
Toward the Rational Design of Organic Catalysts for Organocatalysed Atom Transfer Radical Polymerisation


Abstract
1. Introduction
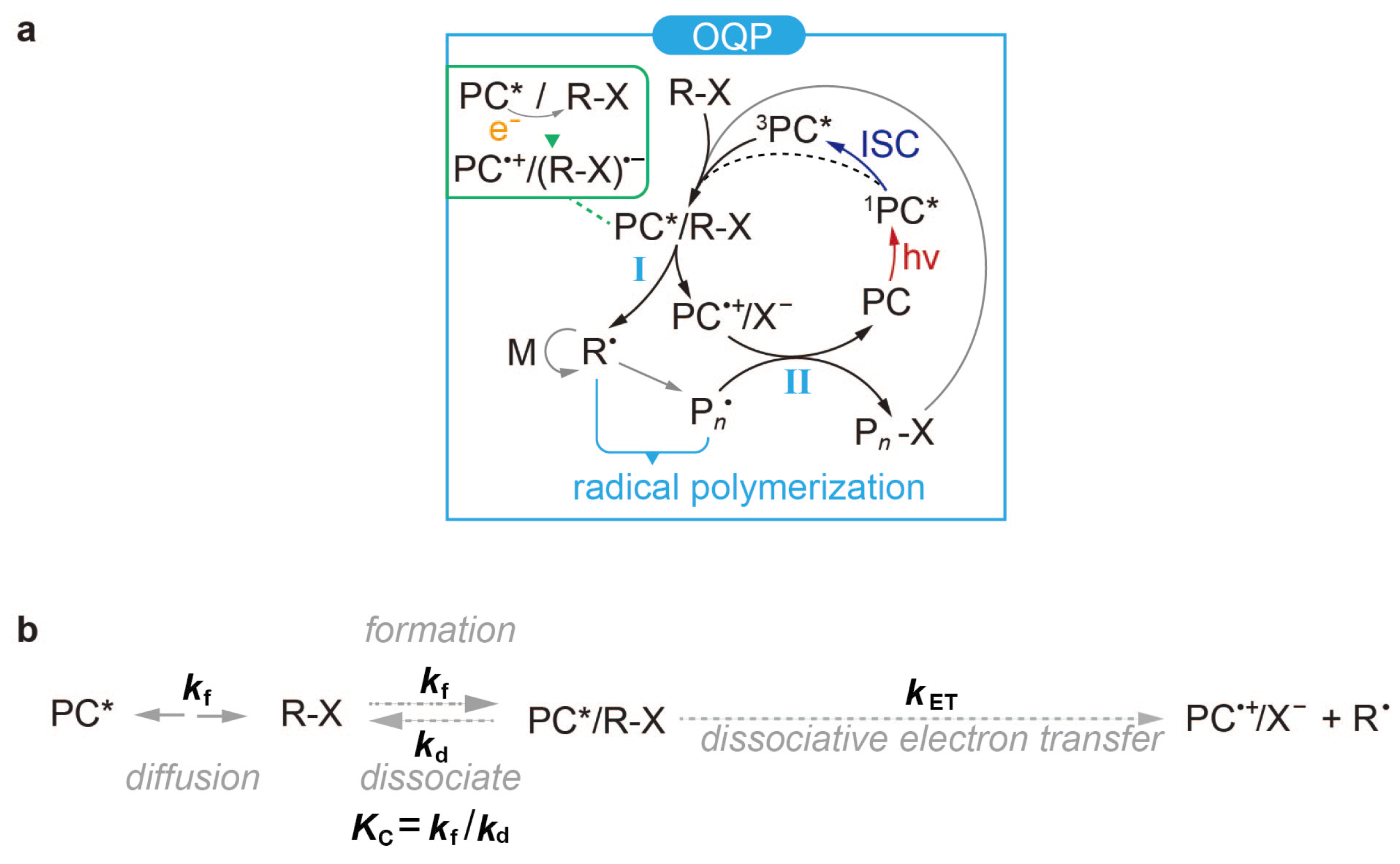
2. Theoretical Background
3. Methods
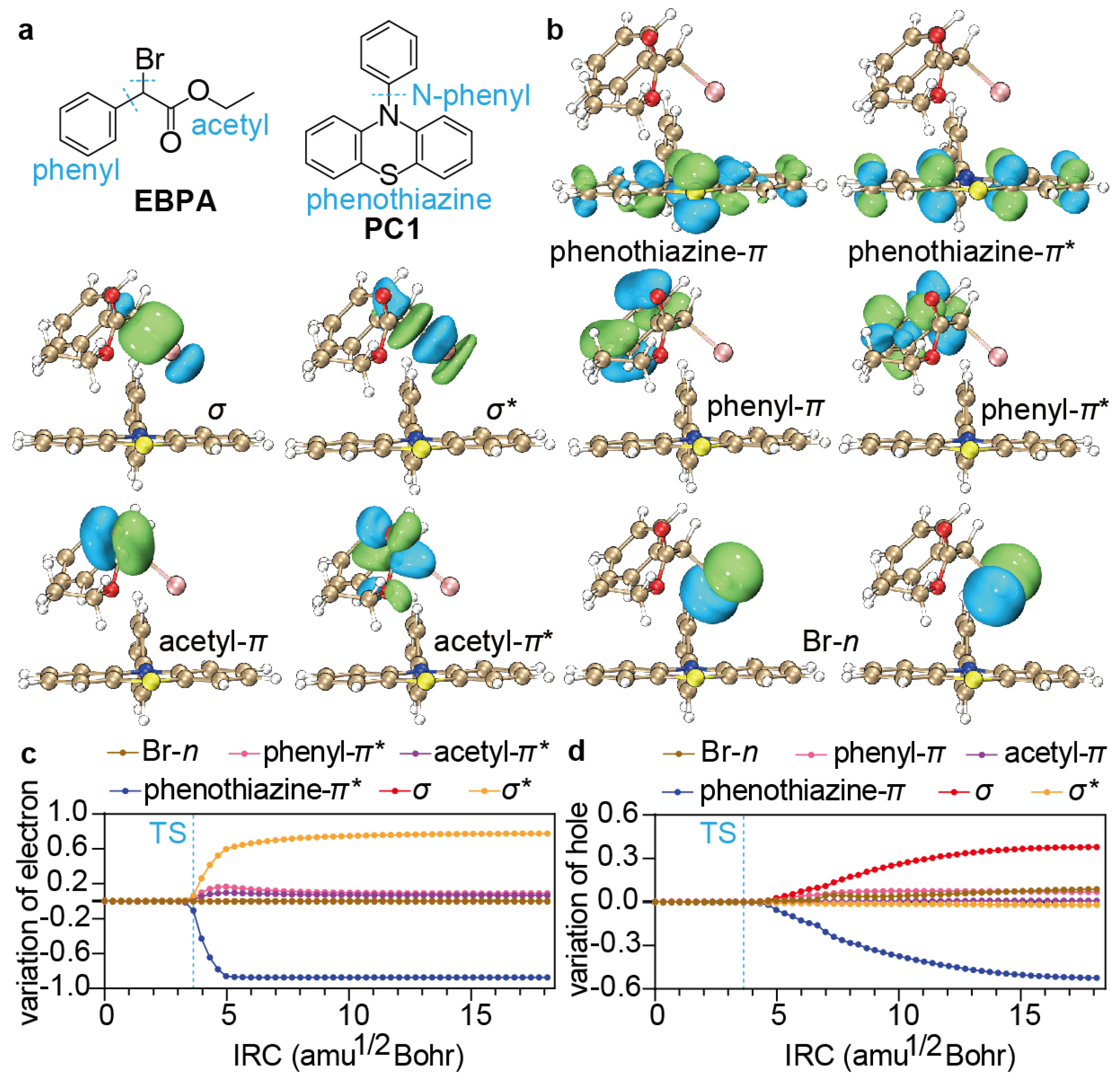
3.1. Reaction Path Analysis
3.2. Determination of Descriptors
3.3. Visualisation
4. Results and Discussion
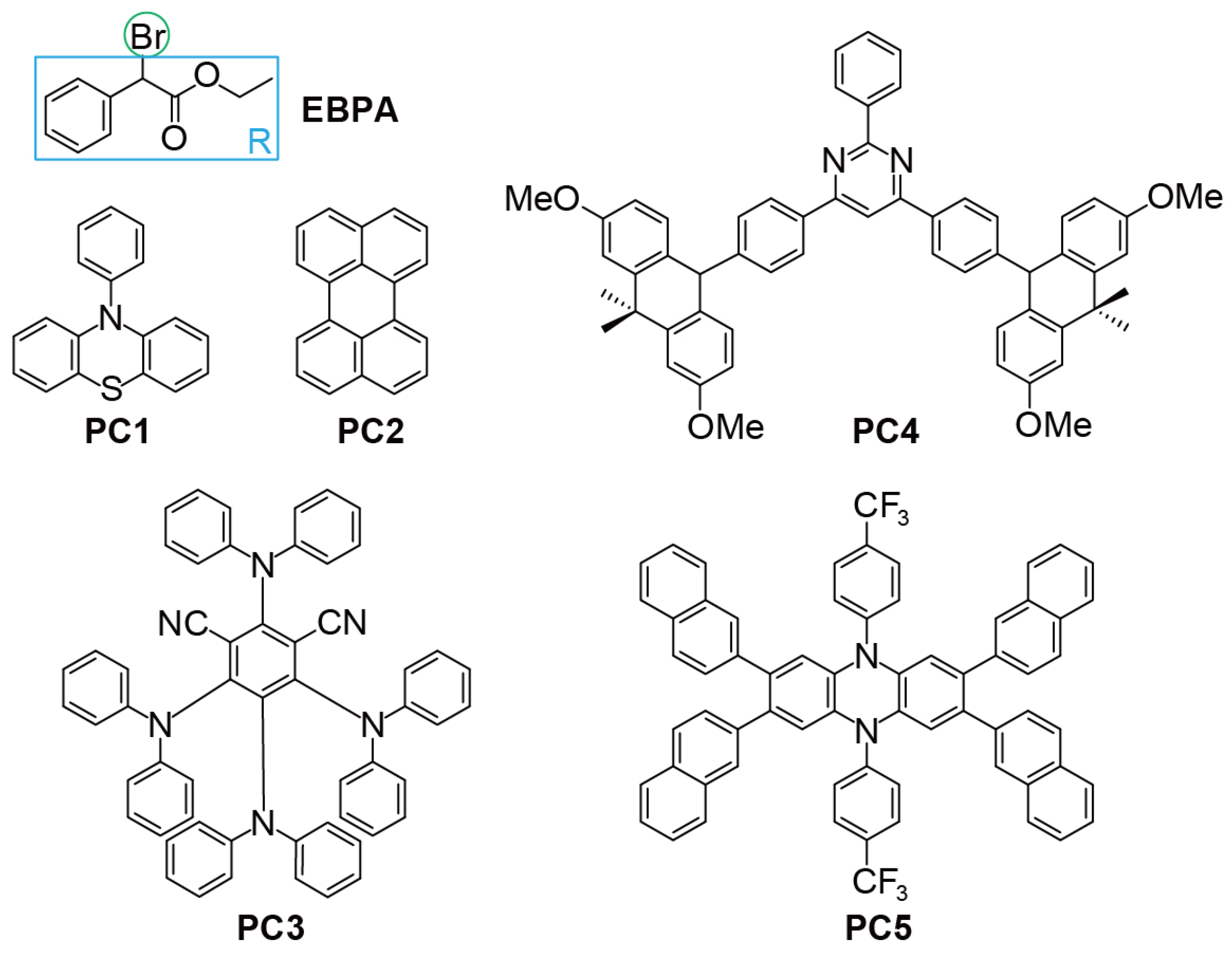
4.1. Model Systems
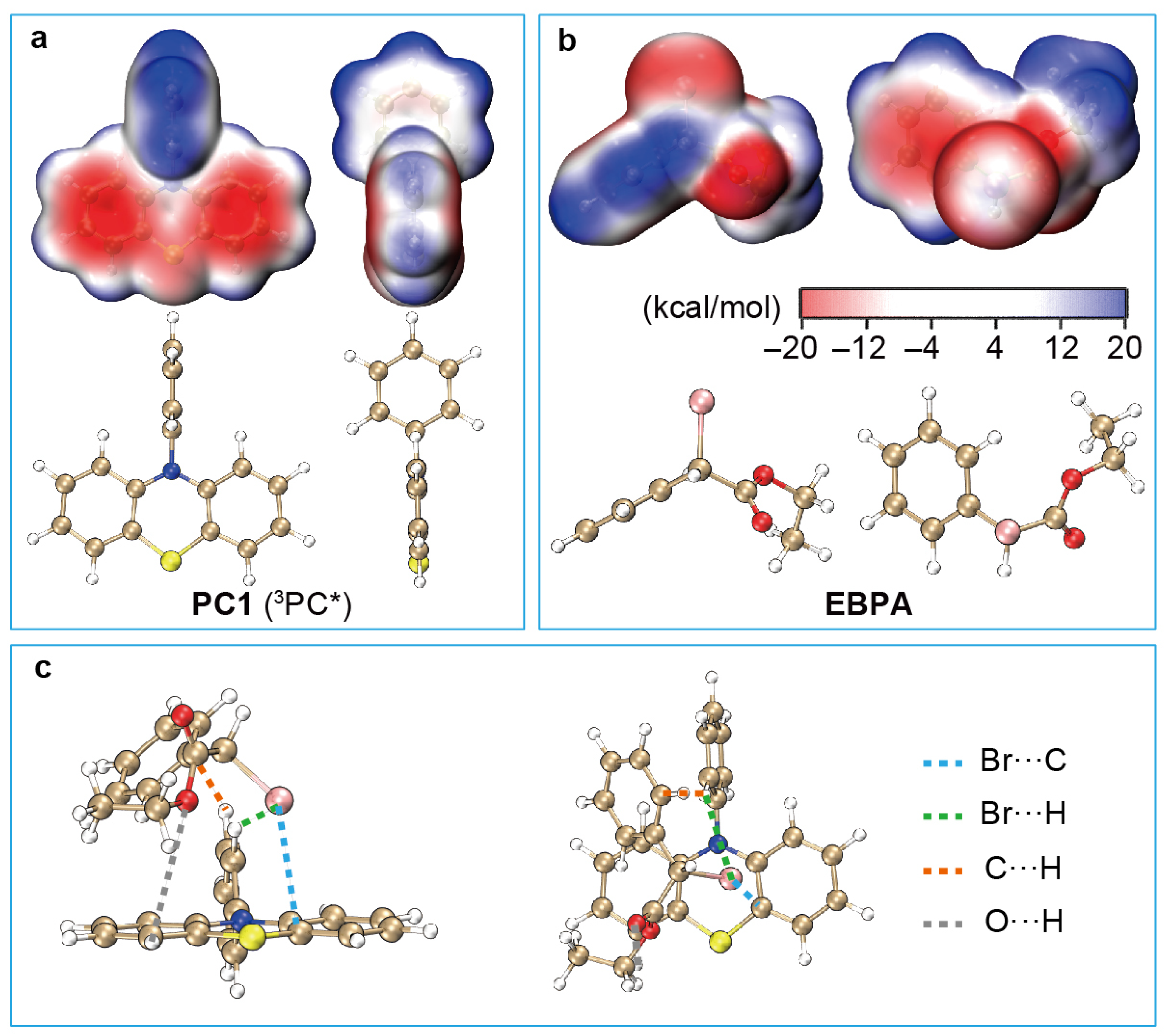
4.2. Formation of Encounter Exciplex
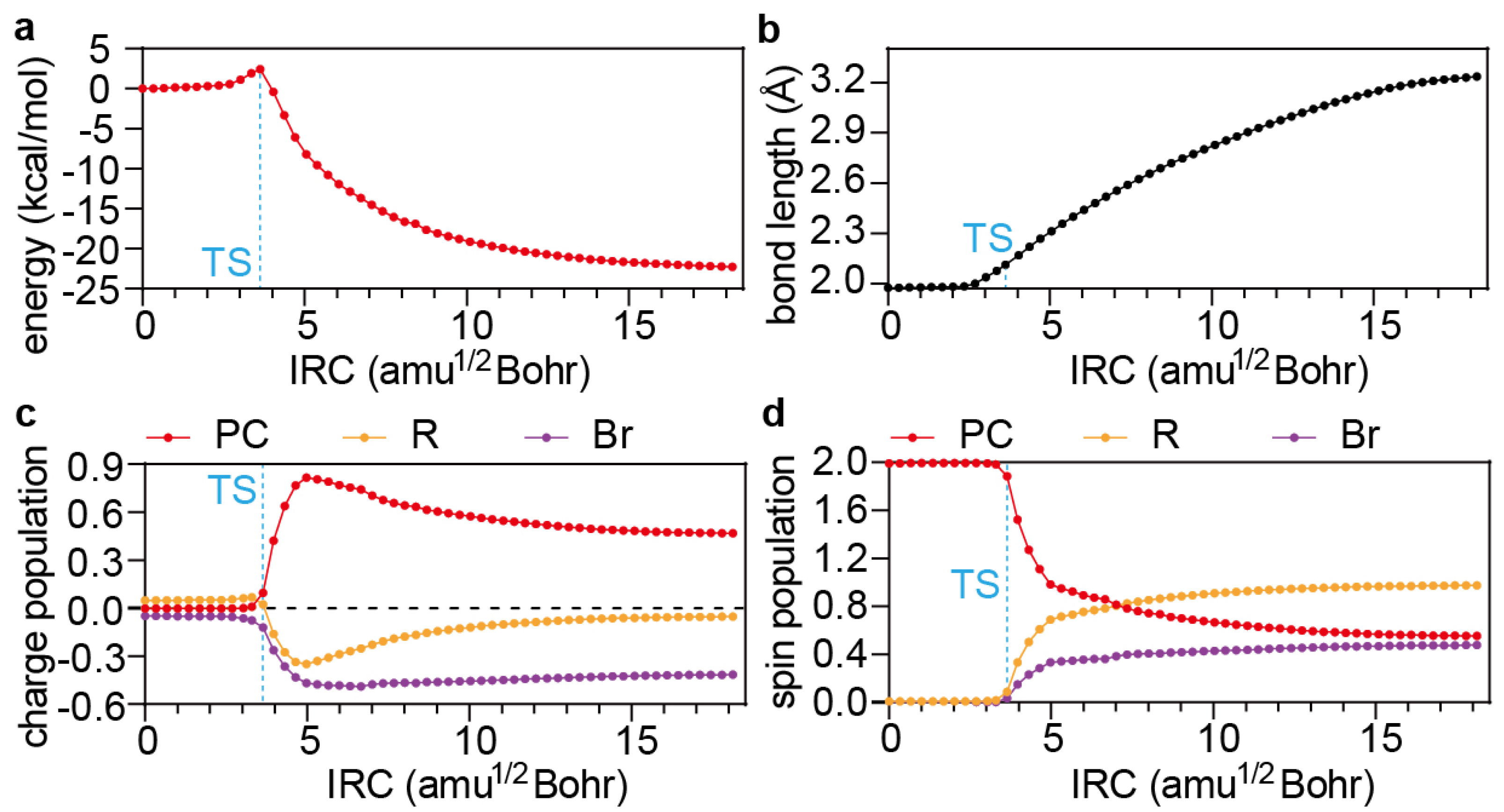
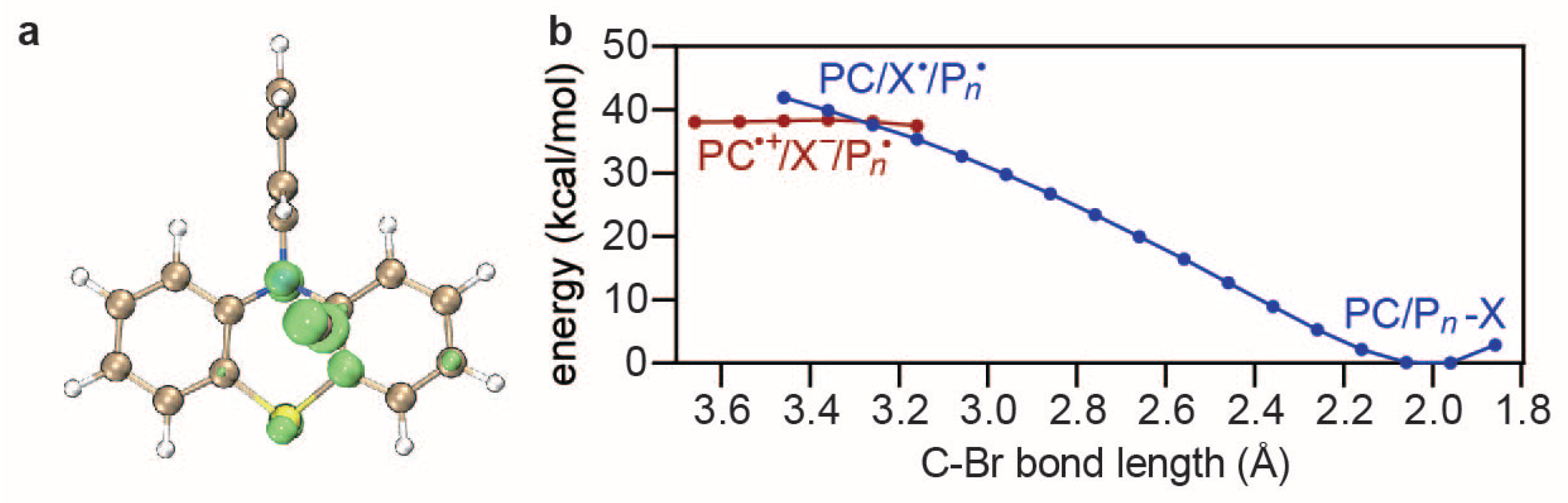
4.3. Dissociative Electron Transfer
4.4. Deactivation and Side Reactions
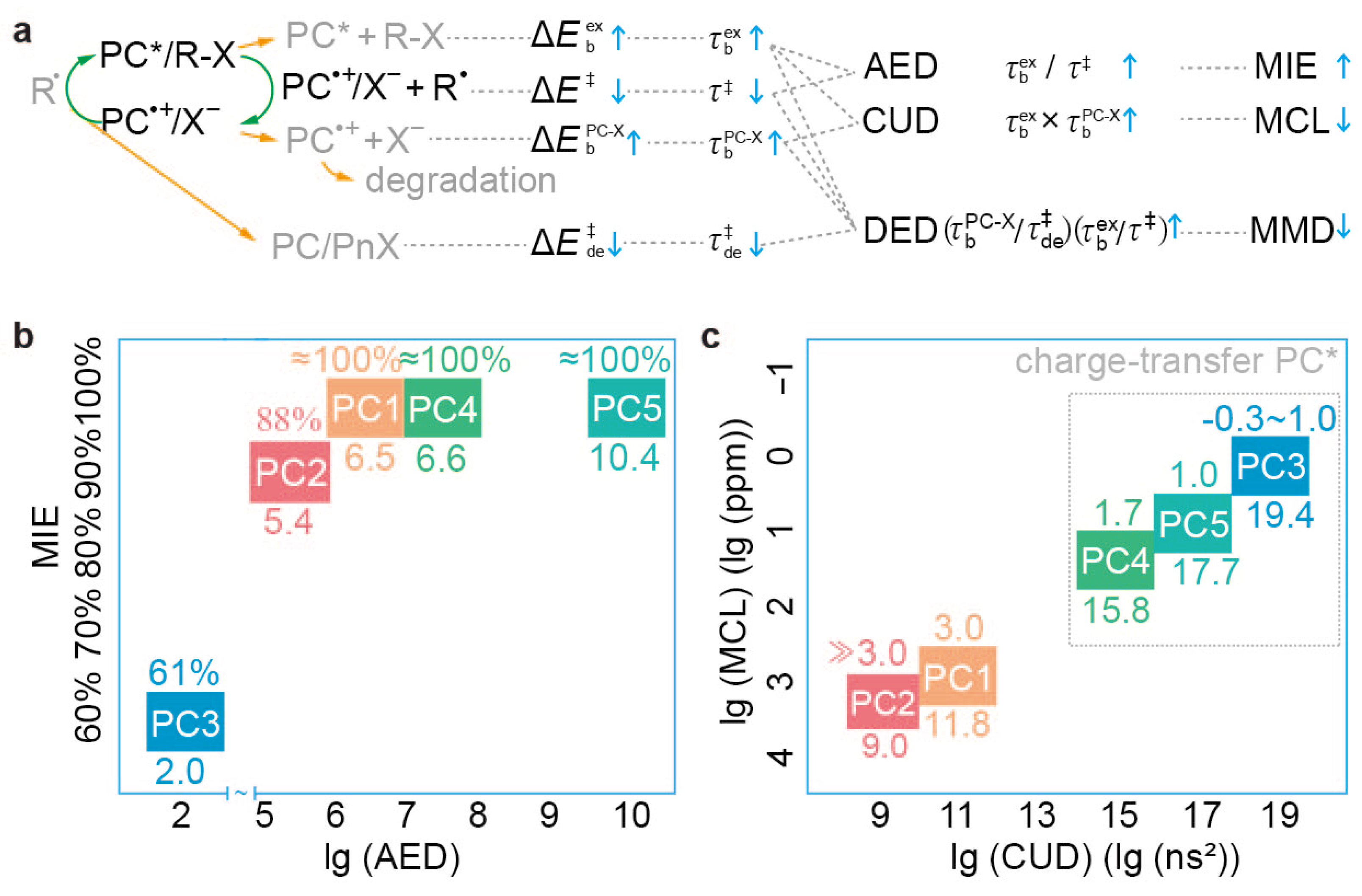
4.5. Validity of New Descriptors
- Interaction between 3PC* and R-X: PC5 features a sizable conjugated chromophore core with six conjugated ligands attached in a nearly orthogonal configuration. The attached aryl ligands, being electron-donating compared to the electron-withdrawing dihydrophenazine chromophore core, result in low-lying charge-transfer states, particularly 3PC*, facilitating its interaction with R-X and thereby enhancing . Additionally, the orthogonal arrangement exposes the aryl ligands’ peripheral hydrogen binding sites to both sides of the chromophore plane, allowing R-X to leverage these sites, further boosting .
- Electron affinity balance: The electron affinities and the disparity between the chromophore core and ligands are important for overall analysis. PC5 achieves a delicate balance, ensuring that while constructing low-lying charge-transfer states is feasible, the overall electron affinity is neither excessively strong nor weak. This balance allows 3PC* to donate an electron to R-X, facilitating a low for dissociative electron transfer. Simultaneously, PC·+ maintains sufficient oxidation, ensuring a low enough for the deactivation reaction. If the chromophore core and ligands are too electron-withdrawing or electron-donating, inefficient activation or deactivation occurs, leading to poor polymerisation control.
- Br atom placement in PC·+/X−: Similar to PC3, PC·+/X− in PC5 positions the Br atom within an “H-nest” formed by peripheral H atoms of orthogonal ligands attached to the chromophore core. This configuration results in a high , maximising the stabilisation of PC·+/X− and minimising the loss of reactive PC species within the catalytic cycle. Consequently, this design reduces catalyst loading, contributing to effective polymerisation control.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PC | Photocatalyst |
| O-ATRP | Organocatalysed atom transfer radical polymerisation |
| OQP | Oxidative quenching pathway |
| R-X | Initiator |
| Initiator efficiency | |
| Đ | Molecular weight dispersity |
| PC* | Excited-state PC |
| 1,3PC* | First singlet/triplet excited-state PC |
| S1/T1 | First singlet/triplet state |
| Singlet/triplet quantum yield | |
| Lifetime of first singlet/triplet excited state of PC | |
| Overall rate constant for activation | |
| Rate constant for the diffusion-controlled encounter of PC* and R-X to form an exciplex | |
| Rate constant for the dissociation of the exciplex to PC* and R-X | |
| Formation/dissociation equilibrium constant for the exciplex | |
| Rate constant for dissociative electron transfer in the exciplex | |
| T | Temperature |
| Viscosity of the solvent | |
| Boltzmann constant | |
| h | Planck constant |
| Gibbs free energy barrier for activation | |
| Electronic energy part of | |
| Gibbs free energy change of electron transfer for activation | |
| Sum of the solvent reorganisation energy and the energy required to break the R-X bond | |
| Binding strength between PC* and R-X | |
| Electronic energy barrier for deactivation | |
| Binding strength between PC·+ and X− | |
| EBPA | Ethyl--bromophenylacetate |
| TDDFT | Time-dependent density functional theory |
| LOSC | Localised orbital scaling correction |
| SMD | Density-based implicit solvation model |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| Orbital wavefunction of HOMO/LUMO | |
| IRC | Intrinsic reaction coordinate |
| FLMO | Fragment-localised molecular orbital |
| Lifetime representation for | |
| Lifetime representation for | |
| Lifetime representation for | |
| Lifetime representation for | |
| MIE | Maximum initiator efficiency |
| AED | Activation efficiency descriptor |
| MCL | Minimum catalyst loading |
| CUD | Catalyst utilisation descriptor |
| MMD | Minimum molecular weight dispersity |
| DED | Deactivation efficiency descriptor |
References
- Abd-El-Aziz, A.S.; Antonietti, M.; Barner-Kowollik, C.; Binder, W.H.; Böker, A.; Boyer, C.; Buchmeiser, M.R.; Cheng, S.Z.D.; D’Agosto, F.; Floudas, G.; et al. The Next 100 Years of Polymer Science. Macromol. Chem. Phys. 2020, 221, 2000216. [Google Scholar] [CrossRef]
- Whitfield, R.; Truong, N.P.; Messmer, D.; Parkatzidis, K.; Rolland, M.; Anastasaki, A. Tailoring Polymer Dispersity and Shape of Molecular Weight Distributions: Methods and Applications. Chem. Sci. 2019, 10, 8724–8734. [Google Scholar] [CrossRef] [PubMed]
- Gentekos, D.T.; Sifri, R.J.; Fors, B.P. Controlling Polymer Properties through the Shape of the Molecular-Weight Distribution. Nat. Rev. Mater. 2019, 4, 761–774. [Google Scholar] [CrossRef]
- Meier, M.A.R.; Barner-Kowollik, C. A New Class of Materials: Sequence-Defined Macromolecules and Their Emerging Applications. Adv. Mater. 2019, 31, 1806027. [Google Scholar] [CrossRef]
- De Neve, J.; Haven, J.J.; Maes, L.; Junkers, T. Sequence-Definition from Controlled Polymerization: The next Generation of Materials. Polym. Chem. 2018, 9, 4692–4705. [Google Scholar] [CrossRef]
- Hawker, C.J. Architectural Control in “Living” Free Radical Polymerizations: Preparation of Star and Graft Polymers. Angew. Chem. Int. Ed. 1995, 34, 1456–1459. [Google Scholar] [CrossRef]
- Barner, L.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Complex Macromolecular Architectures by Reversible Addition Fragmentation Chain Transfer Chemistry: Theory and Practice. Macromol. Rapid Commun. 2007, 28, 539–559. [Google Scholar] [CrossRef]
- Aydogan, C.; Yilmaz, G.; Shegiwal, A.; Haddleton, D.M.; Yagci, Y. Photoinduced Controlled/Living Polymerizations. Angew. Chem. Int. Ed. 2022, 61, e202117377. [Google Scholar] [CrossRef]
- Pan, X.; Tasdelen, M.A.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated Controlled Radical Polymerization. Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar] [CrossRef]
- Leibfarth, F.A.; Mattson, K.M.; Fors, B.P.; Collins, H.A.; Hawker, C.J. External Regulation of Controlled Polymerizations. Angew. Chem. Int. Ed. 2013, 52, 199–210. [Google Scholar] [CrossRef]
- Jung, K.; Corrigan, N.; Ciftci, M.; Xu, J.; Seo, S.E.; Hawker, C.J.; Boyer, C. Designing with Light: Advanced 2D, 3D, and 4D Materials. Adv. Mater. 2020, 32, 1903850. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, C.; Sakakibara, K.; Tsujii, Y.; Goto, A. Facile Fabrication of Concentrated Polymer Brushes with Complex Patterning by Photocontrolled Organocatalyzed Living Radical Polymerization. Angew. Chem. Int. Ed. 2018, 57, 13504–13508. [Google Scholar] [CrossRef] [PubMed]
- Pester, C.W.; Narupai, B.; Mattson, K.M.; Bothman, D.P.; Klinger, D.; Lee, K.W.; Discekici, E.H.; Hawker, C.J. Engineering Surfaces through Sequential Stop-Flow Photopatterning. Adv. Mat. 2016, 28, 9292–9300. [Google Scholar] [CrossRef] [PubMed]
- Fors, B.P.; Poelma, J.E.; Menyo, M.S.; Robb, M.J.; Spokoyny, D.M.; Kramer, J.W.; Waite, J.H.; Hawker, C.J. Fabrication of Unique Chemical Patterns and Concentration Gradients with Visible Light. J. Am. Chem. Soc. 2013, 135, 14106–14109. [Google Scholar] [CrossRef] [PubMed]
- Buss, B.L.; Miyake, G.M. Photoinduced Controlled Radical Polymerizations Performed in Flow: Methods, Products, and Opportunities. Chem. Mater. 2018, 30, 3931–3942. [Google Scholar] [CrossRef] [PubMed]
- Zaquen, N.; Rubens, M.; Corrigan, N.; Xu, J.; Zetterlund, P.B.; Boyer, C.; Junkers, T. Polymer Synthesis in Continuous Flow Reactors. Prog. Polym. Sci. 2020, 107, 101256. [Google Scholar] [CrossRef]
- Wu, C.; Corrigan, N.; Lim, C.H.; Liu, W.; Miyake, G.; Boyer, C. Rational Design of Photocatalysts for Controlled Polymerization: Effect of Structures on Photocatalytic Activities. Chem. Rev. 2022, 122, 5476–5518. [Google Scholar] [CrossRef] [PubMed]
- Corbin, D.A.; Miyake, G.M. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-ATRP): Precision Polymer Synthesis Using Organic Photoredox Catalysis. Chem. Rev. 2022, 122, 1830–1874. [Google Scholar] [CrossRef]
- Lorandi, F.; Fantin, M.; Matyjaszewski, K. Atom Transfer Radical Polymerization: A Mechanistic Perspective. J. Am. Chem. Soc. 2022, 144, 15413–15430. [Google Scholar] [CrossRef]
- Chen, M.; Zhong, M.; Johnson, J. Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem. Rev. 2016, 116, 10167–10211. [Google Scholar] [CrossRef]
- Saveant, J.M. A Simple Model for the Kinetics of Dissociative Electron Transfer in Polar Solvents. Application to the Homogeneous and Heterogeneous Reduction of Alkyl Halides. J. Am. Chem. Soc. 1987, 109, 6788–6795. [Google Scholar] [CrossRef]
- Discekici, E.H.; Anastasaki, A.; Read de Alaniz, J.; Hawker, C.J. Evolution and Future Directions of Metal-Free Atom Transfer Radical Polymerization. Macromolecules 2018, 51, 7421–7434. [Google Scholar] [CrossRef]
- Wang, J.; Matyjaszewski, K. Controlled/“Living” Radical Polymerization. Atom Transfer Radical Polymerization in the Presence of Transition-Metal Complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.K.K.; Coote, M.L. A Mechanistic Perspective on Atom Transfer Radical Polymerization. Polym. Int. 2021, 70, 918–926. [Google Scholar] [CrossRef]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Barton, B.E.; Read de Alaniz, J.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Yu, C.; Badgujar, S.; Kim, Y.; Kwon, Y.; Kim, D.; Lee, J.; Akhter, T.; Thangavel, G.; Park, L.S.; et al. Highly Efficient Organic Photocatalysts Discovered via a Computer-Aided-Design Strategy for Visible-Light-Driven Atom Transfer Radical Polymerization. Nat. Catal. 2018, 1, 794–804. [Google Scholar] [CrossRef]
- Cole, J.P.; Federico, C.R.; Lim, C.H.; Miyake, G.M. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization Using Low ppm Catalyst Loading. Macromolecules 2019, 52, 747–754. [Google Scholar] [CrossRef]
- Ma, Q.; Song, J.; Zhang, X.; Jiang, Y.; Ji, L.; Liao, S. Metal-free Atom Transfer Radical Polymerization with ppm Catalyst Loading under Sunlight. Nat. Commun. 2021, 12, 429. [Google Scholar] [CrossRef]
- Sartor, S.M.; McCarthy, B.G.; Pearson, R.M.; Miyake, G.M.; Damrauer, N.H. Exploiting Charge-Transfer States for Maximizing Intersystem Crossing Yields in Organic Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 4778–4781. [Google Scholar] [CrossRef]
- Sartor, S.M.; Lattke, Y.M.; McCarthy, B.G.; Miyake, G.M.; Damrauer, N.H. Effects of Naphthyl Connectivity on the Photophysics of Compact Organic Charge-Transfer Photoredox Catalysts. J. Phys. Chem. A 2019, 123, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Sartor, S.M.; Chrisman, C.H.; Pearson, R.M.; Miyake, G.M.; Damrauer, N.H. Designing High-Triplet-Yield Phenothiazine Donor–Acceptor Complexes for Photoredox Catalysis. J. Phys. Chem. A 2020, 124, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Price, M.J.; Puffer, K.O.; Kudisch, M.; Knies, D.; Miyake, G.M. Structure–property Relationships of Core-substituted Diaryl Dihydrophenazine Organic Photoredox Catalysts and Their Application in O-ATRP. Polym. Chem. 2021, 12, 6110–6122. [Google Scholar] [CrossRef]
- Corbin, D.A.; Puffer, K.O.; Chism, K.A.; Cole, J.P.; Theriot, J.C.; McCarthy, B.G.; Buss, B.L.; Lim, C.; Lincoln, S.R.; Newell, B.S.; et al. Radical Addition to N,N-Diaryl Dihydrophenazine Photoredox Catalysts and Implications in Photoinduced Organocatalyzed Atom Transfer Radical Polymerization. Macromolecules 2021, 54, 4507–4516. [Google Scholar] [CrossRef]
- Lattke, Y.M.; Corbin, D.A.; Sartor, S.M.; McCarthy, B.G.; Miyake, G.M.; Damrauer, N.H. Interrogation of O-ATRP Activation Conducted by Singlet and Triplet Excited States of Phenoxazine Photocatalysts. J. Phys. Chem. A 2021, 125, 3109–3121. [Google Scholar] [CrossRef]
- Li, C.; Zheng, X.; Su, N.; Yang, W. Localized Orbital Scaling Correction for Systematic Elimination of Delocalization Error in Density Functional Approximations. Natl. Sci. Rev. 2017, 5, 203–215. [Google Scholar] [CrossRef]
- Mei, Y.; Yu, J.; Chen, Z.; Su, N.; Yang, W. LibSC: Library for Scaling Correction Methods in Density Functional Theory. J. Chem. Theory. Comput. 2022, 18, 840–850. [Google Scholar] [CrossRef]
- Wu, F.; Liu, W.; Zhang, Y.; Li, Z. Linear-Scaling Time-Dependent Density Functional Theory Based on the Idea of “From Fragments to Molecule”. J. Chem. Theory Comput. 2011, 7, 3643–3660. [Google Scholar] [CrossRef]
- Li, H.; Liu, W.; Suo, B. Localization of Open-Shell Molecular Orbitals via Least Change from Fragments to Molecule. J. Chem. Phys. 2017, 146, 104104. [Google Scholar] [CrossRef]
- Koyama, D.; Dale, H.J.A.; Orr-Ewing, A.J. Ultrafast Observation of a Photoredox Reaction Mechanism: Photoinitiation in Organocatalyzed Atom-Transfer Radical Polymerization. J. Am. Chem. Soc. 2018, 140, 1285–1293. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Sneha, M.; Lewis-Borrell, L.; Amoruso, G.; Oliver, T.A.A.; Tyler, J.; Clark, I.P.; Orr-Ewing, A.J. Singlet and Triplet Contributions to the Excited-State Activities of Dihydrophenazine, Phenoxazine, and Phenothiazine Organocatalysts Used in Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2021, 143, 3613–3627. [Google Scholar] [CrossRef]
- Sneha, M.; Bhattacherjee, A.; Lewis-Borrell, L.; Clark, I.P.; Orr-Ewing, A.J. Structure-Dependent Electron Transfer Rates for Dihydrophenazine, Phenoxazine, and Phenothiazine Photoredox Catalysts Employed in Atom Transfer Radical Polymerization. J. Phys. Chem. B 2021, 125, 7840–7854. [Google Scholar] [CrossRef]
- Pearson, R.M.; Lim, C.H.; McCarthy, B.G.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. [Google Scholar] [CrossRef]
- Buss, B.L.; Lim, C.H.; Miyake, G.M. Dimethyl Dihydroacridines as Photocatalysts in Organocatalyzed Atom Transfer Radical Polymerization of Acrylate Monomers. Angew. Chem. Int. Ed. 2020, 59, 3209–3217. [Google Scholar] [CrossRef]
- Rehm, D.; Weller, A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. III. Applications to Data on the Rates of Organic Redox Reactions. J. Chem. Phys. 1957, 26, 872–877. [Google Scholar] [CrossRef]
- Miyake, G.M.; Theriot, J.C. Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules 2014, 47, 8255–8261. [Google Scholar] [CrossRef]
- Di Savino, A.; Foerster, J.M.; La Haye, T.; Blok, A.; Timmer, M.; Ullmann, G.M.; Ubbink, M. Efficient Encounter Complex Formation and Electron Transfer to Cytochrome c Peroxidase with an Additional, Distant Electrostatic Binding Site. Angew. Chem. Int. Ed. 2020, 59, 23239–23243. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.H.; Ryan, M.; McCarthy, B.G.; Theriot, J.C.; Sartor, S.M.; Damrauer, N.H.; Musgrave, C.B.; Miyake, G.M. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalysts for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2017, 139, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Corbin, D.A.; McCarthy, B.G.; van de Lindt, Z.; Miyake, G.M. Radical Cations of Phenoxazine and Dihydrophenazine Photoredox Catalysts and Their Role as Deactivators in Organocatalyzed Atom Transfer Radical Polymerization. Macromolecules 2021, 54, 4726–4738. [Google Scholar] [CrossRef]
- Pan, X.; Fang, C.; Fantin, M.; Malhotra, N.; So, W.Y.; Peteanu, L.A.; Isse, A.A.; Gennaro, A.; Liu, P.; Matyjaszewski, K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 2411–2425. [Google Scholar] [CrossRef]
- Polgar, A.M.; Huang, S.H.; Hudson, Z.M. Donor modification of thermally activated delayed fluorescence photosensitizers for organocatalyzed atom transfer radical polymerization. Polym. Chem. 2022, 13, 3892–3903. [Google Scholar] [CrossRef]
- Liu, W.; Hong, G.; Dai, D.; Li, L.; Dolg, M. The Beijing Four-Component Density Functional Program Package (BDF) and Its Application to EuO, EuS, YbO and YbS. Theoret. Chem. Acc. 1997, 96, 75–83. [Google Scholar] [CrossRef]
- Liu, W.; Wang, F.; Li, L. The Beijing Density Functional (BDF) Program Package: Methodologies and Applications. J. Theoret. Comput. Chem. 2003, 2, 257–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Suo, B.; Wang, Z.; Zhang, N.; Li, Z.; Lei, Y.; Zou, W.; Gao, J.; Peng, D.; Pu, Z.; et al. BDF: A Relativistic Electronic Structure Program Package. J. Chem. Phys. 2020, 152, 064113. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Harabuchi, Y.; Ono, Y.; Taketsugu, T.; Morokuma, K. Intrinsic Reaction Coordinate: Calculation, Bifurcation, and Automated Search. Int. J. Quantum Chem. 2015, 115, 258–269. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wei, Y.; Yuan, Y.; Guo, Q. Charge Transfer in Excited Donor-acceptor Phenothiazine Derivatives. Chin. J. Chem. 2005, 23, 1430–1436. [Google Scholar] [CrossRef]
- Rodrigues, A.C.B.; Pina, J.; Seixas de Melo, J.S. Structure-Relation Properties of N-substituted Phenothiazines in Solution and Solid State: Photophysical, Photostability and Aggregation-Induced Emission Studies. J. Mol. Liq. 2020, 317, 113966. [Google Scholar] [CrossRef]
- Torres, E.; Berberan-Santos, M.N.; Brites, M.J. Synthesis, Photophysical and Electrochemical Properties of Perylene Dyes. Dyes Pigm. 2015, 112, 298–304. [Google Scholar] [CrossRef]
- Song, Y.; Kim, Y.; Noh, Y.; Singh, V.K.; Behera, S.K.; Abudulimu, A.; Chung, K.; Wannemacher, R.; Gierschner, J.; Lüer, L.; et al. Organic Photocatalyst for ppm-Level Visible-Light-Driven Reversible Addition–Fragmentation Chain-Transfer (RAFT) Polymerization with Excellent Oxygen Tolerance. Macromolecules 2019, 52, 5538–5545. [Google Scholar] [CrossRef]
- Li, Z.; Suo, B.; Zhang, Y.; Xiao, Y.; Liu, W. Combining Spin-adapted Open-shell TD-DFT with Spin–Orbit Coupling. Mol. Phys. 2013, 111, 3741–3755. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Zhang, Y.; Liu, W. Analytic Energy Gradients of Spin-Adapted Open-Shell Time-Dependent Density Functional Theory. J. Chem. Phys 2020, 153, 164109. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Suo, B.; Liu, W. Localization of Molecular Orbitals: From Fragments to Molecule. Acc. Chem. Res. 2014, 47, 2758–2767. [Google Scholar] [CrossRef] [PubMed]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymerisation a | Descriptor b | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Catalyst | Loading ppm | c | Đ | kcal/mol | ns | kcal/mol | ns | kcal/mol | ns | kcal/mol | ns |
| 1 | PC1 [26,52] | 1000 | 116% | 1.30 | 11.2 | × | 2.4 | × | 15.7 | × | 0.4 | × |
| 2 | PC2 [48] | 1000 | 45% | 1.39 | 9.7 | × | 2.4 | × | 13.4 | × | 0.0 | × |
| 3a | PC3 [27] | 10 | 61% | 1.30 | 15.0 | × | 12.3 | × | 22.2 | × | 1.7 | × |
| 3b | 0.5 | 53% | 1.39 | |||||||||
| 4 | PC4 [53] | 50 | 91% | 1.29 | 16.3 | × | 7.3 | × | 16.1 | × | 1.9 | × |
| 5a | PC5 [28] | 10 | 92% | 1.27 | 17.5 | × | 3.3 | × | 17.4 | × | 3.9 | × |
| 5b | 50 | 106% | 1.14 | |||||||||
| Catalyst Unit | nm | L/mol·cm |
ns |
ns | ||
|---|---|---|---|---|---|---|
| PC1 e [65,66] | 319 | 4900 | 28% | 3.4 | 72% | |
| PC2 [67] | 440 | 34,000 | ∼100% | 4.0 | ∼0% | – |
| PC3 [27,68] | 468 | 13,900 | 23% | 2.4 | 77% | |
| PC4 [53] | 398 | 3900 | 35% | 18 | 65% | |
| PC5 [28] | 388 | 20,000 | – | – | – | – |
| Catalyst | Solvent | (PC·+/1PC*)/(PC·+/3PC*) | ||
|---|---|---|---|---|
| Experiment | This Work a | This Work ab | ||
| PC1 | cyclohexane | −1.92/−1.70 [52] | −2.17/−1.57 | −2.91/−2.31 |
| PC2 | toluene | −1.87/−0.70 [48] | −1.70/−0.41 | −2.12/−1.05 |
| PC3 | acetonitrile | −1.58/−1.41 [27] | −1.77/−1.58 | −1.77/−1.59 |
| PC4 | chloroform | −1.79/−1.76 [53] | −1.94/−1.94 | −2.22/−2.21 |
| PC5 | toluene | −1.84 [28]/– | −2.03/−1.58 | −2.52/−2.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wu, C.; Liu, W. Toward the Rational Design of Organic Catalysts for Organocatalysed Atom Transfer Radical Polymerisation. Polymers 2024, 16, 323. https://doi.org/10.3390/polym16030323
Wang Z, Wu C, Liu W. Toward the Rational Design of Organic Catalysts for Organocatalysed Atom Transfer Radical Polymerisation. Polymers. 2024; 16(3):323. https://doi.org/10.3390/polym16030323
Chicago/Turabian StyleWang, Zhilei, Chenyu Wu, and Wenjian Liu. 2024. "Toward the Rational Design of Organic Catalysts for Organocatalysed Atom Transfer Radical Polymerisation" Polymers 16, no. 3: 323. https://doi.org/10.3390/polym16030323
APA StyleWang, Z., Wu, C., & Liu, W. (2024). Toward the Rational Design of Organic Catalysts for Organocatalysed Atom Transfer Radical Polymerisation. Polymers, 16(3), 323. https://doi.org/10.3390/polym16030323