
Statistical and Block Copolymers of n-Dodecyl and Allyl Isocyanate via Titanium-Mediated Coordination Polymerization: A Route to Polyisocyanates with Improved Thermal Stability

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Homopolymer PDDIC
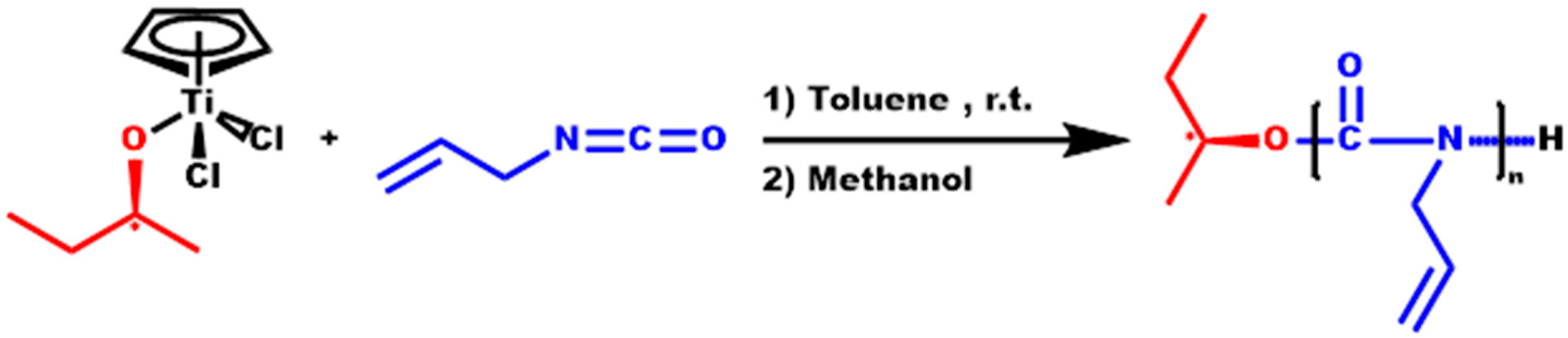
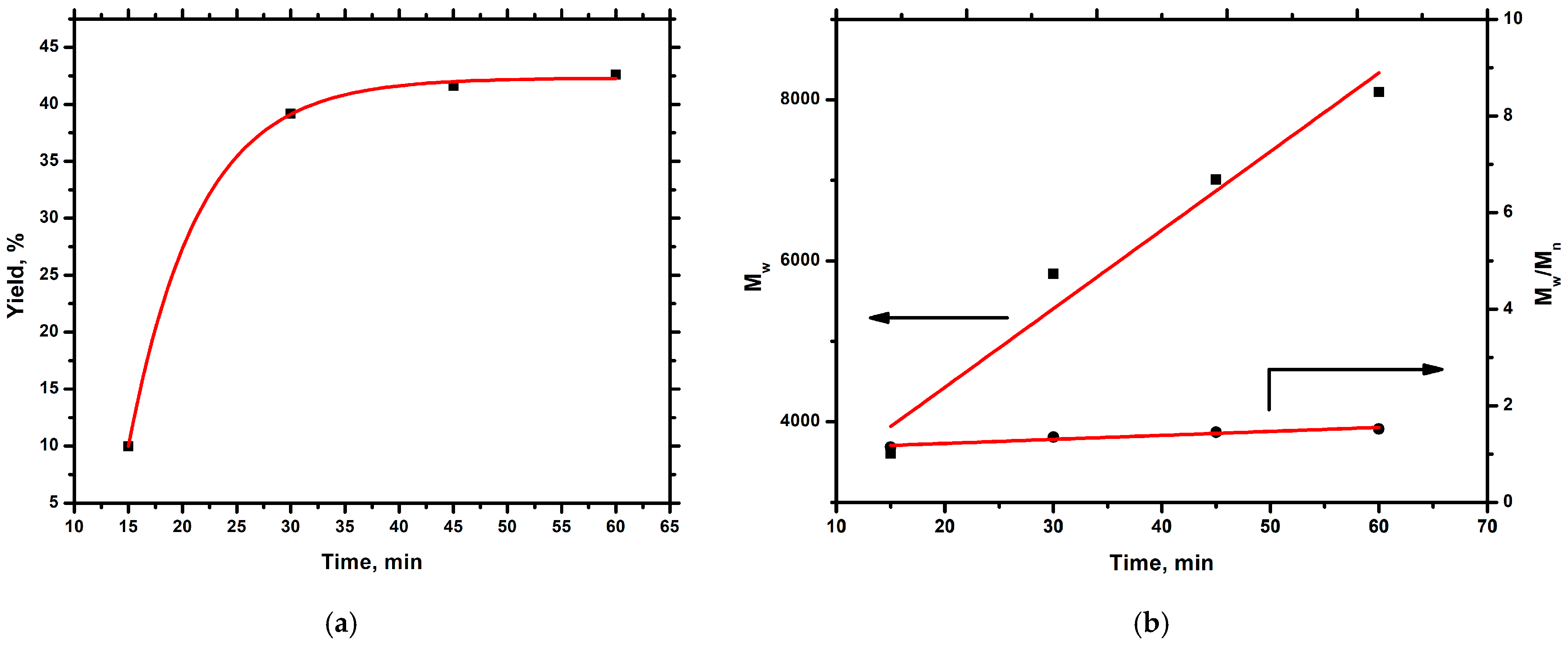
2.3. Synthesis of Homopolymer PALIC and Kinetic Study
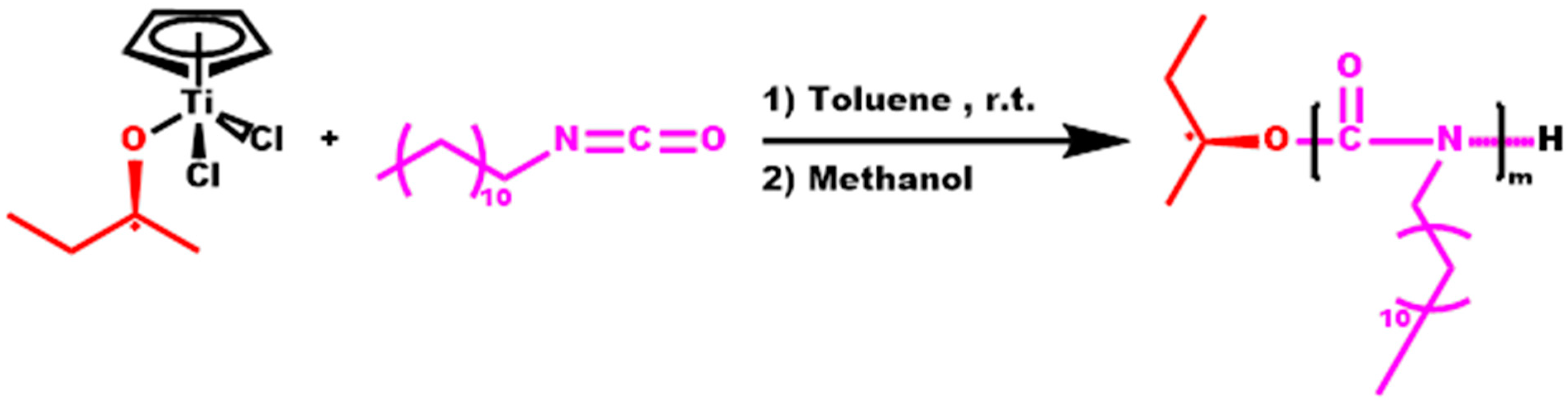
2.4. Synthesis of Statistical Copolymers, P(ALIC-co-DDIC)
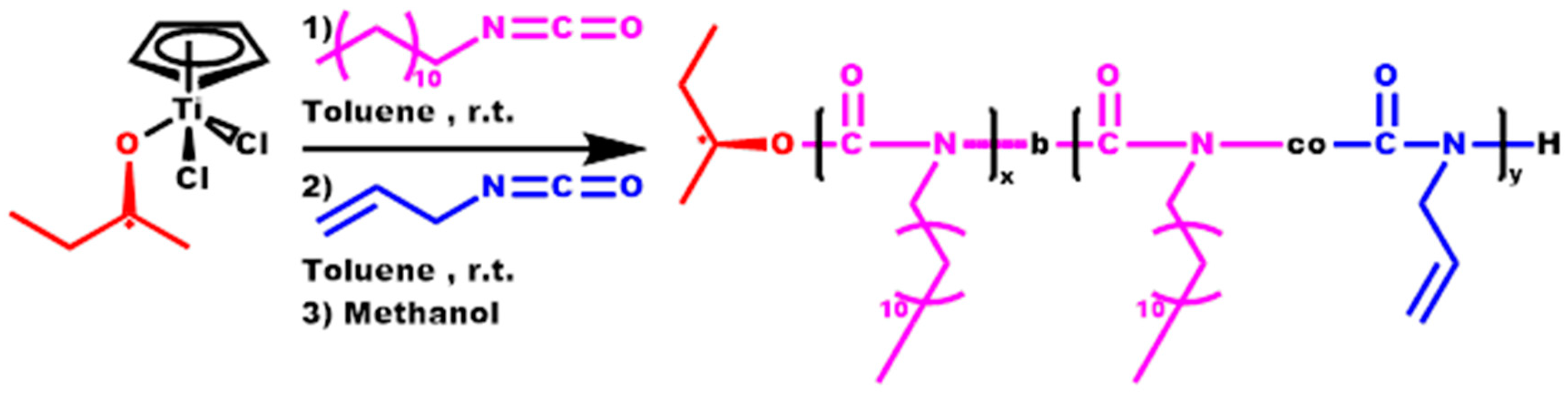
2.5. Synthesis of Block Copolymers, P[DDIC-b-(DDIC-co-ALIC)]
2.6. Synthesis of P3PETPIC and P(3PETPIC-co-DDIC)
2.7. Characterization
3. Results and Discussion
3.1. Homopolymerization of ALIC and Kinetic Study
3.2. Homopolymerization of DDIC
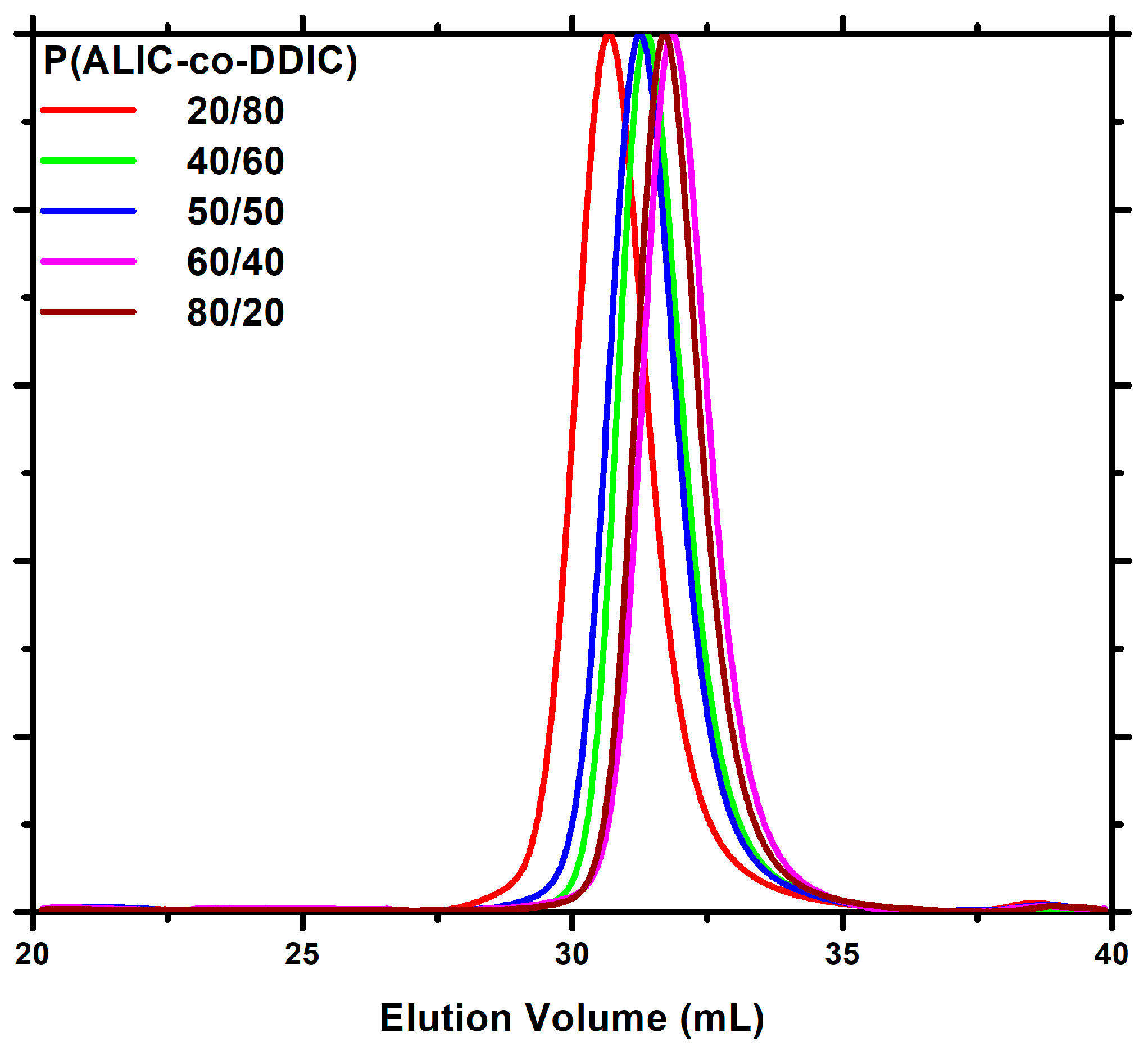
3.3. Statistical Copolymers, P(ALIC-co-DDIC)
3.4. Monomer Reactivity Ratios and Statistical Analysis of the Copolymers
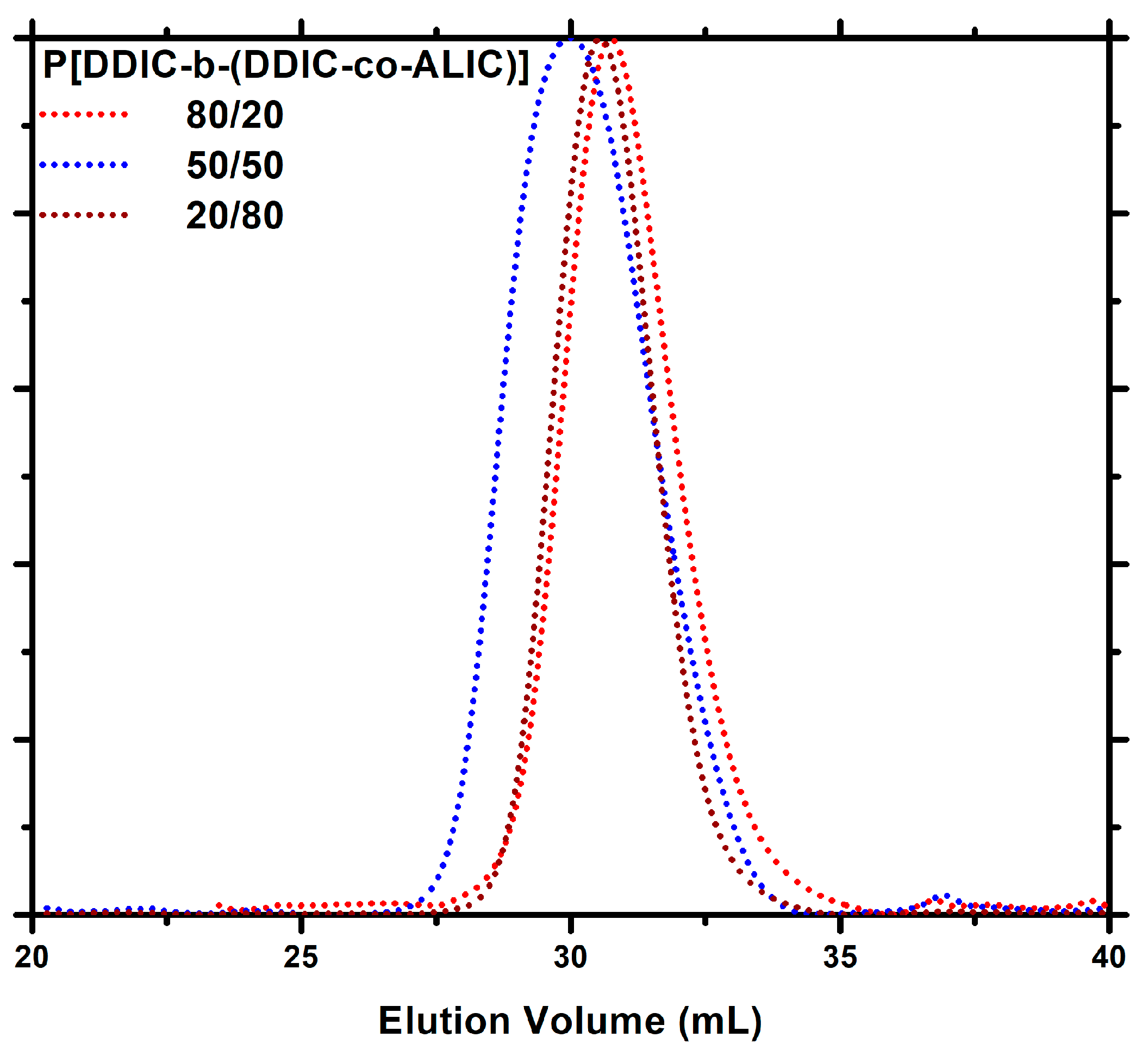
3.5. Block Copolymers, P[DDIC-b-(DDIC-co-ALIC)]
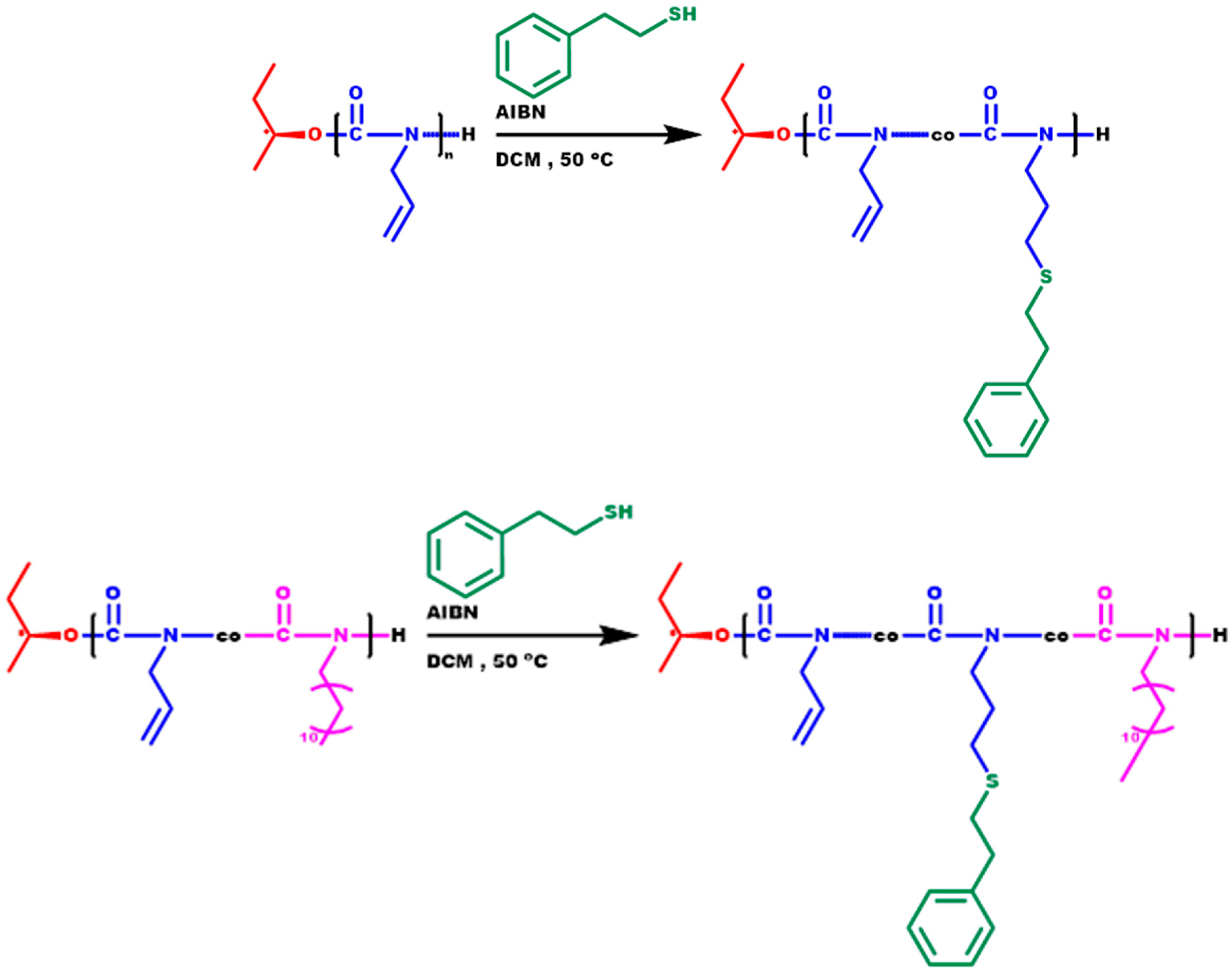
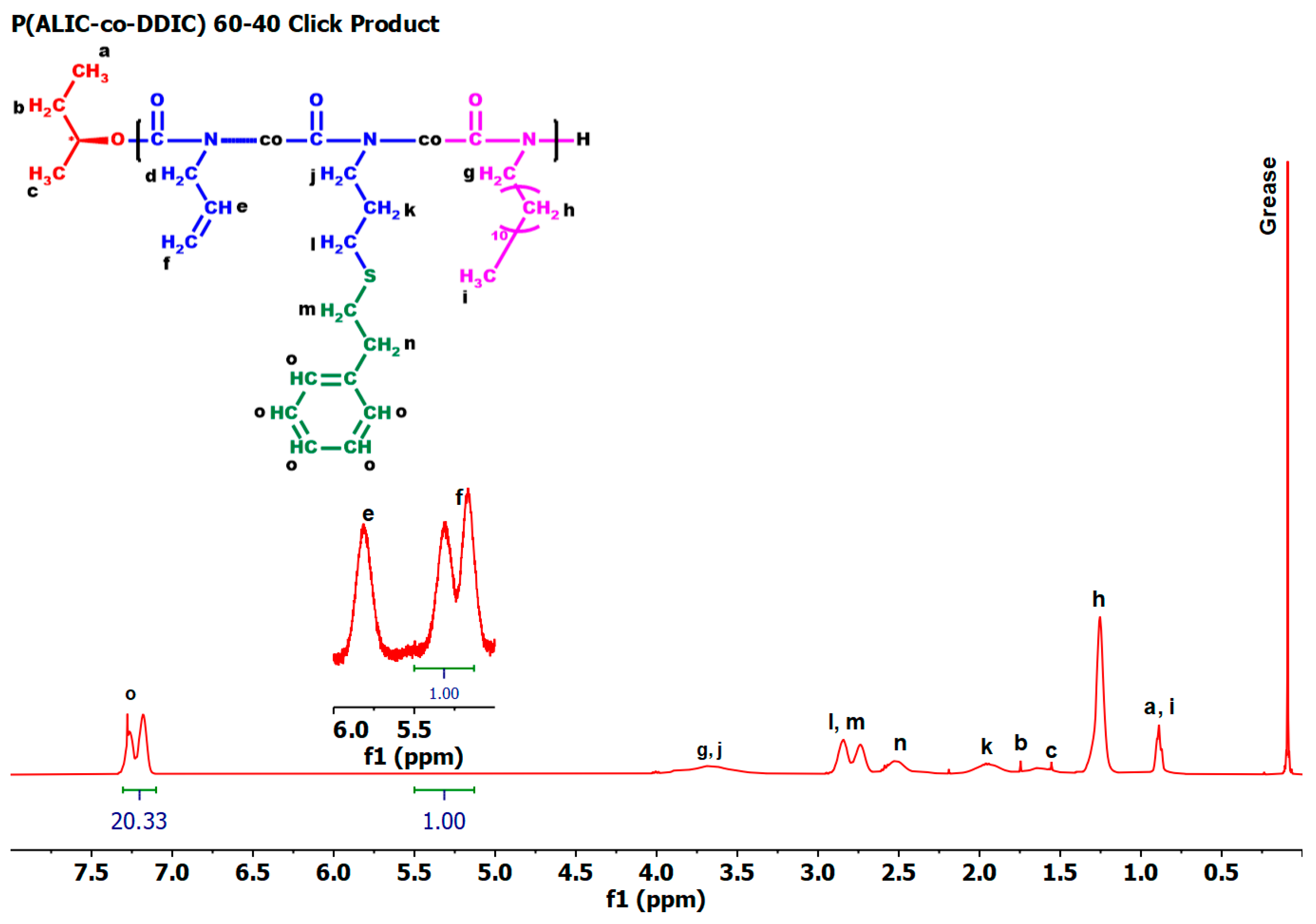
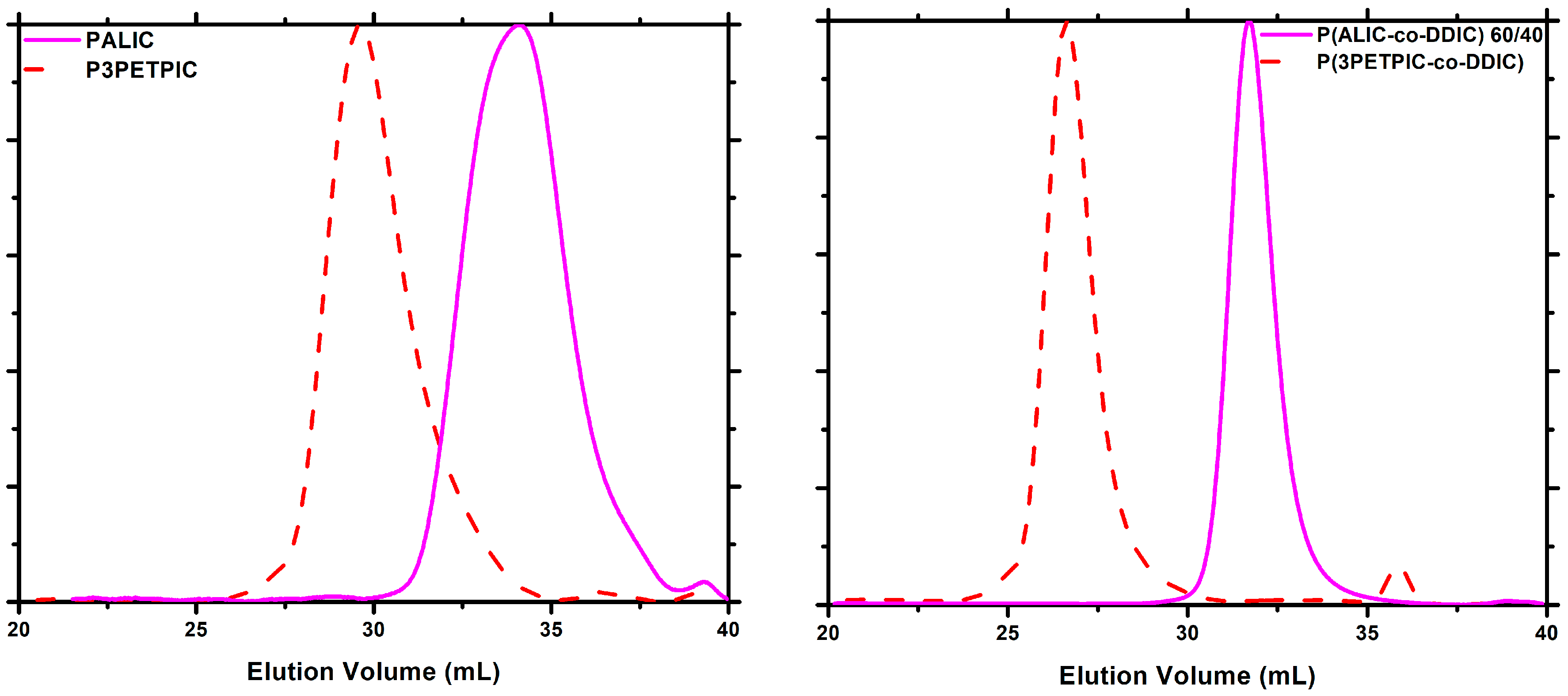
3.6. Synthesis of P3PETPIC and P(3PETPIC-co-DDIC) via Thiol-Ene Click Reaction
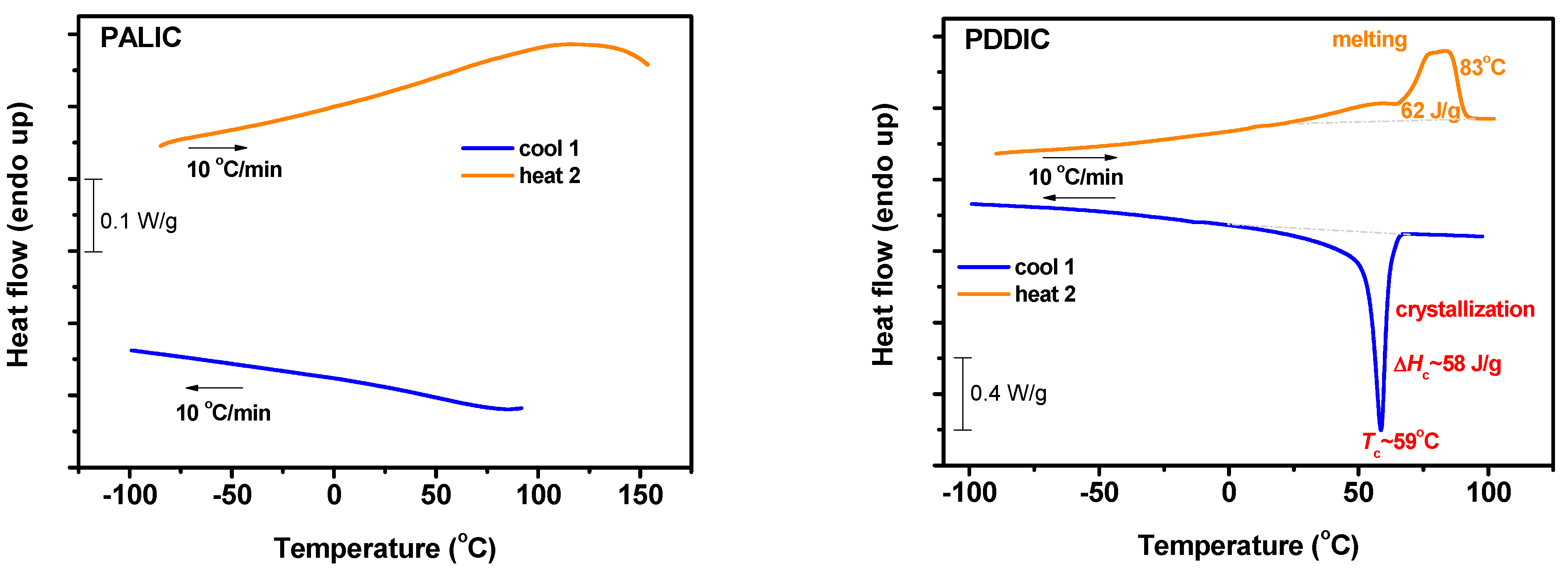
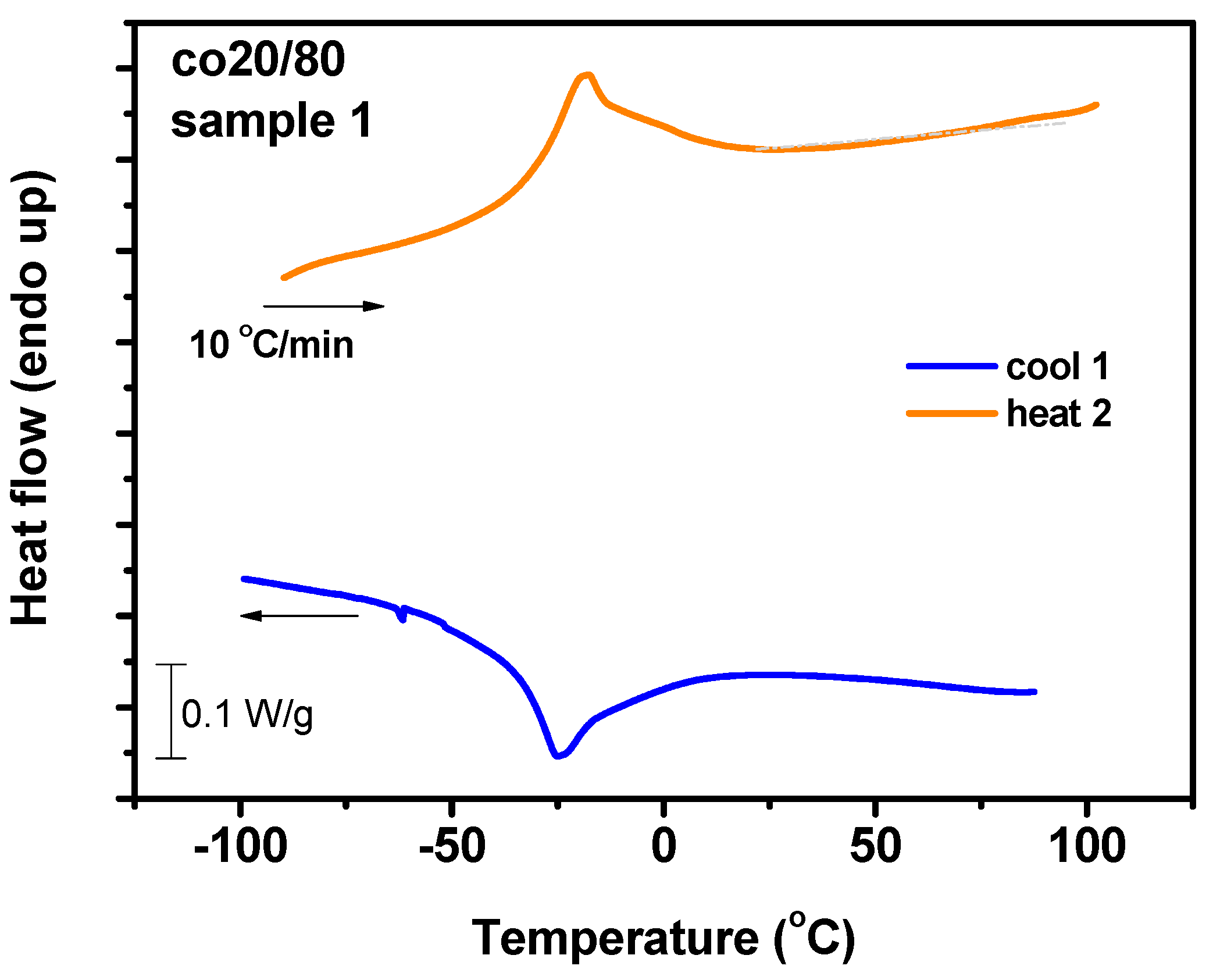
3.7. Thermal Analysis
3.7.1. DSC Analysis
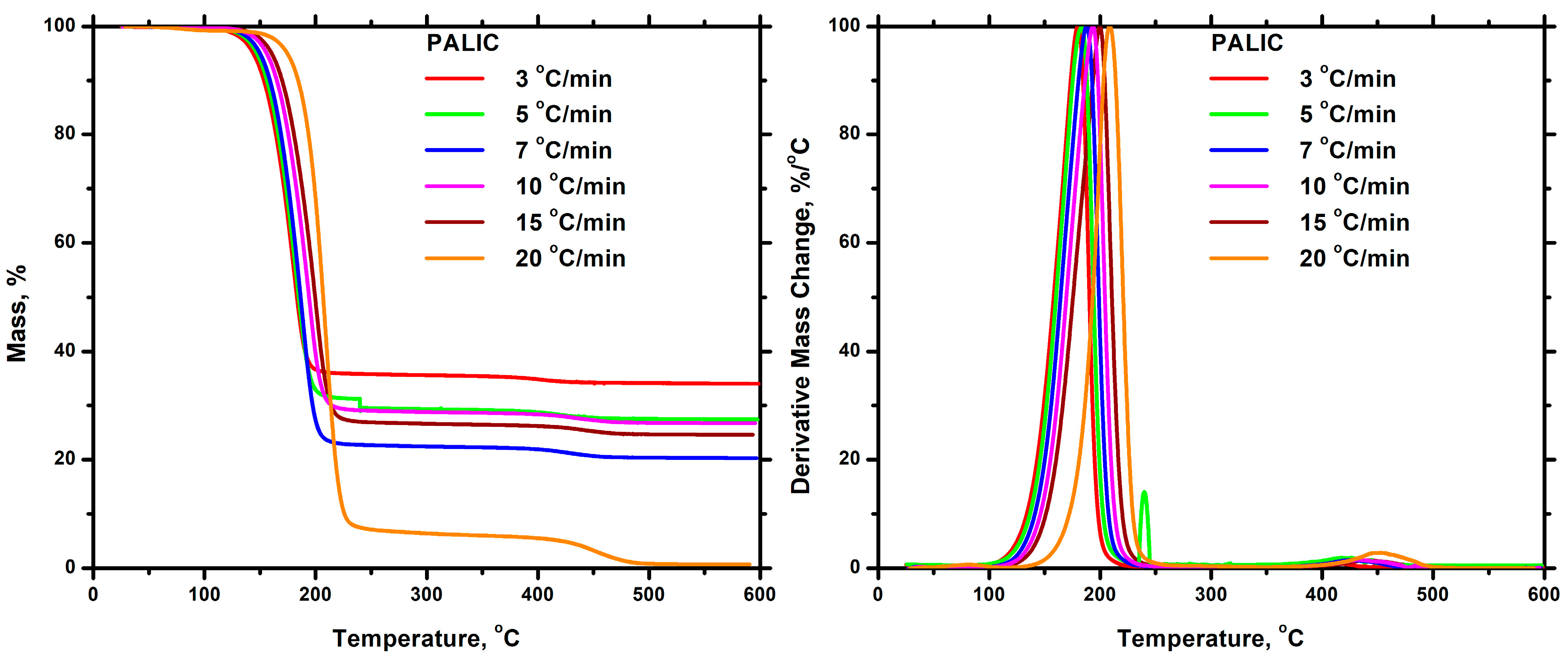
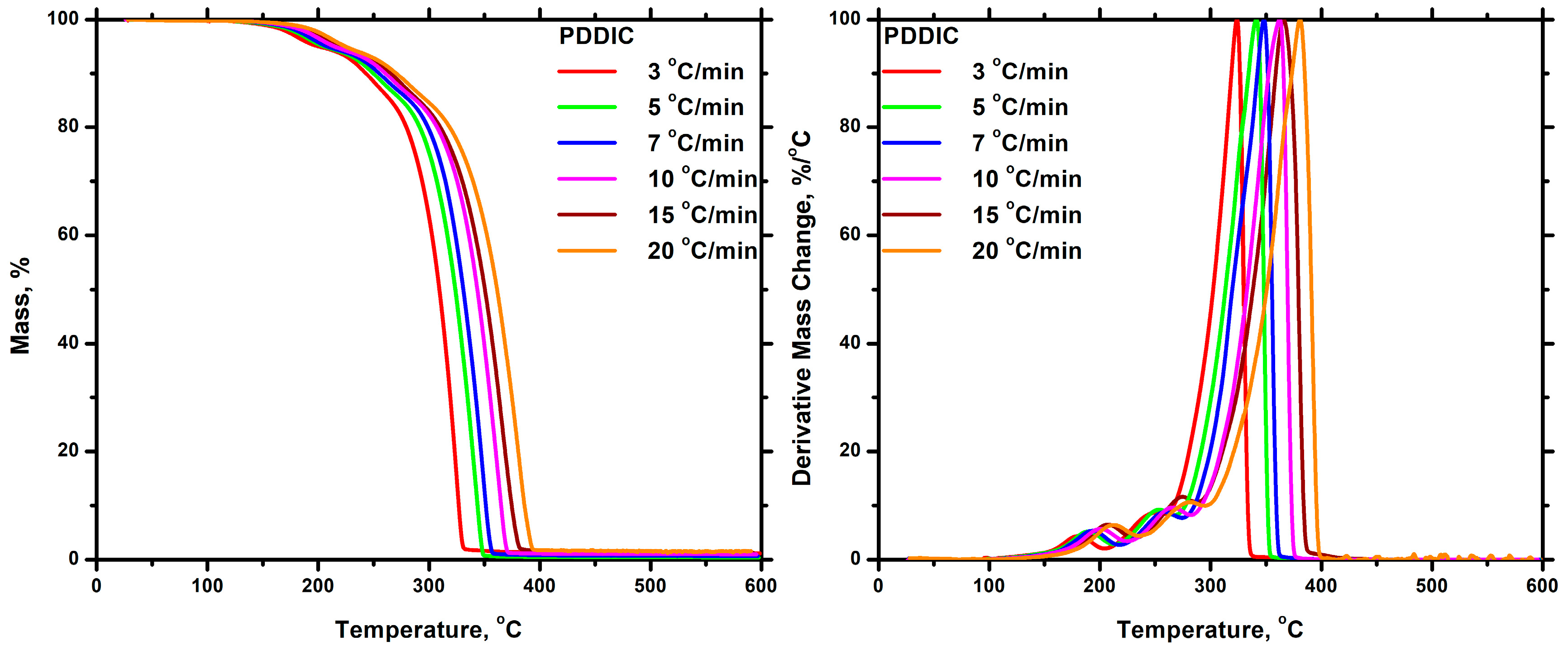
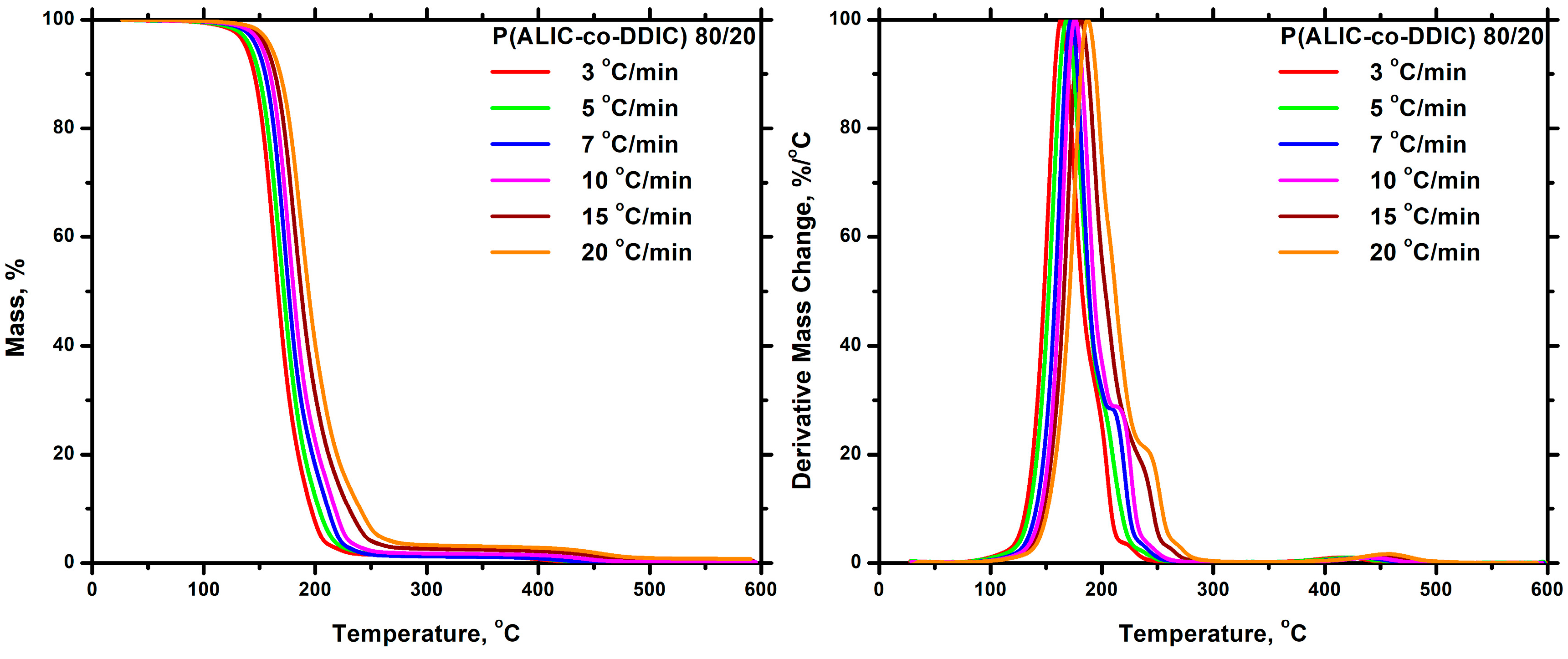
3.7.2. Thermal Decomposition
Homopolymers
Statistical Copolymers
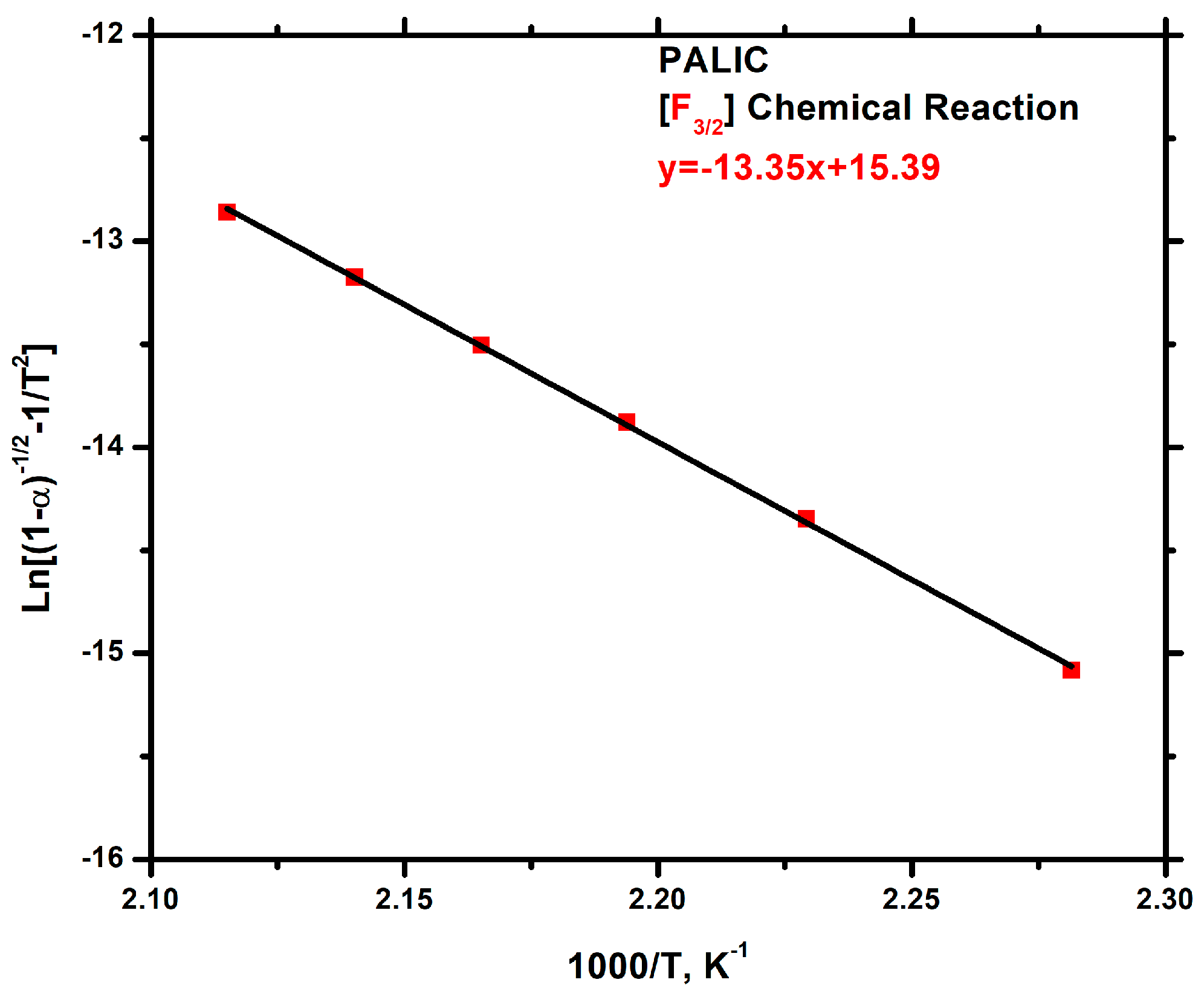
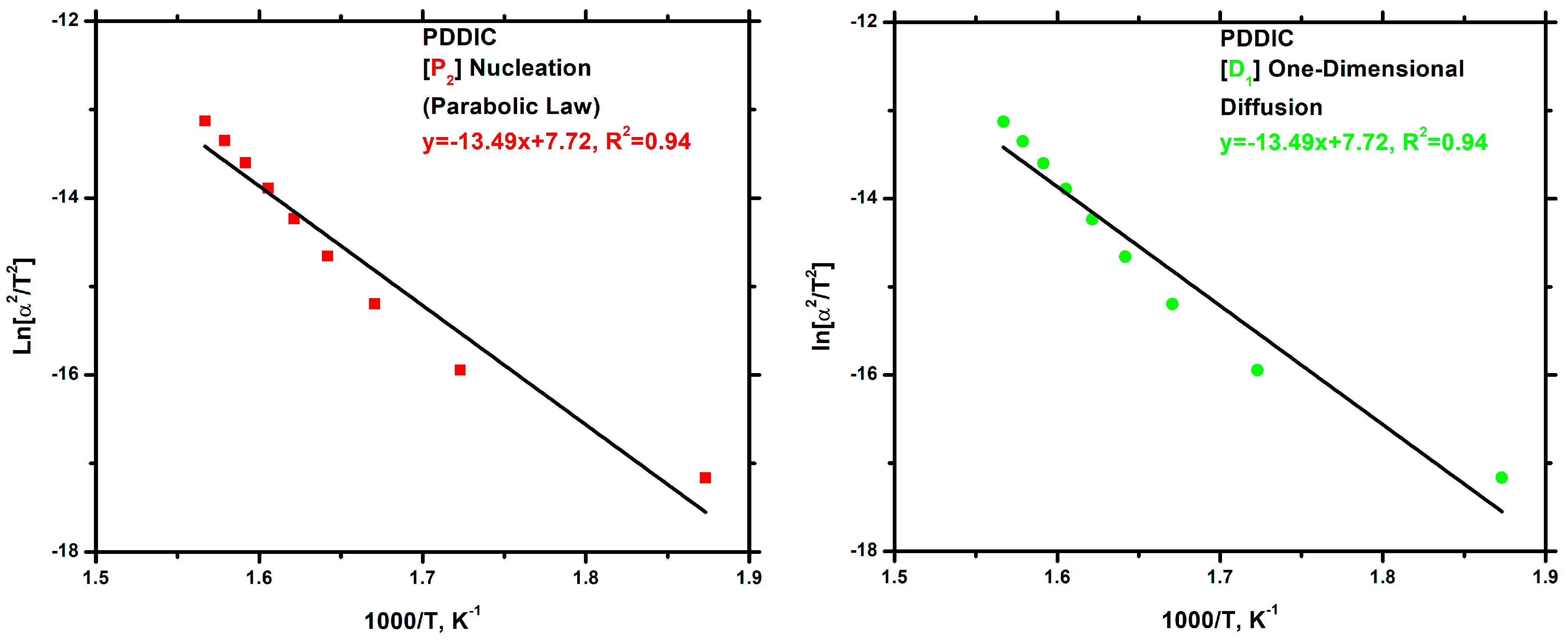
3.7.3. Kinetics of Thermal Degradation of Homopolymers and Statistical Copolymers
3.7.4. Block Copolymers
3.7.5. Thiol-Ene Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kuran, W. Principles of Coordination Polymerization; John Wiley & Sons Ltd.: Chichester, UK, 2001. [Google Scholar]
- Novokshonova, L.A.; Zakharov, V.A. Kinetics of Olefin Polymerization and Active Sites of Heterogeneous Ziegler-Natta Catalysts. Adv. Polym. Sci. 2013, 257, 99–134. [Google Scholar]
- Soga, K.; Shiono, T. Ziegler-Natta catalysts for olefin polymerizations. Prog. Polym. Sci. 1997, 22, 1503–1546. [Google Scholar] [CrossRef]
- Baugh, L.S.; Canich, J.A.M. (Eds.) Stereospecific Polymerization with Single Site Catalysts; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Brintzinger, H.H.; Fischer, D. Development of ansa-Metallocene Catalysts for Isotactic Olefin Polymerization. Adv. Polym. Sci. 2013, 258, 29–42. [Google Scholar]
- Karanikolopoulos, G.; Batis, C.; Pitsikalis, M.; Hadjichristidis, N. The influence of the nature of the catalytic systems on the zirconocenes catalyzed polymerization of alkyl methacrylates. Macromol. Chem. Phys. 2003, 204, 831–840. [Google Scholar] [CrossRef]
- Chen, E.Y.X. Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.E.; Foteinogiannopoulou, G.; Fega, E.; Pitsikalis, M. Statistical Copolymers of 2-Methyl-and 2-Phenyl-oxazoline by Metallocene-Mediated Cationic Ring-Opening Polymerization: Synthesis, Reactivity Ratios, Kinetics of Thermal Decomposition and Self-Assembly Behavior in Aqueous Solutions. J. Macromol. Sci. Part. A 2015, 52, 630–641. [Google Scholar] [CrossRef]
- Batagianni, E.; Marathianos, A.; Koraki, A.; Maroudas, A.-P.; Pitsikalis, M. Metallocene-mediated cationic polymerization of vinyl ethers: Kinetics of polymerization and synthesis and characterization of statistical copolymers. J. Macromol. Sci. Part. A Pure Appl. Chem. 2016, 53, 140–151. [Google Scholar] [CrossRef]
- Zouganelis, S.; Choinopoulos, I.; Goulas, I.; Pitsikalis, M. Statistical copolymers of n-butyl vinyl ether and 2-chloroethyl vinyl ether via metallocene-mediated cationic polymerization. A scaffold for the synthesis of graft copolymers. Polymers 2019, 11, 1510. [Google Scholar] [CrossRef]
- Kostakis, K.; Mourmouris, S.; Karanikolopoulos, G.; Pitsikalis, M.; Hadjichristidis, N. Ring-opening polymerization of lactones using zirconocene catalytic systems. Block copolymerization with methyl methacrylate. J. Polym. Sci. Polym. Chem. Ed. 2007, 45, 3524–3537. [Google Scholar] [CrossRef]
- Khosravi, E.; Szymanska-Buzar, T. (Eds.) Ring Opening Metathesis Polymerization and Realated Chemistry NATO Science Series; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2002; Volume 56. [Google Scholar]
- Grubbs, R.H. (Ed.) Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Giambastiani, G.; Cámpora, J. (Eds.) Olefin Upgrading Catalysis by Nitrogen-Based Metal Complexes I. State of the Art and Perspectives. In Catalysis by Metal Complexes; Springer: Berlin/Heidelberg, Germany, 2017; Volume 35. [Google Scholar]
- Osakada, K. Olefin Polymerization with Non-metallocene Catalysts (Early Transition Metals). Organomet. React. Polym. 2014, 85, 89–117. [Google Scholar]
- Takeuchi, D. Recent progress in olefin polymerization catalyzed by transition metal complexes: New catalysts and new reactions. Dalton Trans. 2010, 39, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tamm, M. Transition metal complexes supported by highly basic imidazolin-2-iminato and imidazolin-2-imine N-donor ligands. Coord. Chem. Rev. 2014, 260, 116–138. [Google Scholar] [CrossRef]
- Batis, C.; Karanikolopoulos, G.; Pitsikalis, M.; Hadjichristidis, N. Complex macromolecular architectures utilizing metallo-cene catalysts. Macromolecules 2003, 36, 9763–9774. [Google Scholar] [CrossRef]
- Wang, Y.; Lai, J.; Gou, Q.Q.; Gao, R.; Zheng, G.; Zhang, R.D.; Song, Z.H.; Yue, Q.; Guo, Z.F. Development of well-defined olefin block (co)polymers achieved by late transition metal catalysts: Catalyst, synthesis and characterization. Coord. Chem. Rev. 2024, 522, 216195. [Google Scholar] [CrossRef]
- Βur, A.J.; Fetters, L.J. The Chain Structure, Polymerization and Conformation of Polyisocyanates. Chem. Rev. 1976, 76, 727–745. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Organotitanium(IV) compounds as catalysts for the polymerization of isocyanates: The polymerization of isocyanates with functionalized side chains. Macromolecules 1993, 26, 436–439. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. “Living” titanium(IV) catalyzed coordination polymerizations of isocyanates. J. Am. Chem. Soc. 1991, 113, 5065–5066. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Living Organotitanium(IV)-Catalyzed Polymerizations of Isocyanates. J. Am. Chem. Soc. 1996, 118, 1906–1916. [Google Scholar] [CrossRef]
- Chae, C.-G.; Seo, H.-B.; Lee, J.-S. Living Anionic Polymerization of Isocyanates. In Anionic Polymerization. Priciples, Practice, Strength, Consequences and Applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Tokyo, Japan, 2015; pp. 339–386. [Google Scholar]
- Shashoua, V.E.; Sweeny, W.; Tietz, R.F. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc. 1960, 82, 866–873. [Google Scholar] [CrossRef]
- Shashoua, V.E. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc. 1959, 81, 3156. [Google Scholar] [CrossRef]
- Shin, Y.-D.; Ahn, J.-H.; Lee, J.-S. Anionic polymerization of isocyanates with optical functionalities. Polymer 2001, 42, 7979–7985. [Google Scholar] [CrossRef]
- Shin, Y.-D.; Kim, S.-Y.; Ahn, J.-H.; Lee, J.-S. Synthesis of Poly(n-hexyl isocyanate) by Controlled Anionic Polymerization in the Presence of NaBPh4. Macromolecules 2001, 34, 2408–2410. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Shin, Y.-D.; Kim, S.-Y.; Lee, J.-S. Synthesis of well-defined block copolymers of n-hexyl isocyanate with isoprene by living anionic polymerization. Polymer 2003, 44, 3847–3854. [Google Scholar] [CrossRef]
- Lee, J.-S.; Ryu, S.-W. Anionic living polymerization of 3-(triethoxysilyl)propyl isicyanate. Macromolecules 1999, 32, 2085–2087. [Google Scholar] [CrossRef]
- Kang, N.-G.; Kang, B.-G.; Koh, H.-D.; Changez, M.; Lee, J.-S. Block copolymers containing pyridine moieties: Precise synthesis and applications. React. Funct. Polym. 2009, 69, 470–479. [Google Scholar] [CrossRef]
- Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Triblock copolymers and penatblock terpolymers of n-hexyl isocyanate with styrene and isoprene: Synthesis, characterization and thermal properties. J. Polym. Sci. Polym. Chem. Ed. 2003, 41, 3094–3102. [Google Scholar] [CrossRef]
- Zorba, G.; Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Anionic polymerization of n-hexyl isocyanate with monofunctional initiators. Application in the synthesis of diblock copolymers with styrene and isoprene. J. Polym. Sci. Polym. Chem. Ed. 2005, 43, 3533–3542. [Google Scholar] [CrossRef]
- Min, J.; Yoo, H.-S.; Shah, P.N.; Chae, C.-G.; Lee, J.-S. Enolate anionic initiator, sodium deoxybenzoin, for leading living natures by formation of aggregates at the growth chain ends. J. Polym. Sci. Polym. Chem. Ed. 2013, 51, 1742–1748. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Shin, Y.-D.; Nath, G.Y.; Park, S.-Y.; Rahman, M.S.; Samal, S.; Lee, J.-S. Unprecedented Control over Polymerization of n-Hexyl Isocyanate using an Anionic Initiator Having Synchronized Function of Chain-End Protection. J. Am. Chem. Soc. 2005, 127, 4132–4133. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Yoo, H.-S.; Changez, M.; Lee, J.-S. Living anionic polymerization of isocyanate containing a reactive carbamate group. Macromolecules 2009, 42, 3927–3932. [Google Scholar] [CrossRef]
- Zorba, G.; Pitsikalis, M.; Hadjichristidis, N. Novel well-defined star homopolymers and star-block copolymers of poly(n-hexyl isocyanate) by anionic polymerization. J. Polym. Sci. Polym. Chem. Ed. 2007, 45, 2387–2399. [Google Scholar] [CrossRef]
- Wu, J.; Pearce, E.M.; Kwei, T.K. A Novel Rod−Coil Block Copolymer and Its Compatible Blends. Macromolecules 2001, 34, 1828–1836. [Google Scholar] [CrossRef]
- Liu, X.; Deng, J.; Wu, Y.; Zhang, L. Amphiphilic triblock terpolymers consisting of poly(n-hexyl isocyanate) and poly(ethylene glycol): Preparation and characterization. Polymer 2012, 53, 5717–5722. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Complex architectures through living polymerizations. The synthesis of "once-broken worms" and triblock copolymers using bimetallic initiators. Macromolecules 1993, 26, 4067–4069. [Google Scholar] [CrossRef]
- Touris, A.; Kostakis, K.; Mourmouris, S.; Kotzabasakis, V.; Pitsikalis, M.; Hadjichristidis, N. Complex Macromolecular Architectures Based on n-Hexyl Isocyanate and ϵ-Caprolactone Using Titanium-Mediated Coordination Polymerization. Macromolecules 2008, 41, 2426–2438. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Synthesis and Characterization of Wormlike Three-Arm Poly(n-hexyl isocyanate) Star Polymers. Macromolecules 2001, 34, 3849–3855. [Google Scholar]
- Choinopoulos, I.; Patias, G.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of chiral poly(l-lactide-b-hexyl isocyanate) macromonomers with norbornenyl end groups and their homopolymerization through ring opening metathesis polymerization to afford polymer brushes. J. Polym. Sci. Part. A Polym. Chem. 2017, 55, 1102–1112. [Google Scholar] [CrossRef]
- Choinopoulos, I.; Patias, G.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of brush diblock and triblock copolymers bearing polynorbornene backbone and poly(l-lactide) and/or poly(hexyl isocyanate) side chains by a combination of coordination and ring opening metathesis polymerization. J. Polym. Sci. Part. A Polym. Chem. 2017, 55, 3455–3465. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Du, J.; Rainbolt, E.A.; Pathiranage, T.M.S.K.; Huang, P.; Reuther, J.F.; Novak, B.M.; Biewer, M.C.; Stefan, M.C. A semiconducting liquid crystalline block copolymer containing regioregular poly(3-hexylthiophene) and nematic poly(n-hexyl isocyanate) and its application in bulk heterojunction solar cells. J. Mater. Chem. A 2014, 2, 16148–16156. [Google Scholar] [CrossRef]
- Miyake, G.M.; Weitekamp, R.A.; Piunova, V.A.; Grubbs, R.H. Synthesis of isocyanate-based brush block copolymers and their rapid self-assembly to infrared-reflecting photonic crystals. J. Am. Chem. Soc. 2012, 134, 14249–14254. [Google Scholar] [CrossRef]
- Satoh, T.; Nishikawa, N.; Kawato, D.; Suemasa, D.; Jung, S.; Kim, Y.Y.; Ree, M.; Kakuchi, T. Precise synthesis of a rod-coil miktoarm star copolymer containing poly(n-hexyl isocyanate) and aliphatic ester. Polym. Chem. 2014, 5, 588–599. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Mihara, T.; Kikuchi, M.; Lien, L.T.N.; Nagai, K. Synthesis of Methacrylate-Ended Poly(n-hexyl isocyanate) Rodlike Macromonomers and Their Radical Copolymerization Behavior. Macromolecules 2007, 40, 950–958. [Google Scholar] [CrossRef]
- Deike, S.; Binder, W.H. Induction of Chirality in β-Turn Mimetic Polymer Conjugates via Postpolymerization “Click” Coupling. Macromolecules 2017, 50, 2637–2644. [Google Scholar] [CrossRef]
- Schneider, N.S.; Furusaki, S.; Lenz, R.W. Chain stiffness in polyisocyanates. J. Polym. Sci. Part. A Gen. Pap. 1965, 3, 933–948. [Google Scholar] [CrossRef]
- Teramoto, A. Cooperative conformational transitions in linear macromolacules undergoing chiral perturbations. Progr. Polym. Sci. 2001, 26, 667–720. [Google Scholar] [CrossRef]
- Gu, H.; Sato, T.; Teramoto, A.; Varichon, L.; Green, M.M. Molecular Mechanisms for the Optical Activities of Polyisocyanates Induced by Intramolecular Chiral Perturbations. Polym. J. 1997, 29, 77–84. [Google Scholar] [CrossRef]
- Tonelli, A.E. Conformational Characteristics of the Poly(n-alkyl isocyanates). Macromolecules 1974, 7, 628–631. [Google Scholar] [CrossRef]
- Lecomte, L.; Desreux, V. Dielectric properties of poly(4-methylphenylisocyanate) and poly(4-methoxyphenylisocyanate) in solution. Eur. Polym. J. 1976, 12, 741–747. [Google Scholar] [CrossRef]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Mao, G.-P. Self-Assembled Smectic Phases in Rod-Coil Block Copolymers. Science 1996, 273, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Hwang, S.S. Zigzag morphology of a poly(styrene-b-hexyl isocyanate) rod-coil block copolymer. Macromolecules 1995, 28, 1688–1697. [Google Scholar] [CrossRef]
- Vazaios, A.; Touris, A.; Echeverria, M.; Zorba, G.; Pitsikalis, M. Micellization behaviour of linear and nonlinear block copolymers based on poly(n-hexyl isocyanate) in selective solvents. Polymers 2020, 12, 1678. [Google Scholar] [CrossRef]
- Green, M.M.; Peterson, N.C.; Sato, T.; Teramoto, A.; Cook, R.; Lifson, S. A Helical Polymer with a Cooperative Response to Chiral Information. Science 1995, 268, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Matsuda, M.; Nakano, T.; Yashima, E. Asymmetric Polymerization of Isocyanates with Optically Active Anionic Initiators. Polym. J. 1993, 4, 391–396. [Google Scholar] [CrossRef]
- Μayer, S.; Zentel, R. A new chiral polyisocyanate: An optical switch triggered by a small amount of photochromic side groups. Macromol. Chem. Phys. 1998, 199, 1675–1682. [Google Scholar] [CrossRef]
- Baudis, S.; Ligon, S.C.; Seidler, K.; Weigel, G.; Grasl, C.; Bergmeister, H.; Schima, H.; Liska, R. Hard-Block Degradable Thermoplastic Urethane-Elastomers for Electrospun Vascular Prostheses. J. Polym. Sci. Part. A Polym. Chem. 2012, 50, 1272–1280. [Google Scholar] [CrossRef]
- Godfrey, R.A.; Miller, G.W. Block polymers of isocyanates and vinyl monomers by homogeneous anionic polymerization. J. Polym. Sci. Part. A-1 Polym. Chem. 1969, 7, 2387–2404. [Google Scholar] [CrossRef]
- Chae, C.-G.; Shah, P.N.; Min, J.; Seo, H.-B.; Lee, J.-S. Synthesis of Novel Amphiphilic Polyisocyanate Block Copolymer with Hydroxyl Side Group. Macromolecules 2014, 47, 1563–1569. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Well-defined polyisocyanates via organotitanium(IV) catalyzed living polymerization of isocyanates. Makromol. Chem. Macromol. Symp. 1993, 67, 203–211. [Google Scholar] [CrossRef]
- Zhao, W.; Kloczkowski, A.; Mark, J.E.; Erman, B.; Bahar, I. Main-chain lyotropic liquid-crystalline elastomers. 1. Syntheses of cross-linked polyisocyanate gels acquiring liquid-crystalline behavior in the swollen state. Macromolecules 1996, 29, 2796–2804. [Google Scholar] [CrossRef]
- Ratkanthwar, K.; Zhao, J.; Zhang, H.; Hadjichristidis, N.; Mays, J.W. Schlenk Techniques for Anioinic Polymerization. In Anionic Polymerization. Principles, Practice, Strength, Consequences and Applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part. A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Hagiopol, C. Copolymerization: Toward a Systematic Approach; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdos, F. Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method. J. Macromol. Sci. Part A Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Beginn, U. COPOINT—A simple computer program to determine copolymerization parameters by numerical integration. E-Polymers 2005, 5, 759–773. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 155–270. [Google Scholar]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part. B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Elias, H.G. Synthesis, Materials, and Technology; Springer: New York, NY, USA, 1984; Volume 2. [Google Scholar]
- Mantzara, D.; Katara, A.; Panteli, M.; Stavrakaki, I.G.; Plachouras, N.V.; Choinopoulos, I.; Pitsikalis, M. Synthesis and characterization of statistical and block copolymers of n-hexyl isocyanate and 2-chloroethyl isocyanate via coordination polymerization. J. Polym. Sci. 2024, 62, 2484–2502. [Google Scholar] [CrossRef]
- Katara, A.; Mantzara, D.; Panteli, M.; Choinopoulos, I.; Pitsikalis, M. Statistical and Block Copolymers of n-Hexyl Isocyanate and 2–Phenylethyl Isocyanate via Coordination Polymerization. Synthesis, Characterization and Thermal Properties. Eur. Polym. J. 2023, 199, 112441. [Google Scholar] [CrossRef]
- Panteli, M.; Mantzara, D.; Katara, A.; Choinopoulos, I.; Pitsikalis, M. Synthesis and characterization ofStatistical and Block Copolymers of n-Hexyl Isocyanate and 3–(triethoxysilyl) propyl Isocyanate via Coordination Polymerization. Polymers 2023, 15, 4113. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Jordan, E.F., Jr. Side-chain crystallinity. III. Influence of side-chain crystallinity on the glass transition temperatures of selected copolymers incorporating n-octadecyl acrylate or vinyl stearate. J. Polym. Sci. Part A-1 Polym. Chem. 1971, 9, 3367–3378. [Google Scholar] [CrossRef]
- Alig, I.; Jarek, M.; Hellmann, G.P. Restricted segmental mobility in side-chain crystalline comblike polymers, studied by dielectric relaxation measurements. Macromolecules 1998, 31, 2245–2251. [Google Scholar] [CrossRef]
- Ozawa, T. A New Method of Analyzing Thermogravimetric Data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Part. B Polym. Lett. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetic analysis of derivative curves in thermal analysis. J. Therm. Anal. Calorim. 1970, 2, 301–324. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Sbirrazzuoli, N. Isoconversional Kinetic Analysis of Thermally Stimulated Processes in Polymers. Macromol. Rapid Commun. 2006, 27, 1515–1532. [Google Scholar] [CrossRef]
- Doyle, C.D. Kinetic analysis of thermogravimetric data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Kinetic Parameters from Thermogravimetric Data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Trache, D.; Abdelaziz, A.; Siouani, B. A simple and linear isoconversional method to determine the pre-exponential factors and the mathematical reaction mechanism functions. J. Therm. Anal. Calorim. 2017, 128, 335–348. [Google Scholar] [CrossRef]
- Galukhin, A.; Liavitskaya, T.; Vyazovkin, S. Kinetic and mechanistic insights into thermally initiated polymerization of cyanate esters with different bridging groups. Macromol. Chem. Phys. 2019, 220, 1900141. [Google Scholar] [CrossRef]
- Boulkadid, M.K.; Touidjine, S.; Trache, D.; Belkhiri, S. Analytical methods for the assessment of curing kinetics of polyurethane binders for high energy composites. Crit. Rev. Anal. Chem. 2022, 52, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Tarchoun, A.F.; Trache, D.; Klapötke, T.M.; Chelouche, S.; Derradji, M.; Bessa, W.; Mezroua, A. A promising energetic polymer from Posidonia oceanica brown alge: Synthesis, characterization, and kinetic modeling. Macromol. Chem. Phys. 2019, 220, 1900358. [Google Scholar]
- Liqing, L.; Donghua, C. Application of iso-temperature method of multiple rate to kinetic analysis. Dehydration for calcium oxalate monohydrate. J. Therm. Anal. Calorim. 2004, 78, 283–293. [Google Scholar] [CrossRef]
- Lim, A.C.R.; Chin, B.L.F.; Jawad, Z.A.; Hii, K.L. Kinetic analysis of rice husk pyrolysis using Kissinger-Akahira-Sunose (KAS) method. Procedia Eng. 2016, 148, 1247–1251. [Google Scholar] [CrossRef]
- Hayoune, F.; Chelouche, S.; Trache, D.; Zitouni, S.; Grohens, Y. Thermal decomposition kinetics and lifetime prediction of a PP/PLA blend supplemented with iron stearate during artificial aging. Thermochim. Acta 2020, 690, 178700. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Mn a | Mw a | Đa | Yield (%) |
|---|---|---|---|---|
| 15 | 3169 | 3607 | 1.14 | 10.0 |
| 30 | 4328 | 5836 | 1.35 | 39.2 |
| 45 | 4830 | 7012 | 1.45 | 41.6 |
| 60 | 5336 | 8096 | 1.52 | 42.6 |
| Feed Molar Ratio ALIC/DDIC | Yield (%) | Molar Ratio ALIC/DDIC a | Mw b | Đ b |
|---|---|---|---|---|
| 80/20 | 34.0 | 90/10 | 19800 | 1.06 |
| 60/40 | 35.3 | 73/27 | 17600 | 1.07 |
| 50/50 | 30.5 | 63/37 | 16800 | 1.06 |
| 40/60 | 38.5 | 51/49 | 21600 | 1.09 |
| 20/80 | 42.7 | 23/77 | 26100 | 1.08 |
| Method | rALIC | rDDIC |
|---|---|---|
| F-R | 2.63 | 1.14 |
| IF-R | 2.60 | 1.13 |
| K-T | 2.60 | 1.12 |
| ext K-T | 3.04 | 1.08 |
| COPOINT | 2.64 | 1.14 |
| Feed Molar Ratio ALIC/DDIC | Molar Ratio ALIC/DDIC a | Yield (%) | Mw b | Đ b |
|---|---|---|---|---|
| 80/20 | 76/24 | 42.8 | 9200 | 1.11 |
| 50/50 | 44/56 | 38.0 | 11100 | 1.17 |
| 20/80 | 8/91 | 41.7 | 8300 | 1.13 |
| Weight Loss (a) | PALIC | PDDIC | 20/80 | 40/60 | 50/50 | 60/40 | 80/20 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | |
| 0.1 | 94.56 | 87.26 | 162.04 | 153.24 | 136.95 | 129.14 | 117.67 | 110.11 | 111.94 | 104.46 | 143.26 | 135.95 | 122.32 | 115.18 |
| 0.2 | 102.13 | 94.65 | 135.12 | 125.65 | 113.51 | 105.12 | 111.02 | 103.13 | 107.78 | 100.05 | 128.22 | 120.66 | 122.49 | 115.26 |
| 0.3 | 110.94 | 103.29 | 121.49 | 111.77 | 109.03 | 100.30 | 96.15 | 87.84 | 100.63 | 95.65 | 124.15 | 116.42 | 122.66 | 115.26 |
| 0.4 | 120.49 | 112.77 | 116.42 | 106.45 | 112.85 | 103.71 | 93.16 | 84.60 | 88.34 | 80.12 | 119.33 | 111.44 | 121.24 | 113.85 |
| 0.5 | 130.55 | 122.82 | 113.76 | 103.71 | 111.69 | 102.30 | 96.89 | 88.00 | 84.76 | 76.26 | 104.71 | 96.64 | 118.33 | 110.77 |
| 0.6 | 145.43 | 137.53 | 112.27 | 102.05 | 110.77 | 101.22 | 99.97 | 90.83 | 88.50 | 79.77 | 96.31 | 88.09 | 113.60 | 106.04 |
| 0.7 | 110.86 | 100.55 | 111.44 | 101.71 | 99.72 | 90.41 | 92.99 | 83.931 | 95.07 | 86.59 | 107.03 | 99.30 | ||
| 0.8 | 109.28 | 98.89 | 112.68 | 102.71 | 100.14 | 90.58 | 92.90 | 83.60 | 99.39 | 90.66 | 100.14 | 92.24 | ||
| 0.9 | 106.20 | 95.81 | 111.69 | 101.63 | 101.88 | 92.07 | 95.81 | 86.09 | 105.54 | 96.40 | 84.60 | 76.44 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iatrou, M.; Katara, A.; Klonos, P.A.; Kyritsis, A.; Pitsikalis, M. Statistical and Block Copolymers of n-Dodecyl and Allyl Isocyanate via Titanium-Mediated Coordination Polymerization: A Route to Polyisocyanates with Improved Thermal Stability. Polymers 2024, 16, 3537. https://doi.org/10.3390/polym16243537
Iatrou M, Katara A, Klonos PA, Kyritsis A, Pitsikalis M. Statistical and Block Copolymers of n-Dodecyl and Allyl Isocyanate via Titanium-Mediated Coordination Polymerization: A Route to Polyisocyanates with Improved Thermal Stability. Polymers. 2024; 16(24):3537. https://doi.org/10.3390/polym16243537
Chicago/Turabian StyleIatrou, Maria, Aikaterini Katara, Panagiotis A. Klonos, Apostolos Kyritsis, and Marinos Pitsikalis. 2024. "Statistical and Block Copolymers of n-Dodecyl and Allyl Isocyanate via Titanium-Mediated Coordination Polymerization: A Route to Polyisocyanates with Improved Thermal Stability" Polymers 16, no. 24: 3537. https://doi.org/10.3390/polym16243537
APA StyleIatrou, M., Katara, A., Klonos, P. A., Kyritsis, A., & Pitsikalis, M. (2024). Statistical and Block Copolymers of n-Dodecyl and Allyl Isocyanate via Titanium-Mediated Coordination Polymerization: A Route to Polyisocyanates with Improved Thermal Stability. Polymers, 16(24), 3537. https://doi.org/10.3390/polym16243537