diblock copolymers with different compositions and molecular weights. J. Polym. Sci. Part B Polym Phys. 2010, 48, 289–293. [Google Scholar] [CrossRef]
- Flory, A.L.; Ramanathan, T.; Brinson, L.C. Physical aging of single wall carbon nanotube polymer nanocomposites: Effect of functionalization of the nanotube on the enthalpy relaxation. Macromolecules 2010, 43, 4247–4252. [Google Scholar] [CrossRef]
- Babal, A.S.; Gupta, R.; Singh, B.P.; Dhakate, S.R. Depression in glass transition temperature of multiwalled carbon nanotubes reinforced polycarbonate composites: Effect of functionalization. RSC Adv. 2015, 5, 43462–43472. [Google Scholar] [CrossRef]
- Saldaña–Rojas, U.A.; Cortés–Llamas, S.A.; Jiménez–Avalos, J.A.; García–Carvajal, Z.Y.; Olea–Rodríguez, M.A.; Cajero–Zul, L.R.; Nuño–Donlucas, S.M. Evaluation of the toxicity and cisplatin drug-release ability of nanocomposites of polyethylene glycol and carbon nanotubes functionalized with Zn2+ ions. Int. J. Polym. Mater. 2024, 73, 946–960. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Wang, J.; Chen, T.; Wang, Y.; Tan, Y.W.; Su, H.; Chan, K.L.; Chen, H. Probing the kinetics of short–distance drug release from nanocarriers to nanoacceptors. Angew. Chem. Int. Ed. 2010, 49, 8426–8430. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gumy, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Kim, H.; Fassihi, R. Application of binary polymer in drug release rate modulation. 2. Influence of formulation variables and hydrodynamic conditions on release kinetics. J. Pharm. Sci. 1997, 86, 323–328. [Google Scholar] [CrossRef]
- Polli, J.E.; Rekhi, G.S.; Augsburger, L.L.; Shah, V.P. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J. Pharm. Sci. 1997, 86, 690–700. [Google Scholar] [CrossRef]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity upon surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add–in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.X.; Mensik, C.; Krishnaswami, S.; Hartmut, D.; Eichler, H.-G.; Schmetterer, L.; Wolzt, M. Pharmacokinetic–pharmacodynamic profile of systemic nitric oxide–synthase inhibition with L–NMMA in humans. Br. J. Clin. Pharmacol. 1999, 47, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems, 1st ed.; Bruschi, M.L., Ed.; Elsevier: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Shoaib, M.H.; Tazeen, J.; Merchant, H.A.; Yousuf, R.I. Evaluation of drug release kinetics from ibuprofen matrix tablets using HPMC. Pak. J. Pharm. Sci. 2006, 19, 119–124. [Google Scholar]
- Giri, B.R.; Kim, J.S.; Park, J.H.; Jin, S.G.; Kim, K.S.; Din, F.U.; Choi, H.G.; Kim, D.W. Improved bioavailability and high photostability of methotrexate by spray–dried surface–attached solid dispersion with and aqueous medium. Pharmaceutics 2021, 13, 111. [Google Scholar] [CrossRef]
- Erdal, N.B.; Lando, G.A.; Yadav, A.; Srivastava, R.K.; Hakkarainen, M. Hydrolytic degradation of porous crosslinked poly(e–caprolactone) synthesized by high internal phase emulsion templating. Polymers 2020, 12, 1849. [Google Scholar] [CrossRef]
- Yoshioka, T.; Kamada, F.; Kawazoe, N.; Tateishi, T.; Chen, G. Structural changes and biodegradation of PLLA, PCL, and PLGA sponges during in vitro incubation. Polym. Eng. Sci. 2010, 50, 1895–1903. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Legend Assigned to the Prepared PLLA–PCL–PEG/CNTspo Nanocomposites | Name of the PLLA–PCL–PEG Terpolymer Used as the Polymer Matrix of a Nanocomposite | Content of CNTspo (wt.%) |
|---|---|---|
| NC 1 | PLLA–PCL–PEG 2 | 1.0 |
| NC 2 | PLLA–PCL–PEG 2 | 0.5 |
| NC 3 | PLLA–PCL–PEG 4 | 1.0 |
| NC 4 | PLLA–PCL–PEG 4 | 0.5 |
| Homopolymer | Molar Ratio of [M]/[Wet BD] | Mw (g/mol) | Mw/Mn | Yield (%) |
|---|---|---|---|---|
| PLLA 1 | 30/1 | 24,670 | 1.07 | 90 |
| PLLA 2 | 40/1 | 31,110 | 1.08 | 92 |
| PLLA 3 | 50/1 | 34,250 | 1.09 | 95 |
| PLLA 4 | 60/1 | 34,800 | 1.07 | 90 |
| PCL 1 | 30/1 | 10,280 | 1.07 | 90 |
| PCL 2 | 40/1 | 25,650 | 1.10 | 90 |
| PCL 3 | 50/1 | 32,330 | 1.16 | 93 |
| PCL 4 | 60/1 | 35,370 | 1.11 | 91 |
| Copolymer | PLLA | PCL |
|---|---|---|
| PLLA–PCL 1 | 63.0 | 37.0 |
| PLLA–PCL 2 | 52.8 | 47.2 |
| PLLA–PCL 3 | 62.1 | 37.9 |
| PLLA–PCL 4 | 54.7 | 45.3 |
| Terpolymer | PLLA | PCL | PEG |
|---|---|---|---|
| PLLA–PCL–PEG 1 | 68.2 | 22.4 | 9.4 |
| PLLA–PCL–PEG 2 | 46.0 | 40.0 | 14.0 |
| PLLA–PCL–PEG 3 | 51.0 | 38.4 | 10.6 |
| PLLA–PCL–PEG 4 | 48.0 | 43.0 | 9.0 |
| Sample | Tg1 (K) | Tg2 (K) | Tm1 (K) | Tm2 (K) | ΔHm1 J/g | Tm3 (K) | ΔHm2 J/g |
|---|---|---|---|---|---|---|---|
| PLLA–PCL–PEG 2 | 221.15 | 287.15 | 318.45 | – | 8.1 | 398.85 | 35.9 |
| PLLA–PCL–PEG 4 | 223.15 | 285.15 | 317.45 | 323.55 | 8.0 | 398.55 | 36.6 |
| NC 1 | 216.15 | 291.15 | 313.95 | 319.85 | 30.2 | 413.65 | 29.6 |
| NC 2 | 218.15 | 258.15 | 312.65 | 319.45 | 33.6 | 413.35 | 28.5 |
| NC 3 | 219.15 | 290.15 | 305.55 | 314.45 | 26.4 | 405.05 | 19.9 |
| NC 4 | 217.15 | 252.15 | 302.15 | 312.75 | 23.3 | 387.95 | 20.5 |
| Models | Tested Materials | |||
|---|---|---|---|---|
| PLLA–PCL–PEG 2 | NC 1 | NC 2 | ||
| Higuchi | 14.12 ± 0.36 | 11.56 ± 0.29 | 11.51 ± 0.24 | |
| R2 | 0.975 ± 0.013 | 0.963 ± 0.024 | 0.960 ± 0.002 | |
| MSE | 39.95 ± 23.93 | 53.16 ± 33.81 | 59.26 ± 6.58 | |
| MSC | 3.67 ± 0.54 | 3.31 ± 0.70 | 3.11 ± 0.05 | |
| Korsmeyer–Peppas | 10.34 ± 0.71 | 14.87 ± 3.61 | 14.18 ± 2.89 | |
| n | 0.59 ± 0.03 | 0.44 ± 0.05 | 0.45 ± 0.06 | |
| R2 | 0.984 ± 0.008 | 0.972 ± 0.012 | 0.966 ± 0.008 | |
| MSE | 26.57 ± 16.16 | 41.72 ± 17.77 | 51.47 ± 19.53 | |
| MSC | 4.02 ± 0.53 | 3.46 ± 0.46 | 3.26 ± 0.23 | |
| Korsmeyer–Peppas extended1 | 6.92 ± 3.69 | 6.64 ± 0.49 | 6.47 ± 3.36 | |
| n | 0.77 ± 0.24 | 0.78 ± 0.05 | 0.76 ± 0.14 | |
| b | 0.12 ± 0.69 | 0.12 ± 0.37 | 0.12 ± 0.17 | |
| R2 | 0.995 ± 0.003 | 0.993 ± 0.004 | 0.996 ± 0.002 | |
| MSE | 1.19 ± 0.85 | 1.68 ± 0.23 | 0.88 ± 0.73 | |
| MSC | 4.99 ± 0.70 | 4.60 ± 0.54 | 5.17 ± 0.55 | |
| First-order | 0.06 ± 0.003 | 0.05 ± 0.008 | 0.04 ± 0.001 | |
| R2 | 0.991 ± 0.001 | 0.996 ± 0.003 | 0.993 ± 0.005 | |
| MSE | 13.88 ± 3.78 | 5.85 ± 3.91 | 10.01 ± 5.32 | |
| MSC | 4.61 ± 0.16 | 5.53 ± 0.74 | 4.96 ± 0.72 | |
| Hixson–Crowell | 0.015 ± 0.001 | 0.012 ± 0.002 | 0.012 ± 0.001 | |
| R2 | 0.995 ± 0.004 | 0.985 ± 0.010 | 0.983 ± 0.020 | |
| MSE | 6.47 ± 5.28 | 21.18 ± 13.68 | 21.60 ± 25.91 | |
| MSC | 5.56 ± 1.04 | 4.23 ± 0.71 | 4.75 ± 1.93 | |
| Models | Tested Materials | |||
|---|---|---|---|---|
| PLLA–PCL–PEG 4 | NC 3 | NC 4 | ||
| Higuchi | 15.86 ± 0.35 | 11.26 ± 0.38 | 11.56 ± 0.17 | |
| R2 | 0.985 ± 0.002 | 0.966 ± 0.006 | 0.961 ± 0.015 | |
| MSE | 19.58 ± 4.40 | 50.50 ± 13.51 | 51.48 ± 17.69 | |
| MSC | 4.10 ± 0.11 | 3.29 ± 0.18 | 3.16 ± 0.38 | |
| Korsmeyer–Peppas | 17.65 ± 1.93 | 12.71 ± 0.14 | 17.20 ± 2.33 | |
| n | 0.47 ± 0.04 | 0.47 ± 0.01 | 0.40 ± 0.03 | |
| R2 | 0.989 ± 0.006 | 0.967 ± 0.007 | 0.979 ± 0.002 | |
| MSE | 16.33 ± 10.25 | 49.82 ± 14.44 | 28.39 ± 2.04 | |
| MSC | 4.34 ± 0.56 | 3.28 ± 0.20 | 3.70 ± 0.10 | |
| Korsmeyer–Peppas extended1 | 13.75 ± 5.67 | 4.88 ± 0.33 | 11.83 ± 3.21 | |
| n | 0.61 ± 0.14 | 0.86 ± 0.06 | 0.56 ± 0.09 | |
| b | 0.89 ± 2.83 | – 0.86 ± 0.18 | – 0.38 ± 0.51 | |
| R2 | 0.993 ± 0.005 | 0.988 ± 0.003 | 0.997 ± 0.001 | |
| MSE | 2.66 ± 1.66 | 2.43 ± 0.15 | 0.65 ± 0.11 | |
| MSC | 4.76 ± 0.79 | 4.05 ± 0.23 | 5.54 ± 0.23 | |
| First-order | 0.08 ± 0.001 | 0.04 ± 0.004 | 0.05 ± 0.005 | |
| R2 | 0.981 ± 0.015 | 0.996 ± 0.003 | 0.987 ± 0.006 | |
| MSE | 25.01 ± 17.65 | 5.55 ± 3.84 | 16.28 ± 7.78 | |
| MSC | 3.98 ± 0.89 | 5.61 ± 0.85 | 4.34 ± 0.53 | |
| Hixson–Crowell | 0.02 ± 0.001 | 0.01 ± 0.001 | 0.01 ± 0.001 | |
| R2 | 0.964 ± 0.032 | 0.984 ± 0.011 | 0.970 ± 0.010 | |
| MSE | 46.05 ± 37.69 | 21.98 ± 14.72 | 39.53 ± 11.87 | |
| MSC | 3.43 ± 1.05 | 4.23 ± 0.82 | 3.41 ± 0.34 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Iñiguez, K.J.; Figueroa-Ochoa, E.B.; Martínez-Richa, A.; Cajero-Zul, L.R.; Nuño-Donlucas, S.M. Synthesis of Poly(L–lactide)–poly(ε–caprolactone)–poly(ethylene glycol) Terpolymer Grafted onto Partially Oxidized Carbon Nanotube Nanocomposites for Drug Delivery. Polymers 2024, 16, 2580. https://doi.org/10.3390/polym16182580
González-Iñiguez KJ, Figueroa-Ochoa EB, Martínez-Richa A, Cajero-Zul LR, Nuño-Donlucas SM. Synthesis of Poly(L–lactide)–poly(ε–caprolactone)–poly(ethylene glycol) Terpolymer Grafted onto Partially Oxidized Carbon Nanotube Nanocomposites for Drug Delivery. Polymers. 2024; 16(18):2580. https://doi.org/10.3390/polym16182580
Chicago/Turabian StyleGonzález-Iñiguez, Karla J., Edgar B. Figueroa-Ochoa, Antonio Martínez-Richa, Leonardo R. Cajero-Zul, and Sergio M. Nuño-Donlucas. 2024. "Synthesis of Poly(L–lactide)–poly(ε–caprolactone)–poly(ethylene glycol) Terpolymer Grafted onto Partially Oxidized Carbon Nanotube Nanocomposites for Drug Delivery" Polymers 16, no. 18: 2580. https://doi.org/10.3390/polym16182580
APA StyleGonzález-Iñiguez, K. J., Figueroa-Ochoa, E. B., Martínez-Richa, A., Cajero-Zul, L. R., & Nuño-Donlucas, S. M. (2024). Synthesis of Poly(L–lactide)–poly(ε–caprolactone)–poly(ethylene glycol) Terpolymer Grafted onto Partially Oxidized Carbon Nanotube Nanocomposites for Drug Delivery. Polymers, 16(18), 2580. https://doi.org/10.3390/polym16182580