

De- and Re-Structuring of Starch to Control the Melt and Solid State Visco-Elasticity as Method for Getting New Multi Component Compounds with Scalable Properties

 , ,
, ,  ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Designing of New Compounds, Procedure and Materials
2.2. Characterization
2.2.1. Miscibility
Chemical Structure
Morphological Structure
2.2.2. Vasco-Elastic Behavior
In the Melted State
- The Rheological Properties of the Melts
- Extrudates Swelling
In the Solid State
- Dynamic Mechanical Properties (DMA)
- Mechanical Properties
2.3. Other Properties
2.3.1. Density
2.3.2. Hardness
2.3.3. Color
2.3.4. Durability–Qualitative Assessments of Mechanical Properties over Almost 10 Years
3. Results
3.1. Compounding
3.1.1. Deficiencies at Preliminary Compounding of Starch with PVA (Figure 2)

Compounding Using Polymer as Powder with Similar Diameter (Figure 2)
Pre-Conditioning of the Solid and Liquid Components before Melt Compounding (Figure 3)

Drying of Polymers Before Melt Compounding
3.2. Miscibility of Binary Blends
3.2.1. Chemical Structure (Figure 4, Table S1—In Supplementary Data)
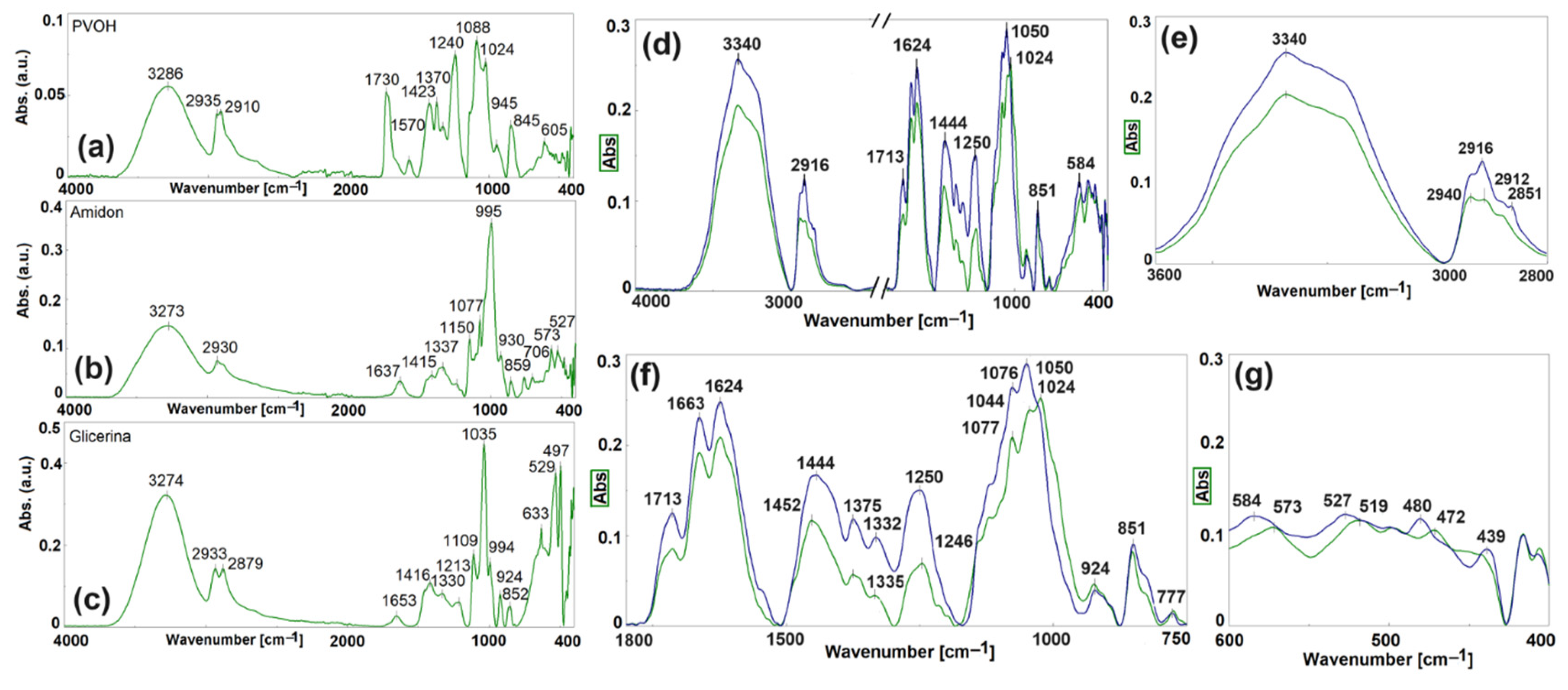
3.2.2. Miscibility—SEM Morphology (Figure 5)

3.3. Visco-Elastic Behavior
3.3.1. The Melt Viscoelasticity
The Viscous Deformation
- The Influence of the Formulation on the Flowing Regime (Shear Stress and Shear Rate)
- Dependence of shear stress (τ) and shear rate () on the starch content (Figure 6)
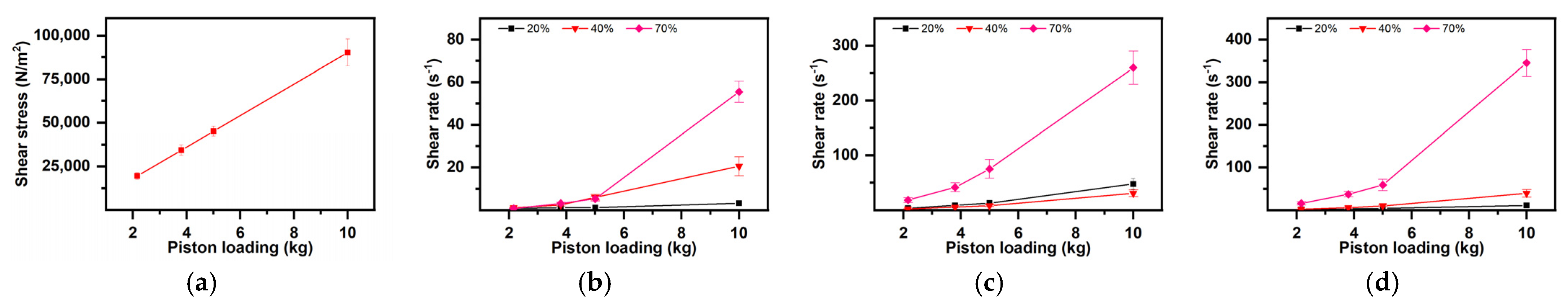
- 2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code/Shear rate, s−1/Temperature and weight range | s−1, 155 °C (Weight_i, kg) for 40 [p] plasticizer/Item for 60 [p] plasticizer | |
| s−1, 125 °C (Weight_i, kg) for 40 [p] plasticizer/Item for 60 [p] plasticizer | ||
| 20% starch | 70% starch | |
| A. kg | 3.8–16 kg | |
| A1—155 °C, s−1 | 28 ÷ 12/60 ÷ 30 | 12 ÷ 6/18 ÷ 8 |
| A2—125 °C, s−1 | 2 ÷ 1/18 ÷ 5 | 3 ÷ 2/4 ÷ 2 |
| A1/A2 | 14 ÷ 12/3 ÷ 5 | 4 ÷ 3/4.5 ÷ 4 |
| B. kg | 5–3.8 kg | |
| B1—155 °C, s−1 | 40 ÷ 30/75÷ 55 | 17 ÷ 12/42 ÷ 18 |
| B2—125 °C, s−1 | 4 ÷ 3/22 ÷ 17 | 7 ÷ 3/20 ÷ 18 |
| B1/B2 | 10 ÷ 10/3.4 ÷ 3.24 | 2.43 ÷ 4/2.1 ÷ 1 |
| C. kg | 10–5 kg | |
| C1—155 °C, s−1 | 93 ÷ 40/180 ÷ 75 | 40 ÷ 17/160 ÷ 42 |
| C2—125 °C, s−1 | 8 ÷ 4/57÷ 20 | 22 ÷ 17/60 ÷ 18 |
| C1/C2 | 11.6 ÷ 10/3.16 ÷ 3.75 | 1.82 ÷ 1/2.66 ÷ 2/33 |
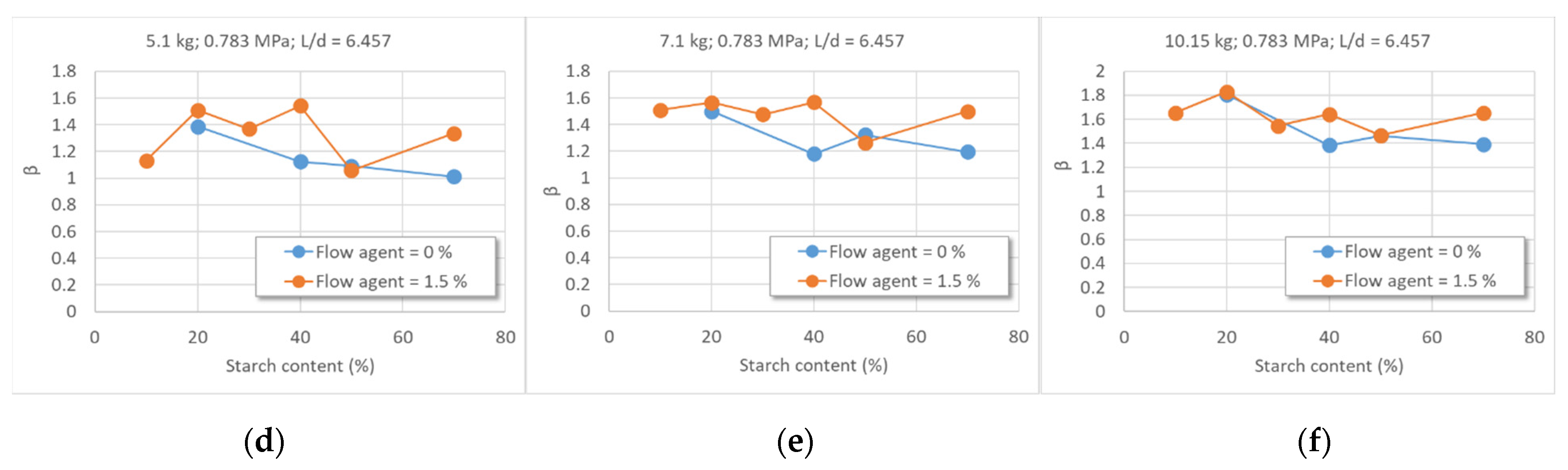
- 3.
- Dependence of the shear speed on the lubrication degree (Figure 8)
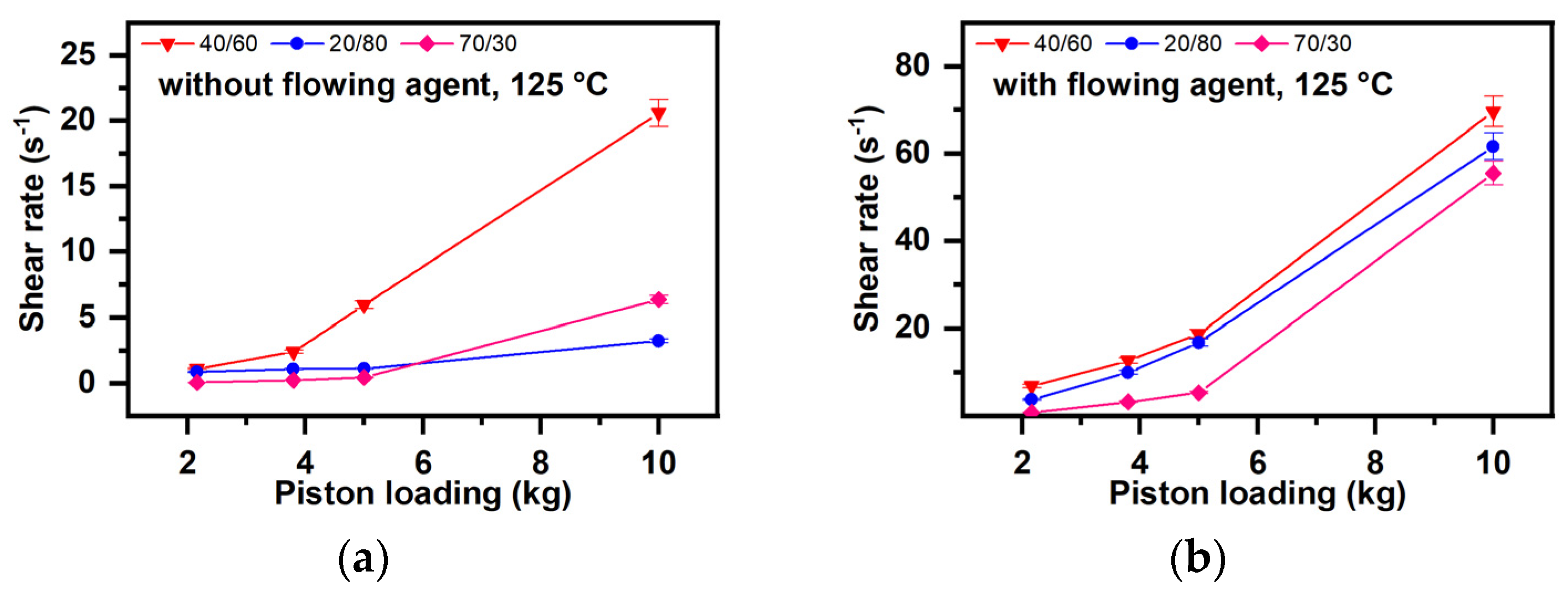
- Influence of the Formulation and the Flow Conditions on the Fluidity
- (i)
- The dependence on the starch quantity (Figure 9)
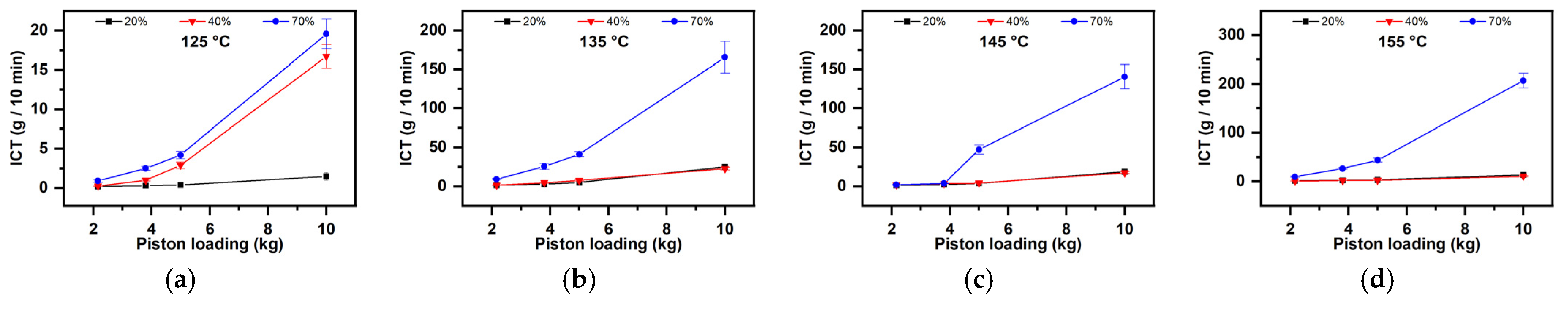
- (ii)
- Dependence on the lubrication degree (Figure 10)
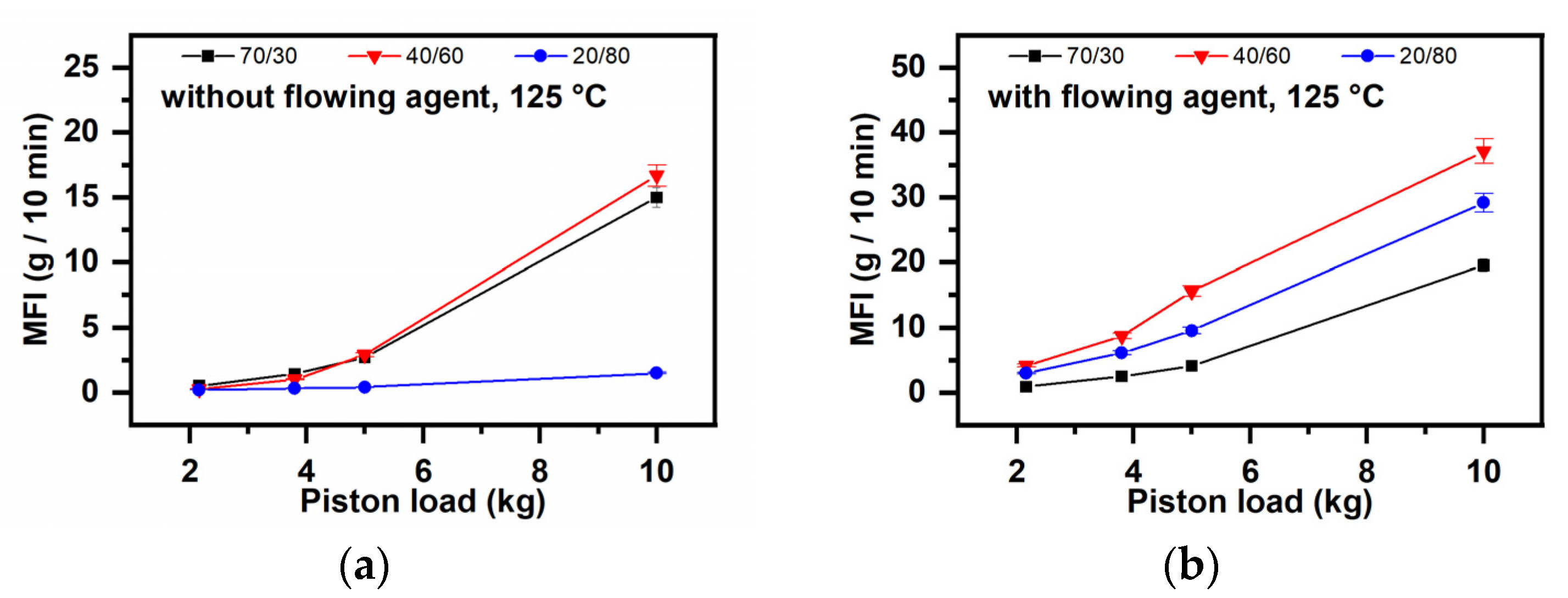
- (i)
- The starch quantity (Figure 11)

- (ii)
- Lubrication degree (Figure 12)
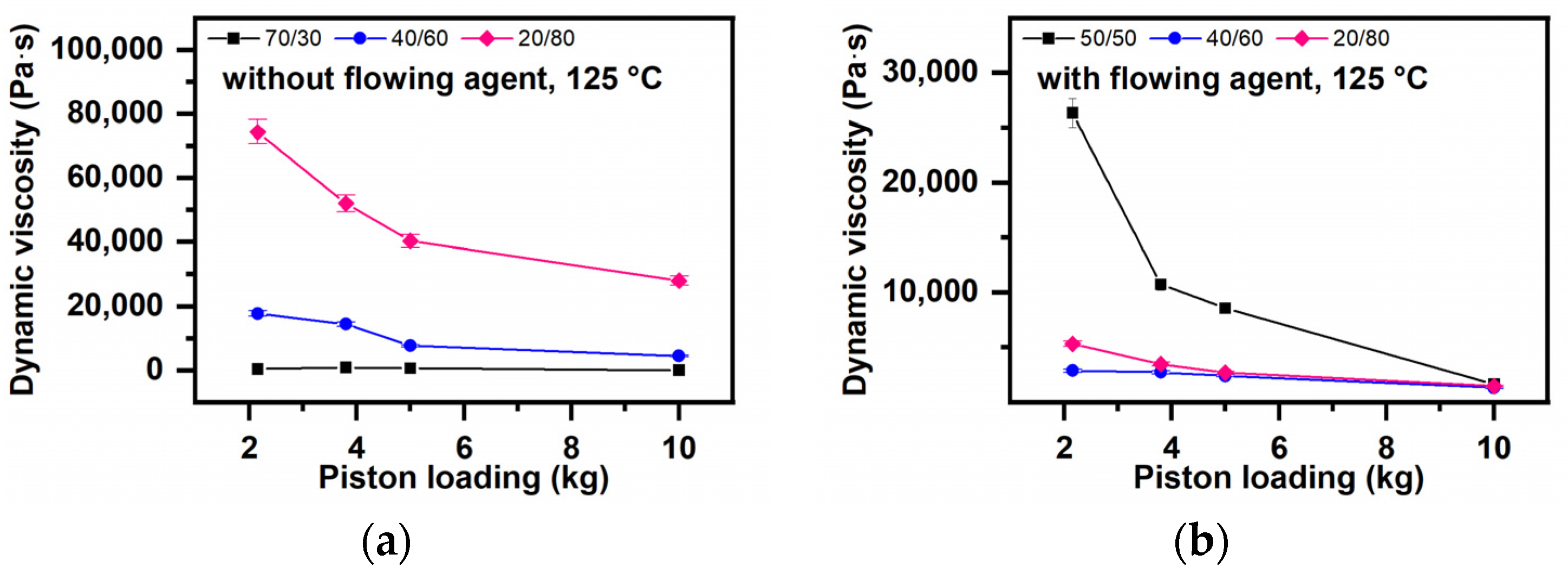
The Melt Elasticity (Extrudate Swelling) (Figure 13)

3.3.2. The Viscoelasticity in the Solid State
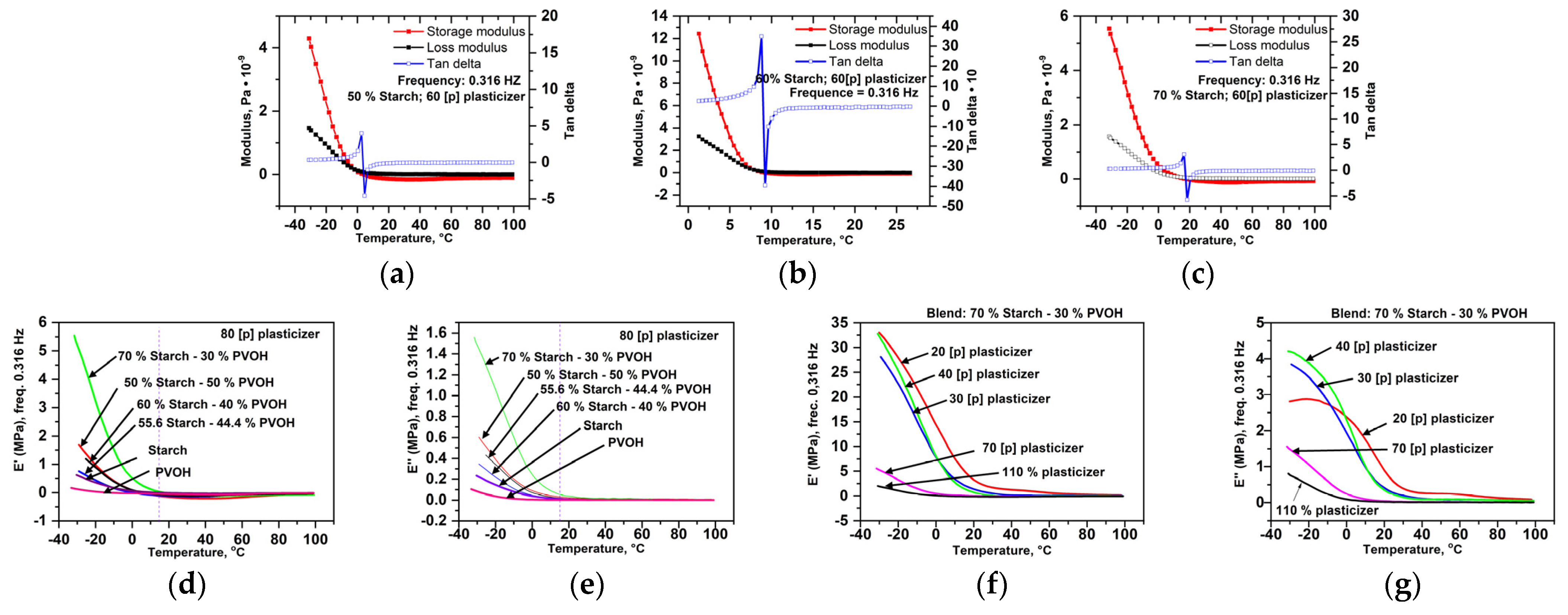
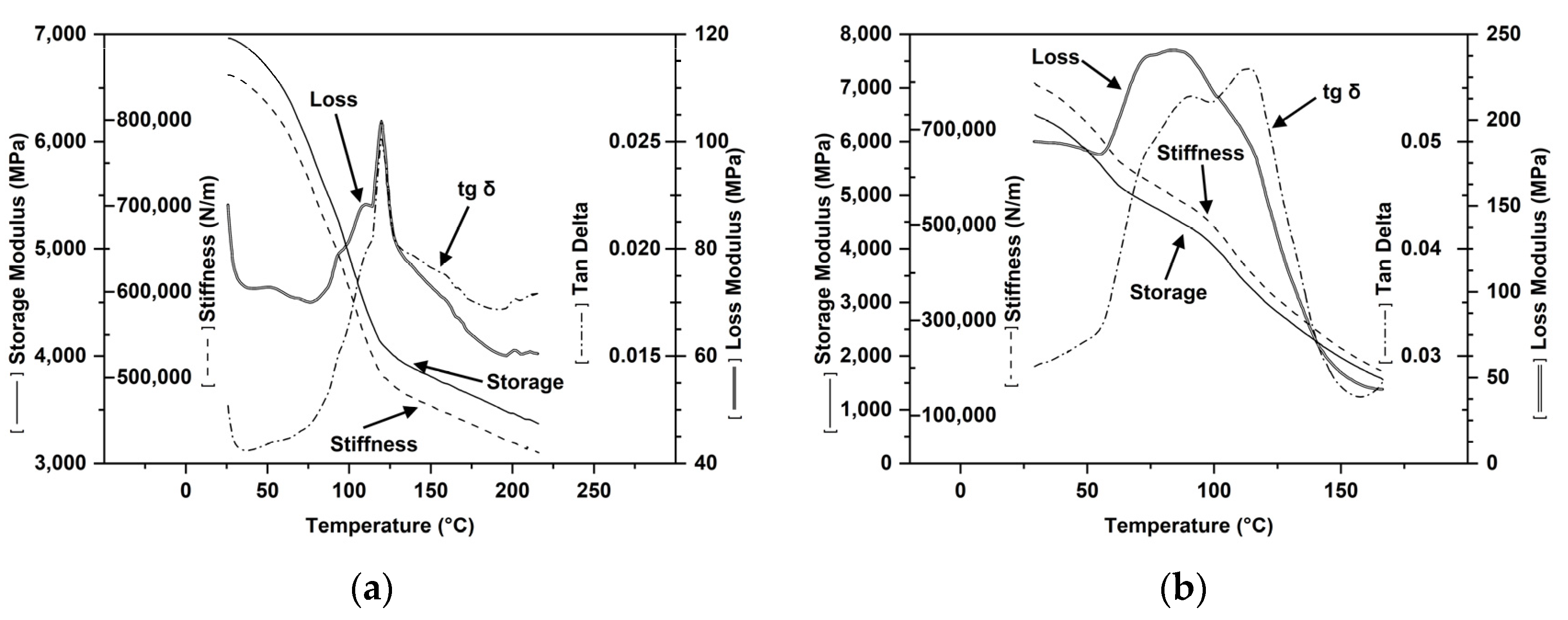
Dynamo-Mechanical Properties (Figure 14)
Mechanical Properties
Other Properties: Density: 1.23–1.31 g/cm3; Measured Values of Hardness Were Ranged as 55–70 °ShA
- Scalable Properties—Thermodynamic Stability (Figure 15)

4. Discussion
5. Conclusions
- The aim of the article was to design and develop new starch-based compounds that are melt-processable into finished product using scalable, classic (extrusion, injection, thermoforming) or 3D printing techniques. In order to achieve this goal, the powder of starch and PVA was blended with a liquid plasticizer in a pre-compounding sequence carried out under selected thermal and mechanical conditions. The melt compounding of these solid blends occurred under conditions that allowed the de-structuring of the two polymers, the melt entanglement of the resulting macromolecules, and the re-structuring of the new, non-degraded compounds. These compounds exhibited high thermodynamically miscibility, good functional properties, and scalable properties. These compounds were selected based on both their molten and solid-state viscoelastic properties and their functional properties. The melt processing of the selected compounds using various techniques demonstrated good scalable properties and a lifetime exceeding 10 years.
- Starting from starch with glass transition at 125 °C and PVA with Tg at 85 °C, following the described working sequence, new starch–PVA compounds with glass transitions between −20 °C and 50 °C were made, showing optimal melt processability using both classic (extrusion, injection) and 3D printing methods. The homogenization degree was controlled by the extent of melt viscous deformations, which depends on shear rates, fluidity, and the melt resistance to flow to prevent polymer degradation. The extrudates swelling was eliminated by managing the melt’s elastic deformability. All these melt rheological properties were influenced by formulation, flow conditions, characteristics of the compounding device, and the dimensional characteristics of the nozzle. Formulations and compounding conditions were selected to allow processing at temperatures below 140 °C.
- The thermodynamic stability will be quantitatively described through comparative FTIR, DSC, etc., analyses, initially and after 10 years, in a future article. Further examination of the time-based thermodynamic stability features of these compounds may pave the way for using starch in medium-life compositions, beyond its current application in short-life products.
- The incomplete melting and rough surfaces were avoided by using only dried polymers before melt compounding, working only with polymers of similar and nearly equal particle diameter, using formulations with flow agents, and controlling deformability in the melted state.
- The FTIR study demonstrated the advanced miscibility of the compounds made using the specified sequence.
- The viscoelastic behavior in the solid state allowed the selection of the formulations with viscous deformability to ensure optimal stretchability for extrusion-blowing and 3D printing processes. The formulations with viscous deformability were selected to ensure good behavior in injection molding and thermoforming processes.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pearce, E.M. Polymeric Multicomponent Materials: An Introduction by L. H. Sperling (Lehigh University). Wiley/VCH: New York. 1997. $59.95. xviii + 397 pp. ISBN 0-471-04138-6. J. Am. Chem. Soc. 1998, 120, 7147–7148. [Google Scholar] [CrossRef]
- van der Vegt, A.K. From Polymers to Plastics; VSSD: Uttar Pradesh, India, 2006. [Google Scholar]
- Gilbert, R.G.; Hess, M.; Jenkins, A.D.; Jones, R.G.; Kratochvil, P.; Stepto, R.F.T. Dispersity in polymer science. Pure Appl. Chem. 2009, 81, 351–353. [Google Scholar]
- Gächter, R.; Müller, H. Plastics Additives Handbook: Stabilizers, Processing Aids, Plasticizers, Filters, Reinforcements, Colorants for Thermoplastics; Hanser Publishers: Munich, Germany, 1987. [Google Scholar]
- Takeshita, H.; Shiomi, T.; Takenaka, K.; Arai, F. Crystallization and higher-order structure of multicomponent polymeric systems. Polymer 2013, 54, 4776–4789. [Google Scholar] [CrossRef]
- Carlsson, A.; Rosen, J.; Dahlqvist, M. Finding stable multi-component materials by combining cluster expansion and crystal structure predictions. Npj Comput. Mater. 2023, 9, 21. [Google Scholar] [CrossRef]
- Taguet, A.; Cassagnau, P.; Lopez-Cuesta, J.M. Structuration, selective dispersion and compatibilizing effect of (nano) fillers in polymer blends. Prog. Polym. Sci. 2014, 39, 1526–1563. [Google Scholar] [CrossRef]
- Dimonie, D.; Petrache, M.; Damian, C.; Anton, L.; Musat, M.; Dima, Ş.-O.; Jinescu, C.; Maria, R. New evidences on the process sensitivity of some renewable blends based on starch considering their melt rheological properties. Int. J. Polym. Sci. 2016, 2016, 7873120. [Google Scholar] [CrossRef]
- Dimonie, D.; Damian, C.; Trusca, R.; Rapa, M. Some aspects conditioning the achieving of filaments for 3D printing from physical modified corn starch. Mater. Plast 2019, 56, 351–359. [Google Scholar] [CrossRef]
- Hindawi, I.A.; Higgins, J.S.; Weiss, R.A. Flow Induced Mixing and Demixing in Polymer Blends. Polymer 1992, 33, 2522–2529. [Google Scholar] [CrossRef]
- Dimonie, D.; Kelnar, I.; Socoteanu, R.; Darie, R.N.; Pop, F.S.; Zaharia, C.; Petrea, C.; Nemteanu, M.; Coserea, R.M. The influence of miscibility and micro-structure on the surface defects of some starch bio-hybrides. Mater. Plast. 2010, 47, 486–491. [Google Scholar]
- Fernandez, M.L. Demixing in polymer blends. Sci. Prog. 1990, 74, 257–277. [Google Scholar]
- Caddeo, C.; Ackermann, J.; Mattoni, A. A theoretical perspective on the thermodynamic stability of polymer blends for solar cells: From experiments to predictive modeling. Sol. RRL 2022, 6, 2200172. [Google Scholar] [CrossRef]
- Utracki, L.A.; Shi, G.-H. Compounding polymer blends. In Polymer Blends Handbook; Springer: Dordrecht, The Netherlands, 2003; p. 577. [Google Scholar]
- Kim, J.H.; Hong, J.S.; Ishigami, A.; Kurose, T.; Ito, H.; Ahn, K.H. Effect of melt-compounding protocol on self-aggregation and percolation in a ternary composite. Polymers 2020, 12, 3041. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Shibayama, M.; Tran-Cong, Q. Structure and Properties of Multiphase Polymeric Materials; CRC Press: Boca Raton, FL, USA, 1998; Volume 46. [Google Scholar]
- Emeje, M.; Blumenberg, M. Chemical Properties of Starch; BoD–Books on Demand: Norderstedt, Germany, 2020. [Google Scholar]
- Salisu, A.A. Preparation and characterization of biodegradable poly (vinyl alcohol)/starch blends. ChemSearch J. 2012, 3, 34–37. [Google Scholar]
- Zanela, J.; Bilck, A.P.; Reis, M.O.; Grossmann, M.V.E.; Yamashita, F. Modified starches on the properties of extruded biodegradable materials of starch and polyvinyl alcohol. J. Polym. Environ. 2020, 28, 3211–3220. [Google Scholar] [CrossRef]
- Jane, J. Starch properties, modifications, and applications. J. Macromol. Sci. Part A Pure Appl. Chem. 1995, 32, 751–757. [Google Scholar] [CrossRef]
- Wang, X.-L.; Yang, K.-K.; Wang, Y.-Z. Properties of starch blends with biodegradable polymers. J. Macromol. Sci. Part C Polym. Rev. 2003, 43, 385–409. [Google Scholar] [CrossRef]
- Meier, M.A.R.; Metzger, J.O.; Schubert, U.S. Plant oil renewable resources as green alternatives in polymer science. Chem. Soc. Rev. 2007, 36, 1788–1802. [Google Scholar] [CrossRef]
- Dimonie, D.; Radovici, C.; Serban, S.; Taranu, A.; Vasilievici, G. Biodegradable compounds from regenerable resources for packings. Mater. Plast. 2007, 44, 148–154. [Google Scholar]
- Dimonie, D.; Radu, S.; Doncea, S.; Pop, F.S.; Petre, C.; Dumitriu, I.; Fierascu, R. The miscibility estimation of some nanocomposites based on starch. e-Polymers 2011, 11, 090. [Google Scholar] [CrossRef]
- Dimonie, D.; Radovici, C.; Coserea, R.M.; Garea, S.; Teodorescu, M. The polymer molecular weight and silicate treatment influence upon the morphology of nanocomposites for food packaging. Rev. Roum. De Chim. 2008, 53, 1017–1026. [Google Scholar]
- Dimonie, D.; Grigorescu, R.M.; Trică, B.; Damian, C.-M.; Vasile, E.; Trusca, R.; Nicolae, C.-A.; Constantinescu-Aruxandei, D.; Oancea, F. Increasing the Efficiency of Multilayered Silicate Melt Incorporation into Starch-Based Polymeric Matrices. J. Compos. Sci. 2024, 8, 72. [Google Scholar] [CrossRef]
- Jan-Georg, R.; Langer, R.; Giovanni, T. Bioplastics for a circular economy. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar]
- Tian, H.; Yan, J.; Rajulu, A.V.; Xiang, A.; Luo, X. Fabrication and properties of polyvinyl alcohol/starch blend films: Effect of composition and humidity. Int. J. Biol. Macromol. 2017, 96, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, J.; Guo, H. Research progress of polyvinyl alcohol water-resistant film materials. Membranes 2022, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, P.S.; Baldone Lara, B.R.; de Souza Nascimento, B.; Dias, M.V. Cassava starch films: Effect of polyvinyl alcohol on films’ water diffusion, permeation and sorption behavior and mechanical properties. Iran. Polym. J. 2022, 31, 1431–1446. [Google Scholar] [CrossRef]
- Guimarães Júnior, M.; Botaro, V.R.; Novack, K.M.; Teixeira, F.G.; Tonoli, G.H.D. High moisture strength of cassava starch/polyvinyl alcohol-compatible blends for the packaging and agricultural sectors. J. Polym. Res. 2015, 22, 192. [Google Scholar] [CrossRef]
- Mao, L.; Imam, S.; Gordon, S.; Cinelli, P.; Chiellini, E. Extruded Cornstarch-Glycerol-Polyvinyl Alcohol Blends: Mechanical Properties, Morphology, and Biodegradability. J. Polym. Environ. 2000, 8, 205–211. [Google Scholar] [CrossRef]
- Zanela, J.; Casagrande, M.; Reis, M.O.; Grossmann, M.V.E.; Yamashita, F. Biodegradable sheets of starch/polyvinyl alcohol (PVA): Effects of PVA molecular weight and hydrolysis degree. Waste Biomass Valorization 2019, 10, 319–326. [Google Scholar] [CrossRef]
- Castro, J.M.; Montalbán, M.G.; Martínez-Pérez, N.; Domene-López, D.; Pérez, J.M.; Arrabal-Campos, F.M.; Fernández, I.; Martín-Gullón, I.; García-Quesada, J.C. Thermoplastic starch/polyvinyl alcohol blends modification by citric acid–glycerol polyesters. Int. J. Biol. Macromol. 2023, 244, 125478. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, J.; Peng, T.; Li, Y.; Lin, D.; Xing, B.; Li, C.; Yang, Y.; Yang, L.; Zhang, L. Preparation and application of starch/polyvinyl alcohol/citric acid ternary blend antimicrobial functional food packaging films. Polymers 2017, 9, 102. [Google Scholar] [CrossRef]
- Yurong, G.; Dapeng, L. Preparation and characterization of corn starch/PVA/glycerol composite films incorporated with ε-polylysine as a novel antimicrobial packaging material. e-Polymers 2020, 20, 154–161. [Google Scholar] [CrossRef]
- Ge, C.; Lansing, B.; Lewis, C.L. Thermoplastic starch and poly (vinyl alcohol) blends centered barrier film for food packaging applications. Food Packag. Shelf Life 2021, 27, 100610. [Google Scholar] [CrossRef]
- Anne, B. Environmental-friendly biodegradable polymers and composites. In Integrated Waste Management; IntechOpen: London, UK, 2011; pp. 341–364. [Google Scholar]
- Xiao, H.; Lin, Q.; Liu, G.-Q.; Wu, Y.; Tian, W.; Wu, W.; Fu, X. Physicochemical properties of chemically modified starches from different botanical origin. Sci. Res. Essays 2011, 6, 4517–4525. [Google Scholar]
- Park, H.M.; Li, X.; Jin, C.Z.; Park, C.Y.; Cho, W.J.; Ha, C.S. Preparation and properties of biodegradable thermoplastic starch/clay hybrids. Macromol. Mater. Eng. 2002, 287, 553–558. [Google Scholar] [CrossRef]
- Jansson, A.; Järnström, L. Barrier and mechanical properties of modified starches. Cellulose 2005, 12, 423–433. [Google Scholar] [CrossRef]
- Chai, W.-L.; Chow, J.-D.; Chen, C.-C. Effects of modified starch and different molecular weight polyvinyl alcohols on biodegradable characteristics of polyvinyl alcohol/starch blends. J. Polym. Environ. 2012, 20, 550–564. [Google Scholar] [CrossRef]
- Nistor, M.-T.; Vasile, C. Influence of the nanoparticle type on the thermal decomposition of the green starch/poly (vinyl alcohol)/montmorillonite nanocomposites. J. Therm. Anal. Calorim. 2013, 111, 1903–1919. [Google Scholar] [CrossRef]
- Chapple, S.; Anandjiwala, R.; Ray, S.S. Mechanical, thermal, and fire properties of polylactide/starch blend/clay composites. J. Therm. Anal. Calorim. 2013, 113, 703–712. [Google Scholar] [CrossRef]
- Li, Z.-Z.; Heo, K.-S.; Xuan, D.-J.; Seol, S.-Y. A study on cooling efficiency using 1-d analysis code suitable for cooling system of thermoforming. J. Mech. Sci. Technol. 2009, 23, 607–613. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, H. Predicting synergistic effects between compounds through their structural similarity and effects on transcriptomes. Bioinformatics 2016, 32, 3782–3789. [Google Scholar] [CrossRef]
- Elias, H.-G.; Elias, H.-G. Additives and Compounding. In Macromolecules: Volume 2: Synthesis, Materials, and Technology; Springer: Berlin/Heidelberg, Germany, 1984; pp. 1123–1146. [Google Scholar]
- Real, L.E.P. Plastic Materials and Additives. In Recycled Materials for Construction Applications: Plastic Products and Composites; Springer: Berlin/Heidelberg, Germany, 2022; pp. 19–33. [Google Scholar]
- Albrecht, T.R.; Dovek, M.M.; Lang, C.A.; Grütter, P.; Quate, C.F.; Kuan, S.W.J.; Frank, C.W.; Pease, R.F.W. Imaging and modification of polymers by scanning tunneling and atomic force microscopy. J. Appl. Phys. 1988, 64, 1178–1184. [Google Scholar] [CrossRef]
- Taghizadeh, M.T.; Abdollahi, R. A kinetics study on the thermal degradation of starch/poly (vinyl alcohol) blend. Chem. Mater. Eng. 2015, 3, 73–78. [Google Scholar] [CrossRef]
- Guarás, M.P.; Ludueña, L.N.; Alvarez, V.A. Recent Advances in Thermoplastic Starch Biodegradable Nanocomposites. In Handbook of Nanomaterials and Nanocomposites for Energy and Environmental Applications; Kharissova, O.V., Martínez, L.M.T., Kharisov, B.I., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–24. [Google Scholar]
- Olabis, O. Polymer-Polymer Miscibility; Elsevier: Amsterdam, The Netherlands, 1978. [Google Scholar]
- Krause, S. Polymer-polymer miscibility. Pure Appl. Chem. 1986, 58, 1553–1560. [Google Scholar] [CrossRef]
- Garton, A. Infrared Spectroscopy of Polymer Blends, Composites and Surfaces; Carl Hanser Verlag: Munich, Germany, 1992. [Google Scholar]
- de Arenaza, I.M.; Meaurio, E.; Sarasua, J.-R. Analysis of the miscibility of polymer blends through molecular dynamics simulations. In Polymerization; IntechOpen: London, UK, 2012. [Google Scholar]
- Bădilescu, S. Polymers and Additives Infrared Spectroscopy; Tehnical Publisher House: Bucharest, Romania, 1982. [Google Scholar]
- Mondal, K.; Sharma, A.; Gupta, M.N. Three phase partitioning of starch and its structural consequences. Carbohydr. Polym. 2004, 56, 355–359. [Google Scholar] [CrossRef]
- Dimonie, D.; Constantin, R.; Vasilievici, G.; Popescu, M.C.; Garea, S. The dependence of the XRD morphology of some bionanocomposites on the silicate treatment. J. Nanomater. 2008, 2008, 1687–4110. [Google Scholar] [CrossRef]
- Bedjaoui, K.; Navarro, R.; Krache, R.; López Valentín, J.; Herrero Calderon, R.; Fernández, A.; Marcos Fernandez, A. Toughening of Immiscible rPS/SAN Blends by SEBS Elastomers: Properties and Morphology; Scientific Research Publishing: Wuhan, China, 2022. [Google Scholar]
- Dixit, M.; Mathur, V.; Gupta, S.; Baboo, M.; Sharma, K.; Saxena, N.S. Investigation of miscibility and mechanical properties of PMMA/PVC blends. J. Optoelectron. Adv. Mater. Rapid Commun. 2009, 3, 1099–1105. [Google Scholar]
- Iqbal, A.; Muhammad, J.; Ahmad, F.; Nawaz, M.; Baloch, M.K.; Mohammad, L. Miscibility and thermal behavior of poly (methyl methacrylate) and polystyrene blend using benzene as a common solvent. Turk. J. Chem. 2022, 46, 2010–2023. [Google Scholar] [CrossRef]
- Abou-Rachid, H.; Lussier, L.S.; Ringuette, S.; Lafleur-Lambert, X.; Jaidann, M.; Brisson, J. On the correlation between miscibility and solubility properties of energetic plasticizers/polymer blends: Modeling and simulation studies. Propellants Explos. Pyrotech. Int. J. Deal. Sci. Technol. Asp. Energetic Mater. 2008, 33, 301–310. [Google Scholar] [CrossRef]
- Meaurio, E.; Hernandez-Montero, N.; Zuza, E.; Sarasua, J.-R. Miscible Blends Based on Biodegradable Polymers. In Characterization of Polymer Blends; Wiley: Hoboken, NJ, USA, 2014; pp. 7–92. [Google Scholar]
- Zanela, J.; Shirai, M.A.; Reis, M.O.; Mali, S.; Grossmann, M.V.E.; Yamashita, F. Mixture design to develop biodegradable sheets with high levels of starch and polyvinyl alcohol. Starch-Stärke 2015, 67, 1011–1019. [Google Scholar] [CrossRef]
- Ruan, Y.; Lu, Y.; An, L.; Wang, Z.-G. Multiple Entanglements between Two Chains in Polymer Melts: An Analysis of Primitive Paths Based on Frenet Trihedron. Macromolecules 2024, 57, 2792–2800. [Google Scholar] [CrossRef]
- Abadi, M.; Serag, M.F.; Habuchi, S. Entangled polymer dynamics beyond reptation. Nat. Commun. 2018, 9, 5098. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.; Hollander, R.W.; Spoor, M.W.; Smith, J.M. Residence time distributions in a plasticating twin screw extruder. AIChE J. 1979, 25, 345–351. [Google Scholar] [CrossRef]
- Rauwendaal, C. Polymer Extrusion; Carl Hanser Verlag GmbH Co KG: München, Germany, 2014. [Google Scholar]
- Tagliavini, G.; Solari, F.; Montanari, R. CFD simulation of a co-rotating twin-screw extruder: Validation of a rheological model for a starch-based dough for snack food. Int. J. Food Eng. 2018, 14, 20170116. [Google Scholar] [CrossRef]
- Hyvärinen, M.; Jabeen, R.; Kärki, T. The Modelling of Extrusion Processes for Polymers—A Review. Polymers 2020, 12, 1306. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B. Understanding the Extruder Processing Zone: The heart of a twin screw extruder. Plast. Addit. Compd. 2008, 10, 30–35. [Google Scholar] [CrossRef]
- Kostic, M.M.; Reifschneider, L.G. Design of extrusion dies. Encycl. Chem. Process. 2006, 10, 633–649. [Google Scholar]
- Karunanithy, C.; Muthukumarappan, K. A Comparative Study on Torque Requirement During Extrusion Pretreatment of Different Feedstocks. BioEnergy Res. 2012, 5, 263–276. [Google Scholar] [CrossRef]
- Willett, J.L.; Finkenstadt, V.L. Comparison of Cationic and Unmodified Starches in Reactive Extrusion of Starch–Polyacrylamide Graft Copolymers. J. Polym. Environ. 2009, 17, 248–253. [Google Scholar] [CrossRef]
- Kuusipalo, J. Starch-Based Polymers in Extrusion Coating. J. Polym. Environ. 2001, 9, 125–135. [Google Scholar] [CrossRef]
- Ben Daly, H.; Sanschagrin, B.; Nguyen, K.T.; Cole, K.C. Effect of polymer properties on the structure of injection-molded parts. Polym. Eng. Sci. 1999, 39, 1736–1751. [Google Scholar] [CrossRef]
- Yilmaz, G.; Ellingham, T.; Turng, L.-S. Improved processability and the processing-structure-properties relationship of ultra-high molecular weight polyethylene via supercritical nitrogen and carbon dioxide in injection molding. Polymers 2017, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Aulova, A.; Oseli, A.; Bek, M.; Prodan, T.; Emri, I. Effect of Pressure on Mechanical Properties of Polymers. In Encyclopedia of Continuum Mechanics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 733–746. [Google Scholar]
- Wang, J. Some Critical Issues for Injection Molding; BoD–Books on Demand: Norderstedt, Germany, 2012. [Google Scholar]
- Zhang, X.; Cheng, X.; Stelson, K.A.; Bhattacharya, M.; Sen, A.; Voller, V.R. Approximate model of thermal residual stress in an injection molded part. J. Therm. Stress. 2002, 25, 523–538. [Google Scholar] [CrossRef]
- Tabi, T.; Kovacs, J.G. Examination of injection moulded thermoplastic maize starch. Express Polym. Lett. 2007, 1, 804–809. [Google Scholar] [CrossRef]
- Htoon, A.; Shrestha, A.K.; Flanagan, B.M.; Lopez-Rubio, A.; Bird, A.R.; Gilbert, E.P.; Gidley, M.J. Effects of processing high amylose maize starches under controlled conditions on structural organisation and amylase digestibility. Carbohydr. Polym. 2009, 75, 236–245. [Google Scholar] [CrossRef]
- Shujun, W.; Jiugao, Y.; Jinglin, Y. Preparation and Characterization of Compatible and Degradable Thermoplastic Starch/Polyethylene Film. J. Polym. Environ. 2006, 14, 65–70. [Google Scholar] [CrossRef]
- Mousia, Z.; Farhat, I.A.; Pearson, M.; Chesters, M.A.; Mitchell, J.R. FTIR microspectroscopy study of composition fluctuations in extruded amylopectin–gelatin blends. Biopolym. Orig. Res. Biomol. 2001, 62, 208–218. [Google Scholar] [CrossRef]
- Mano, J.F.; Koniarova, D.; Reis, R.L. Thermal properties of thermoplastic starch/synthetic polymer blends with potential biomedical applicability. J. Mater. Sci. Mater. Med. 2003, 14, 127–135. [Google Scholar] [CrossRef]
- Flett, M.S.C. Characteristic Frequencies of Chemical Groups in the Infra-Red; Elsevier: Amsterdam, The Netherlands, 1963. [Google Scholar]
- ISO 527; Plastics—Determination of tensile properties. International Organization for Standardization: Geneva, Switzerland, 2012.
- ASTM 792; Standard Specification for Steel Sheet, 55 % Aluminum-Zinc Alloy-Coated by the HotDip Process. ASTM International: West Conshohocken, PA, USA, 2012.
- ASTM 785-23; Standard Test Method for Rockwell Hardness of Plastics and Electrical Insulating Materials. ASTM International: West Conshohocken, PA, USA, 2023.
- Dimonie, D.; Petre, D.; Vasilievici, G. Polyvinyl alcohol melt processing. J. Elastomers Plast. 2007, 39, 181–194. [Google Scholar] [CrossRef]
- Dimonie, D.; Dina, P.; Vasilievici, G. Biodegradable Composites for Non-Food Packages and Process for Preparing the. Same. Patent RO121692B1, 28 February 2008. [Google Scholar]
- Ismail, H.; Nordin, R.; Ahmad, Z.; Rashid, A. Processability and miscibility of linear low-density polyethylene/poly (vinyl alcohol) blends: In situ compatibilization with maleic acid. Iran. Polym. J. 2010, 19, 297–308. [Google Scholar]
- Coserea, R.M.; Dimonie, D.; Dimonie, M.; Hubca, G. Some aspects concerning the rheology of biodegradable starch based materials. Univ. Politeh. Buchar. Sci. Bull. Ser. B-Chem. Mater. Sci. 2012, 74, 37–48. [Google Scholar]
- Quy, R.B. The effect of the interior particle structure of PVC suspension resin on the behavior of plasticizer absorption. J. Macromol. Sci. Part B Phys. 1981, 20, 235–256. [Google Scholar] [CrossRef]
- Daniels, P.H. A brief overview of theories of PVC plasticization and methods used to evaluate PVC-plasticizer interaction. J. Vinyl Addit. Technol. 2009, 15, 219–223. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers. Wiley: Hoboken, NJ, USA, 1980. [Google Scholar]
- Ferry, J.D. Some reflections on the early development of polymer dynamics: Viscoelasticity, dielectric dispersion, and self-diffusion. Macromolecules 1991, 24, 5237–5245. [Google Scholar] [CrossRef]
- Prentice, P. Rheology and Its Role in Plastics Processing; iSmithers Rapra Publishing: Shrewsbury, UK, 1995; Volume 84. [Google Scholar]
- Ibanescu, C. Reologia Sistemelor Polimerice Multifazice. Available online: http://www.didactic.icpm.tuiasi.ro/cv/ibanescuconstanta/cursuri%20web/RSM.pdf (accessed on 1 September 2024).
- Narimissa, E.; Gupta, R.K.; Kao, N.; Bhattacharya, S. Shear and extensional rheological evaluation of polylactide-nanographite platelet nanocomposites through constitutive equations. In Proceedings of the Society of Plastics Engineers ANTEC 2013 Conference, Cincinnati, OH, USA, 22–24 April 2013; pp. 125–132. [Google Scholar]
- Han, C.D. Rheology in Polymer Processing; Academic Press: Cambridge, MA, USA, 1976. [Google Scholar]
- Macosko, C.W. Rheology Principles, Measurements, and Applications; VCH Publ. Inc.: New York, NY, USA, 1994. [Google Scholar]
- Cogswell, F.N. Measuring the extensional rheology of polymer melts. Trans. Soc. Rheol. 1972, 16, 383–403. [Google Scholar] [CrossRef]
- Vlachopoulos, J.; Strutt, D. Rheology of molten polymers. In Multilayer Flexible Packaging; Elsevier: Amsterdam, The Netherlands, 2016; pp. 77–96. [Google Scholar]
- Polychronopoulos, N.D.; Vlachopoulos, J. Polymer processing and rheology. In Functional Polymers; Jafar Mazumder, M., Sheardown, H., Al-Ahmed, A., Eds.; Polymers and Polymeric Composites: A Reference Series; Springer Nature: Berlin/Heidelberg, Germany, 2018; pp. 133–180. [Google Scholar]
- Shenoy, A.V.; Saini, D.R.; Nadkarni, V.M. Melt rheology of polymer blends from melt flow index. Int. J. Polym. Mater. 1984, 10, 213–235. [Google Scholar] [CrossRef]
- Mertz, A.; Mix, A.; Baek, H.; Giacomin, A. Understanding melt index and ASTM D1238. J. Test. Eval. 2012, 41, 50–62. [Google Scholar] [CrossRef]
- Astm, D. 1238-10. Standard test method for melt flow rates of thermoplastics by extrusion plastometer. ICS Number Code 2010, 83, 20. [Google Scholar]
- Zepnik, S.; Kabasci, S.; Kopitzky, R.; Radusch, H.-J.; Wodke, T. Extensional flow properties of externally plasticized cellulose acetate: Influence of plasticizer content. Polymers 2013, 5, 873–889. [Google Scholar] [CrossRef]
- Sadiku-Agboola, O.; Sadiku, E.R.; Adegbola, A.T.; Biotidara, O.F. Rheological properties of polymers: Structure and morphology of molten polymer blends. Mater. Sci. Appl. 2011, 2, 30. [Google Scholar] [CrossRef]
- Jinescu, V. Proprietatile Fizice si Termodinamica Materialelor Plastice; Editura Tehnica: Bucharest, Romania, 1979. [Google Scholar]
- Barnes, H.A.A. Handbook of Elementary Rheology; Institute of Non-Newtonian Fluid Mechanics, University of Wales: Aberystwyth, UK, 2000. [Google Scholar]
- Vlachopoulos, J.; Wagner, J.R.; Society of Plastics, E. SPE Guide on Extrusion Technology and Troubleshooting; Society for Plastic Engineers: Brookfiled, CT, USA, 2001. [Google Scholar]
- Cox, W.P.; Merz, E.H. Correlation of dynamic and steady flow viscosities. J. Polym. Sci. 1958, 28, 619–622. [Google Scholar] [CrossRef]
- Tang, D.; Marchesini, F.H.; Cardon, L.; D’Hooge, D.R. State of the-Art for Extrudate Swell of Molten Polymers: From Fundamental Understanding at Molecular Scale toward Optimal Die Design at Final Product Scale. Macromol. Mater. Eng. 2020, 305, 2000340. [Google Scholar] [CrossRef]
- Colon, A.R.; Kazmer, D.O.; Peterson, A.M.; Seppala, J.E. Characterization of die-swell in thermoplastic material extrusion. Addit. Manuf. 2023, 73, 103700. [Google Scholar] [CrossRef]
- Rauwendaal, C. Polymer Extrusion; Hanse Gardner Publications: Cincinnati, OH, USA, 2001. [Google Scholar]
- ISO 11443; Plastics—Determination of the Fluidity of Plastics Using Capillary and Slit-Die Rheometers. Measurement of Extrudate Swelling. ISO: Geneva, Switzerland, 2014.
- Vlček, J. Swelling of an extrudate. Polym. Mech. 1978, 14, 273–276. [Google Scholar] [CrossRef]
- Kim, B.; Min, J. Residual stress distributions and their influence on post-manufacturing deformation of injection-molded plastic parts. J. Mater. Process. Technol. 2017, 245, 215–226. [Google Scholar] [CrossRef]
- Guerra, N.B.; Reis, T.M.; Scopel, T.; de Lima, M.S.; Figueroa, C.A.; Michels, A.F. Influence of process parameters and post-molding condition on shrinkage and warpage of injection-molded plastic parts with complex geometry. Int. J. Adv. Manuf. Technol. 2023, 128, 479–490. [Google Scholar] [CrossRef]
- Magnier, A.; Scholtes, B.; Niendorf, T. Analysis of residual stress profiles in plastic materials using the hole drilling method–influence factors and practical aspects. Polym. Test. 2017, 59, 29–37. [Google Scholar] [CrossRef]
- Butler, T.I. Predicting Blown Film Residual Stress Levels Influence on Properties. 2006. Available online: https://www.tappi.org/content/enewsletters/eplace/2006/06PLA71.pdf (accessed on 5 September 2024).
- Guo, C.Q.; Pei, Z.L.; Fan, D.; Liu, R.D.; Gong, J.; Sun, C. Predicting multilayer film’s residual stress from its monolayers. Mater. Des. 2016, 110, 858–864. [Google Scholar] [CrossRef]
- De, A.; DebRoy, T. A perspective on residual stresses in welding. Sci. Technol. Weld. Join. 2011, 16, 204–208. [Google Scholar] [CrossRef]
- Riggins, A.W.; Dadmun, M.D. Controlling residual stress in material extrusion 3D printing through material formulation. Addit. Manuf. 2023, 73, 103678. [Google Scholar] [CrossRef]
- DMA—A Beginner’s Guide, Frequently Asked Questions. Available online: https://resources.perkinelmer.com/lab-solutions/resources/docs/GDE_IntroductionToDMA.pdf?_gl=1*10483gt*_gcl_au*MjAxNjM1MjQ5OS4xNzMwMzkzNjMx*_ga*NTk0MzY1MzA0LjE3MzAzOTM2MzE.*_ga_W34ZJ1Z1Q1*MTczMDM5MzYzMS4xLjEuMTczMDM5NDU0MC42MC4wLjA (accessed on 10 September 2024).
- Shenoy, A.V.; Saini, D.R. An approach to the estimation of polymer melt elasticity. Rheol. Acta 1984, 23, 608–616. [Google Scholar] [CrossRef]
- Cristea, M.; Ionita, D.; Simionescu, B.C. A new insight in the dynamo-mechanical behavior of poly(ethylene terephthalate). Eur. Polym. J. 2010, 46, 2005–2012. [Google Scholar] [CrossRef]
- Dimonie, D.; Trica, B.; Damian, C.; Trusca, R. Embedded Target Filler and Natural Fibres as Interface Agents in Controlling the Stretchability of New Starch and PVOH-Based Materials for Rethinked Sustainable Packaging. Materials 2022, 15, 1377. [Google Scholar] [CrossRef] [PubMed]
- Dimonie, D.; Musat, M.; Doncea, S.M.; Damian, C.M.; Anton, L.; Vasile, E.; Trusca, R.; Râpă, M. Controlling the Melt Resistance to Flow as a Possibility of Improving the Miscibility and the Time Behavior of Some Blends Based on Starch. Int. J. Polym. Sci. 2015, 2015, 582901. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimonie, D.; Grigorescu, R.-M.; Trică, B.; Raduly, M.; Damian, C.-M.; Trusca, R.; Mustatea, A.-E.; Dima, S.-O.; Oancea, F. De- and Re-Structuring of Starch to Control the Melt and Solid State Visco-Elasticity as Method for Getting New Multi Component Compounds with Scalable Properties. Polymers 2024, 16, 3063. https://doi.org/10.3390/polym16213063
Dimonie D, Grigorescu R-M, Trică B, Raduly M, Damian C-M, Trusca R, Mustatea A-E, Dima S-O, Oancea F. De- and Re-Structuring of Starch to Control the Melt and Solid State Visco-Elasticity as Method for Getting New Multi Component Compounds with Scalable Properties. Polymers. 2024; 16(21):3063. https://doi.org/10.3390/polym16213063
Chicago/Turabian StyleDimonie, Doina, Ramona-Marina Grigorescu, Bogdan Trică, Monica Raduly, Celina-Maria Damian, Roxana Trusca, Alina-Elena Mustatea, Stefan-Ovidiu Dima, and Florin Oancea. 2024. "De- and Re-Structuring of Starch to Control the Melt and Solid State Visco-Elasticity as Method for Getting New Multi Component Compounds with Scalable Properties" Polymers 16, no. 21: 3063. https://doi.org/10.3390/polym16213063
APA StyleDimonie, D., Grigorescu, R.-M., Trică, B., Raduly, M., Damian, C.-M., Trusca, R., Mustatea, A.-E., Dima, S.-O., & Oancea, F. (2024). De- and Re-Structuring of Starch to Control the Melt and Solid State Visco-Elasticity as Method for Getting New Multi Component Compounds with Scalable Properties. Polymers, 16(21), 3063. https://doi.org/10.3390/polym16213063