

Physicochemical Characterization and In Vitro Activity of Poly(ε-Caprolactone)/Mycophenolic Acid Amorphous Solid Dispersions

 ,
,  ,
,  and
and 
Abstract
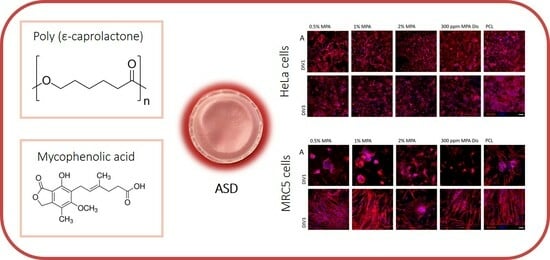
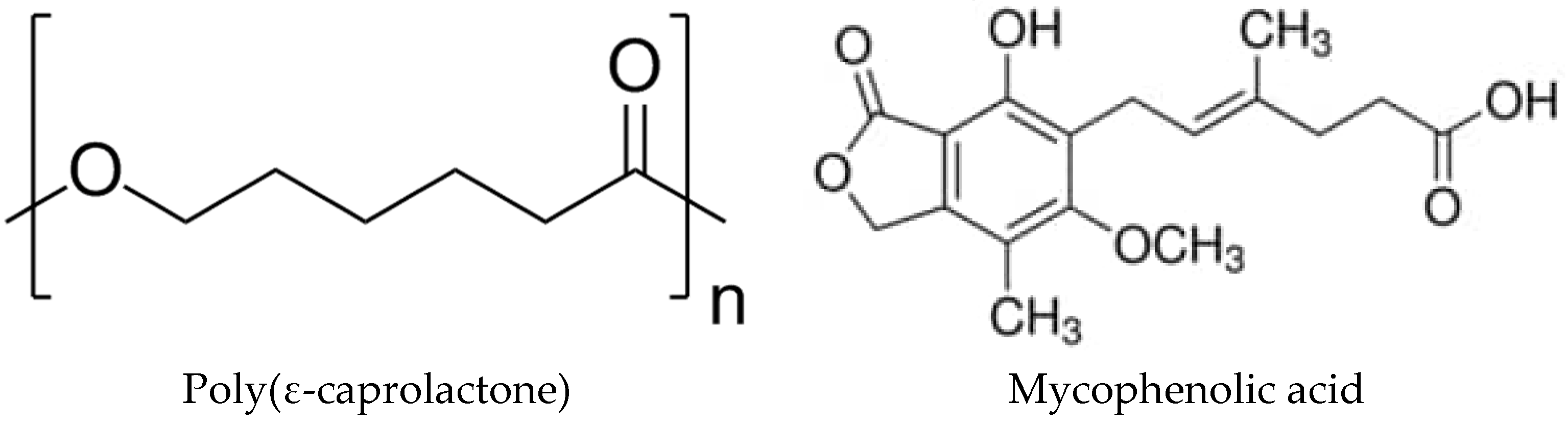
1. Introduction
2. Experimental Section
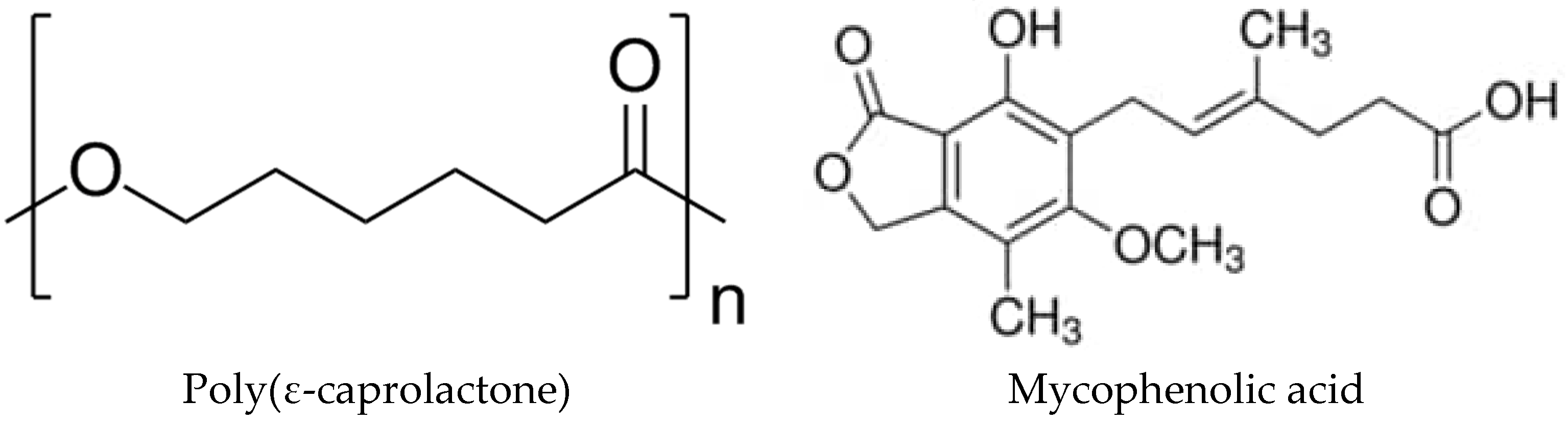
2.1. Starting Materials
2.2. Blend Preparation
2.3. Differential Scanning Calorimetry (DSC)
2.4. Melting Point Depression Analysis
2.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.6. In Vitro Drug Release
2.7. In Vitro Cell Culture Experiments
2.8. Immunostaining
2.9. Cell Count and Statistical Analysis
3. Results and Discussion
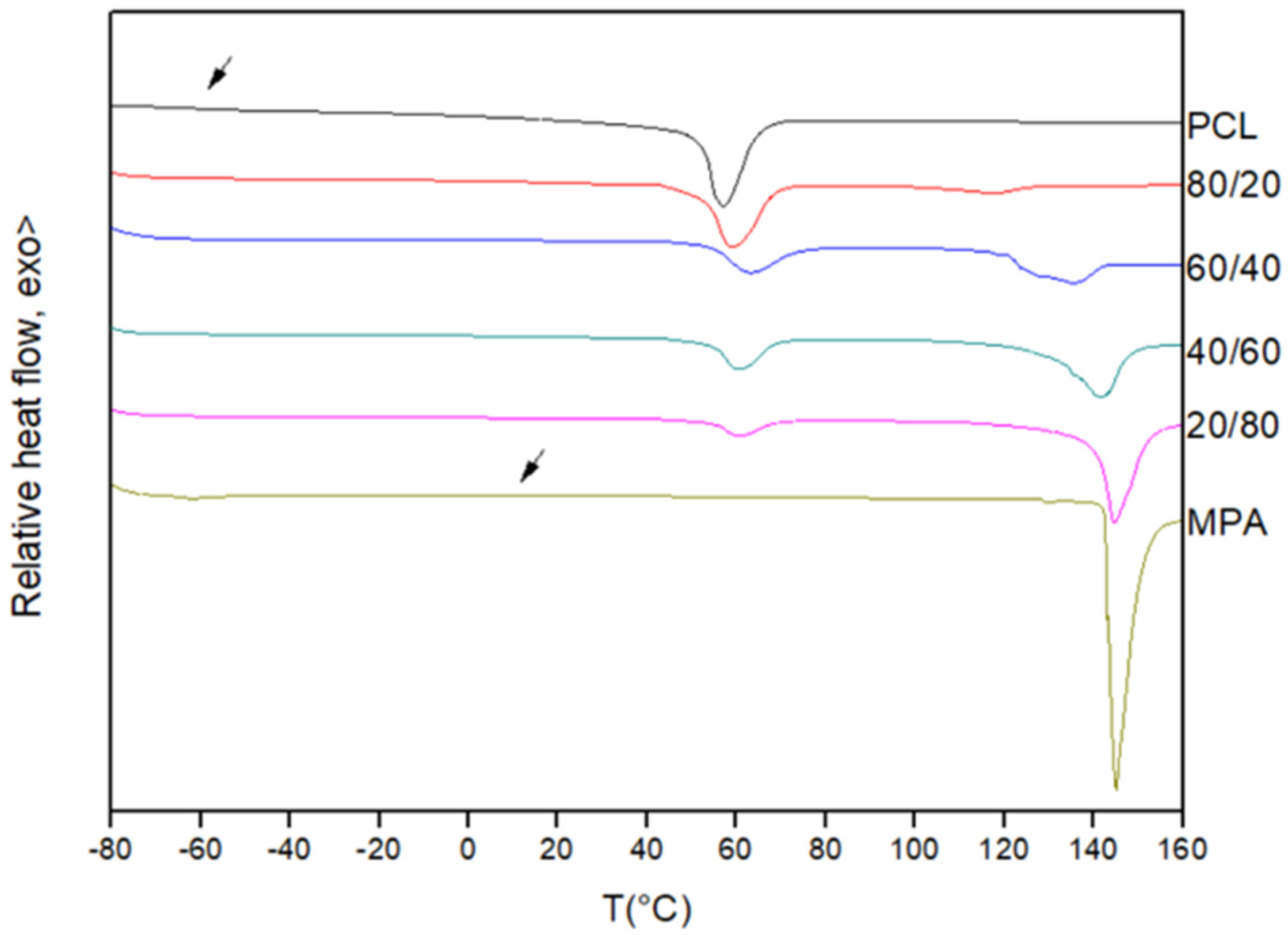
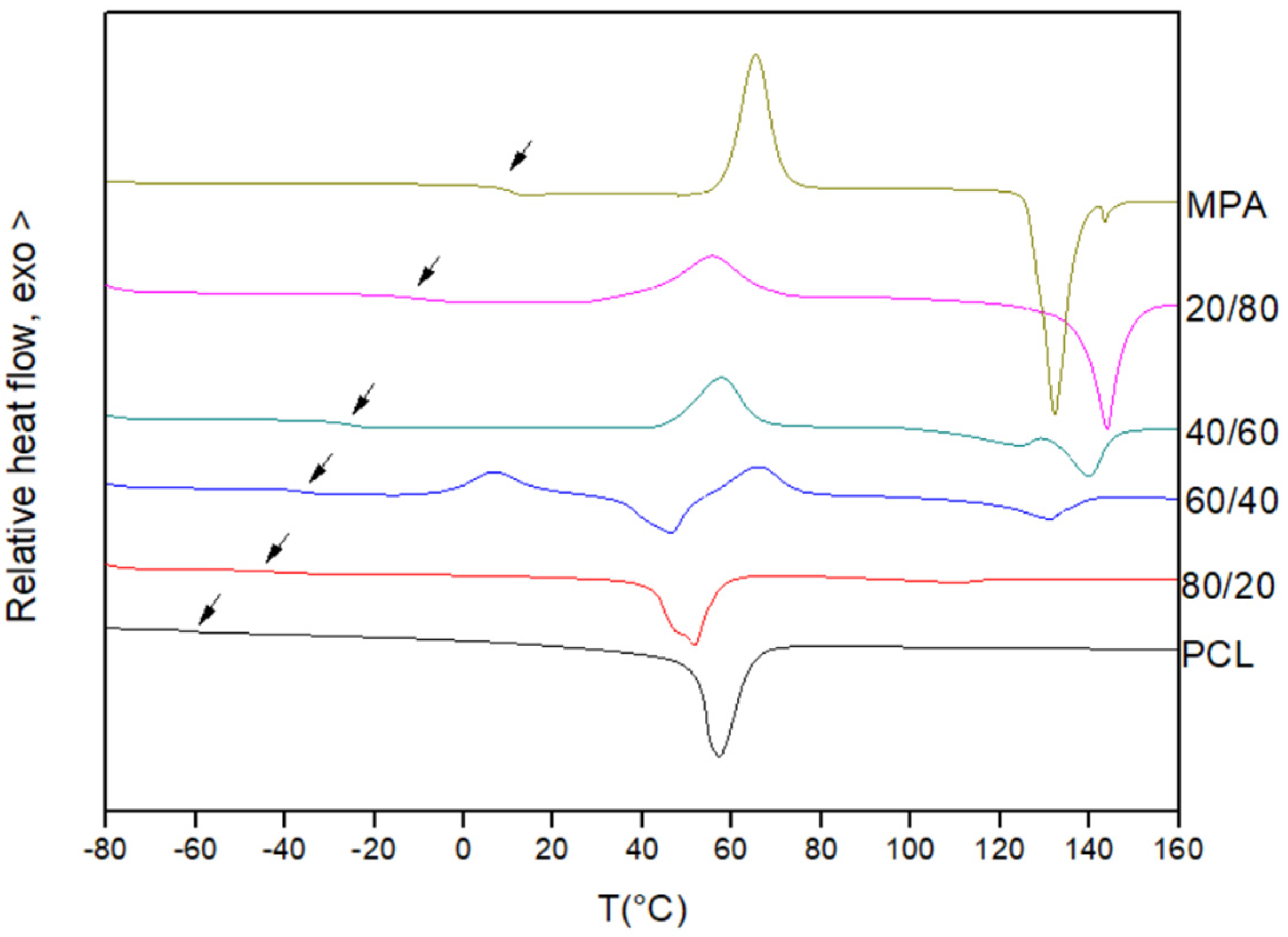
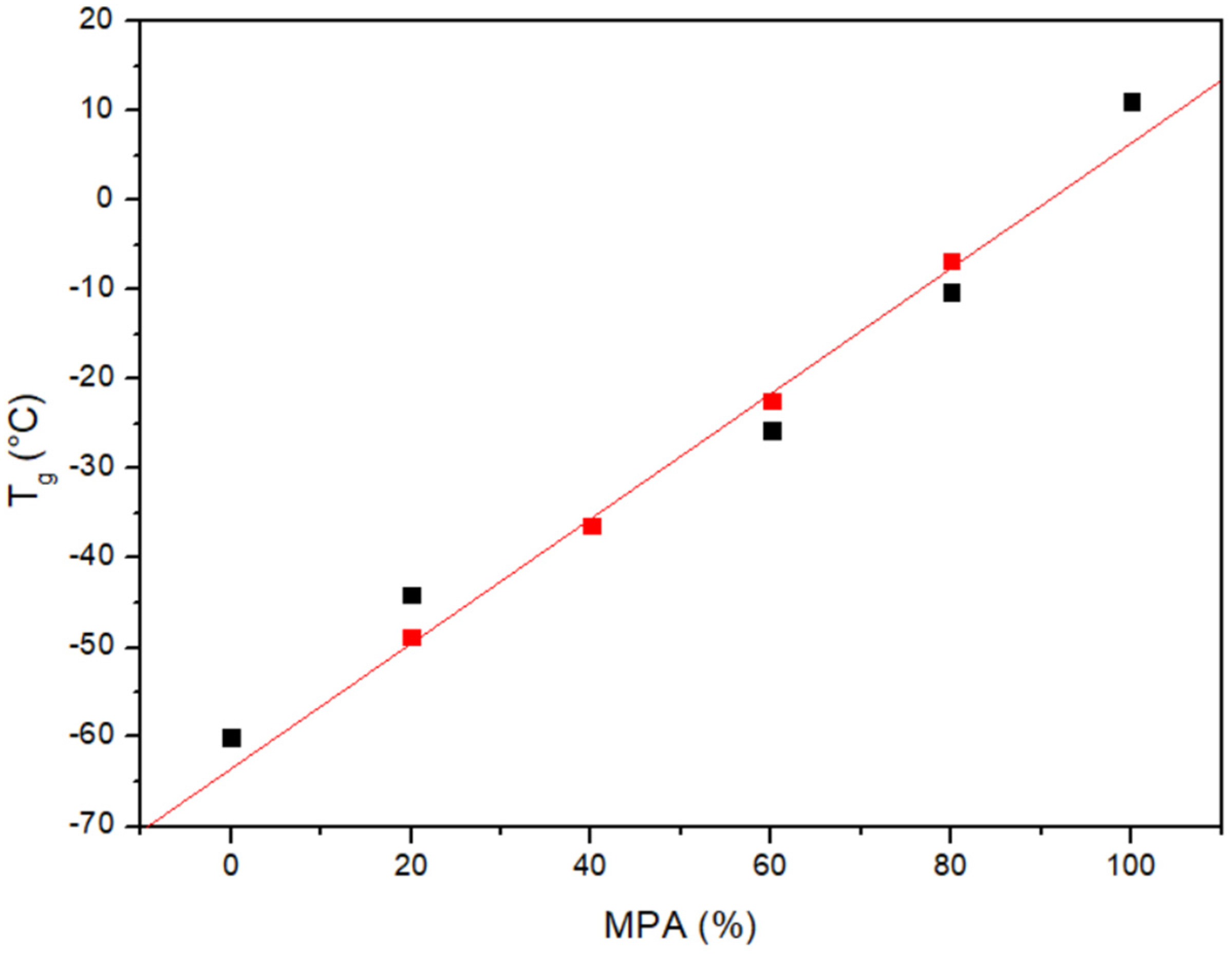
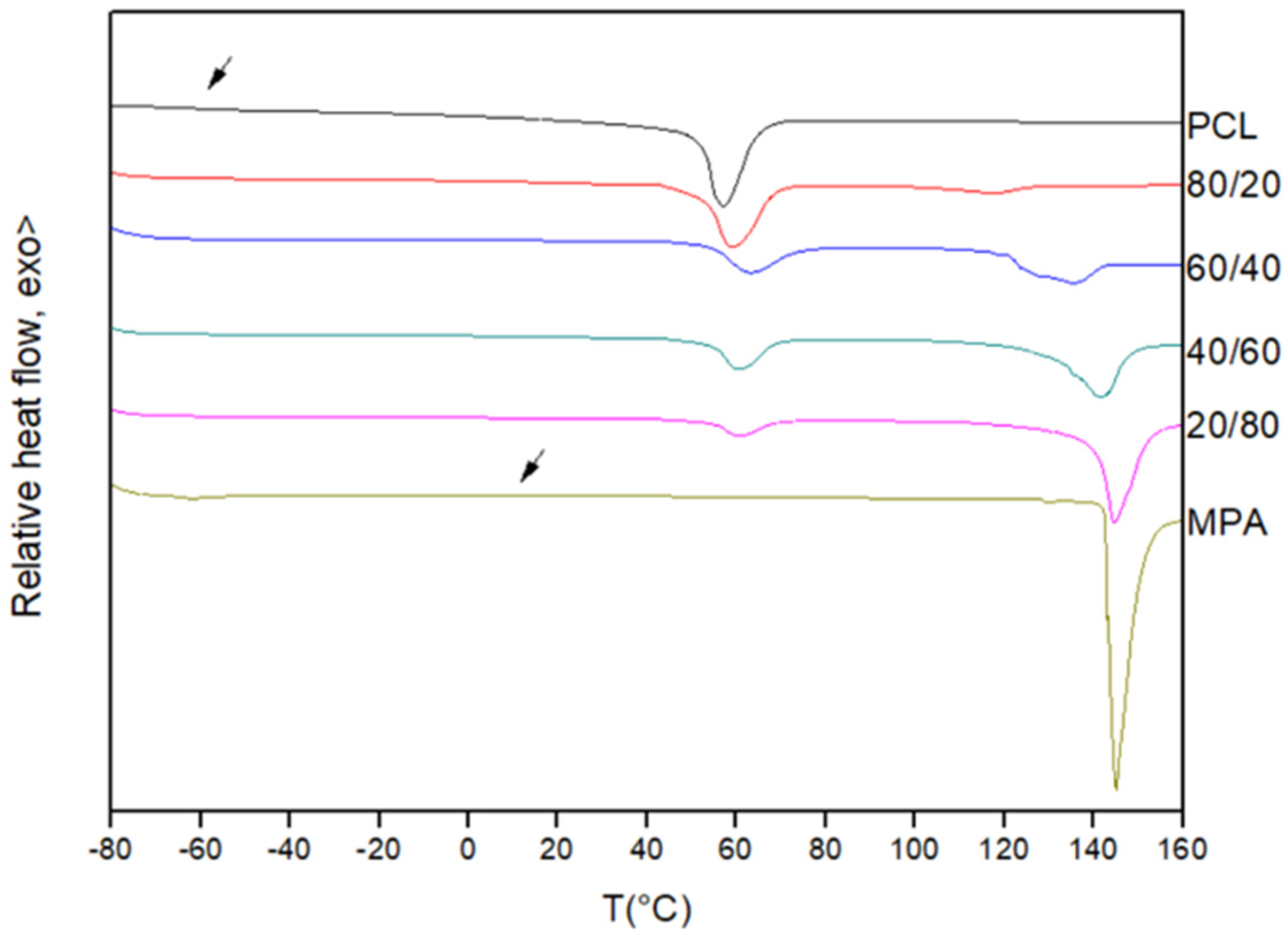
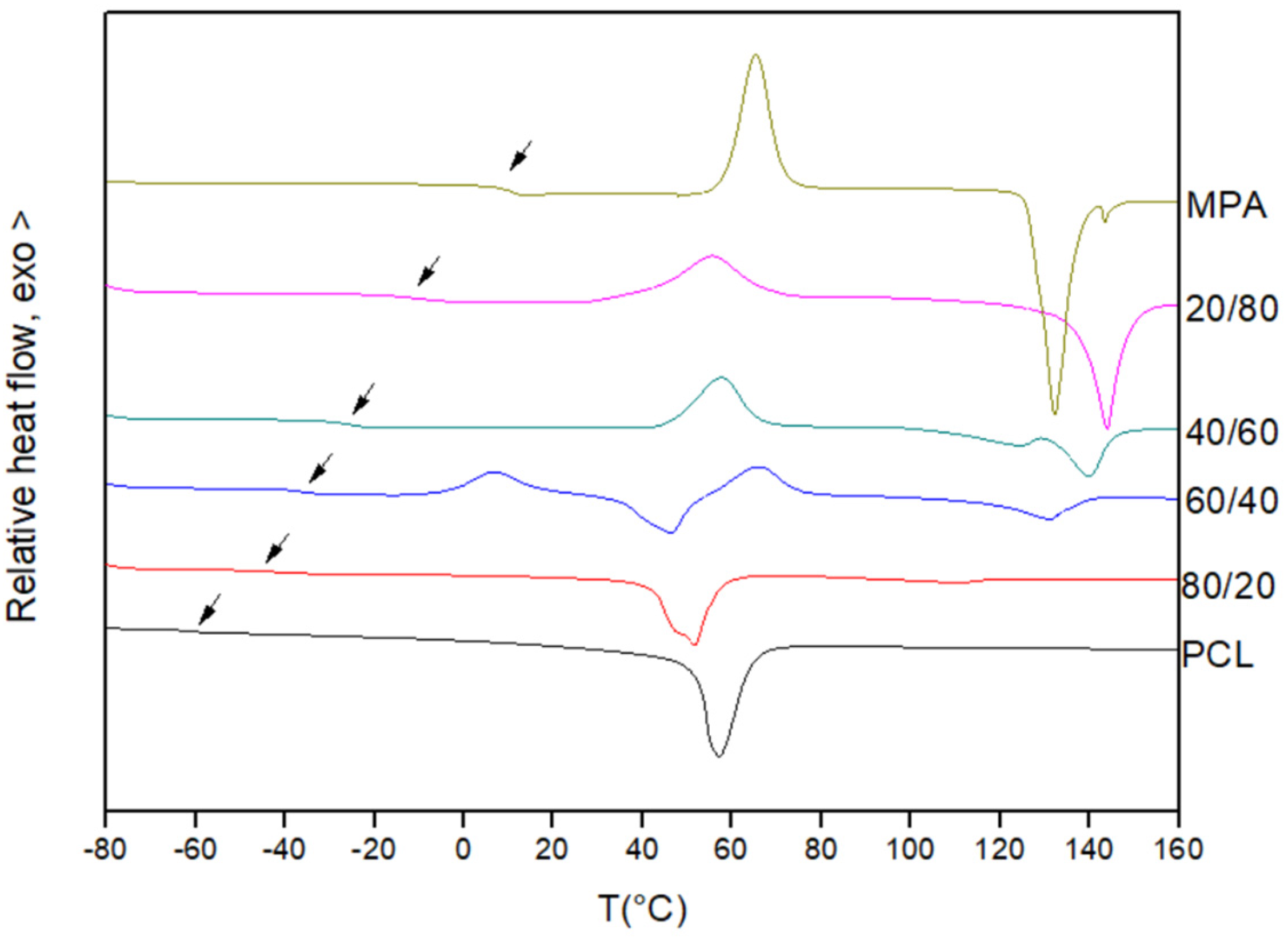
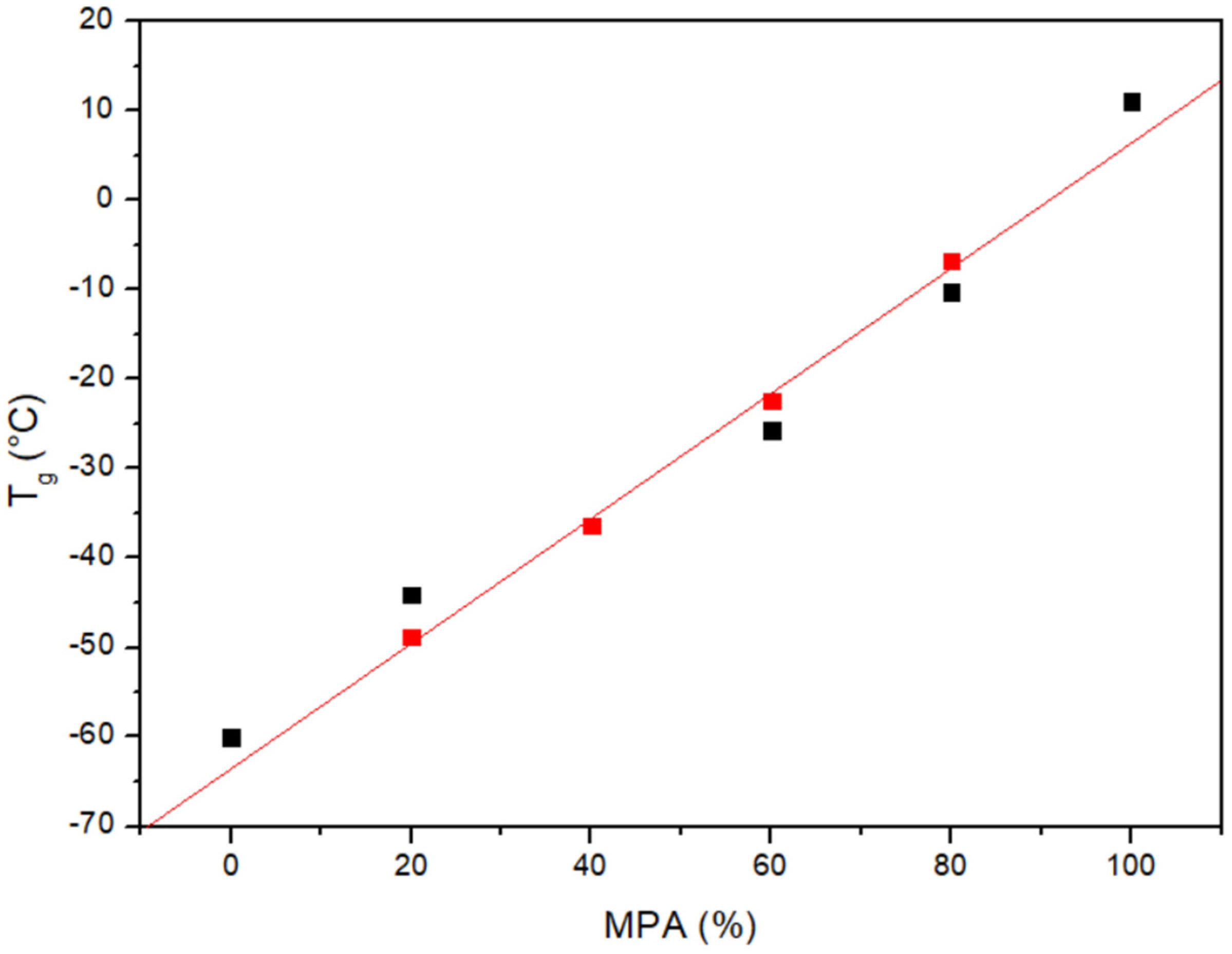
3.1. Miscibility Analysis by Differential Scanning Calorimetry (DSC)
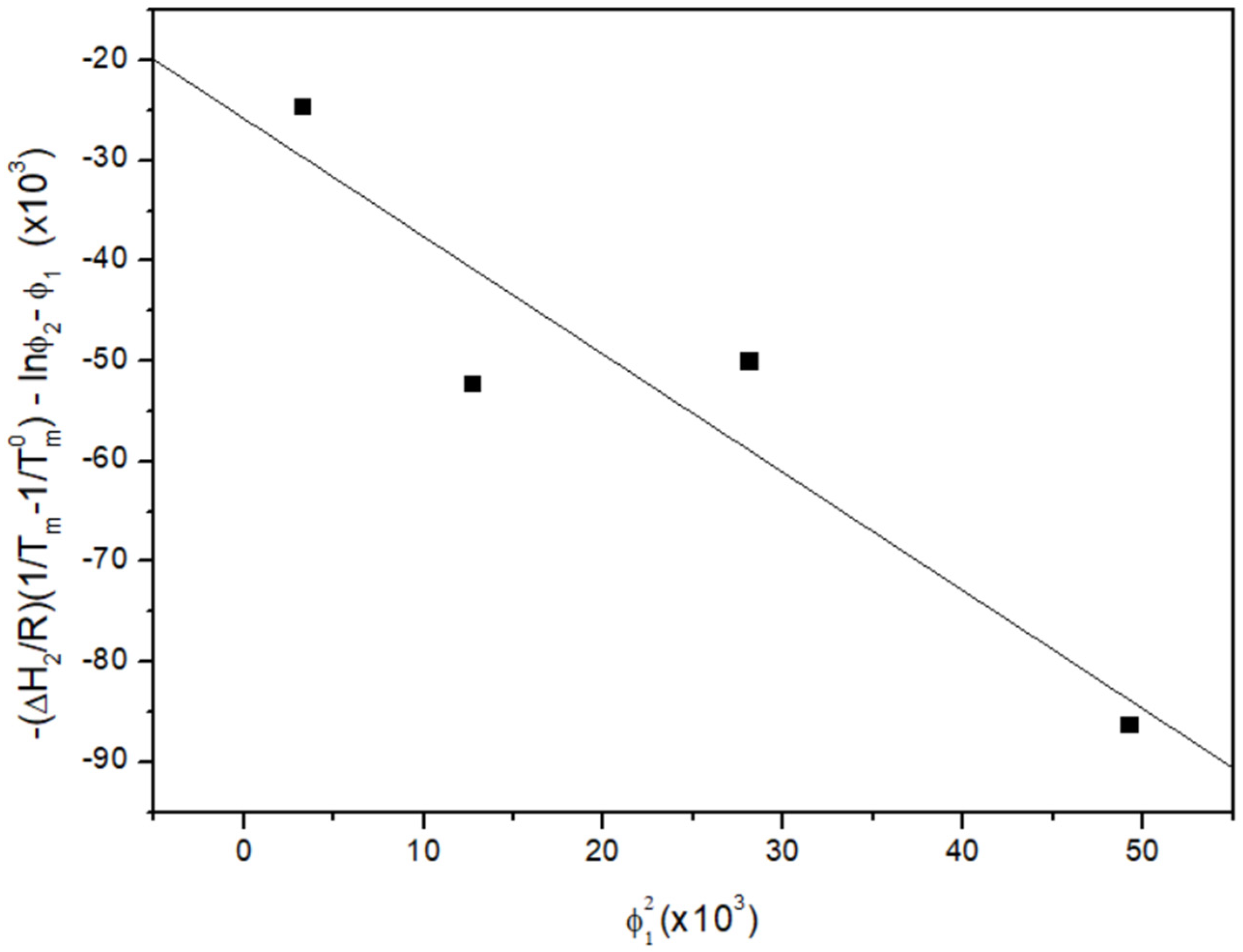
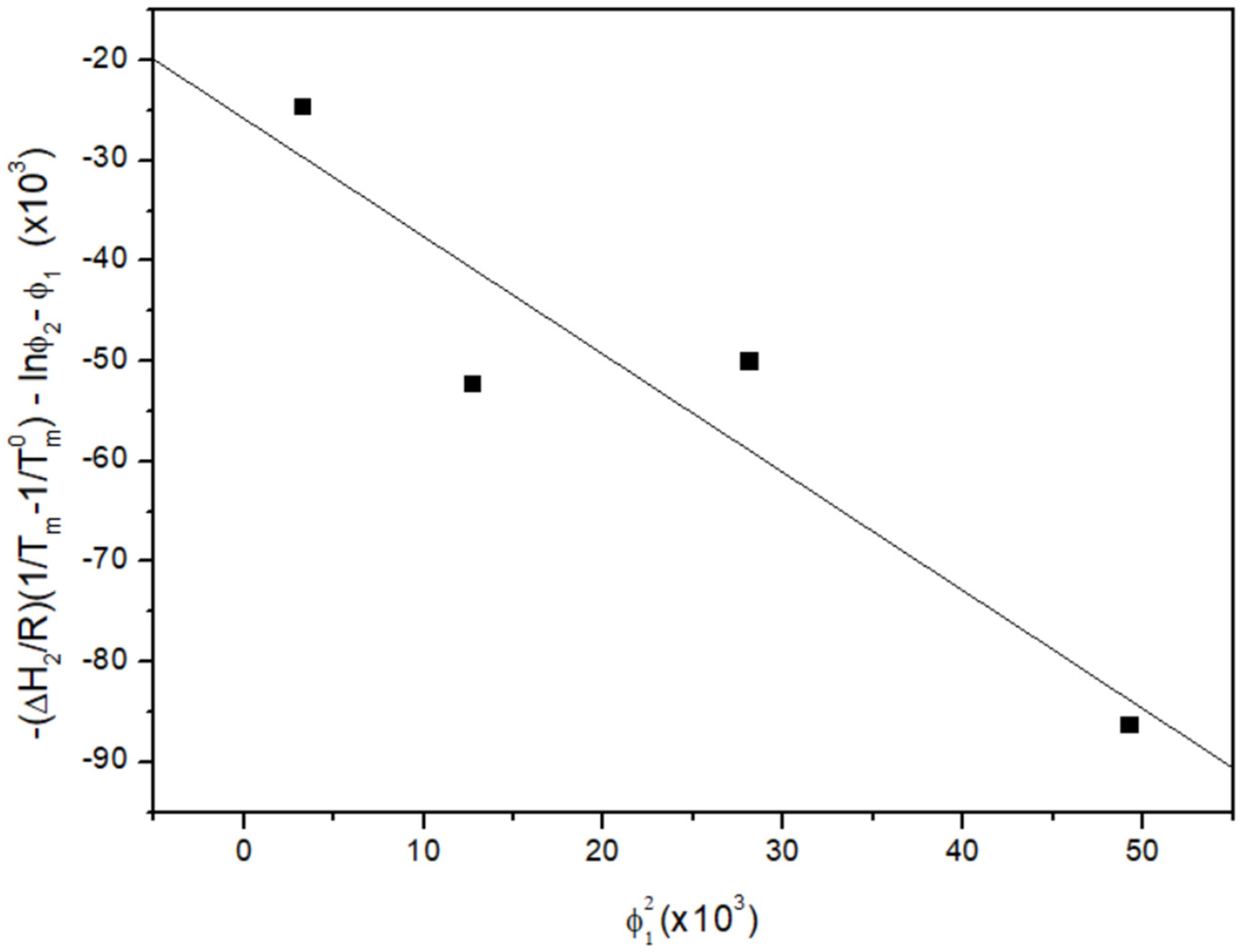
3.2. Melting Point Depression Analysis
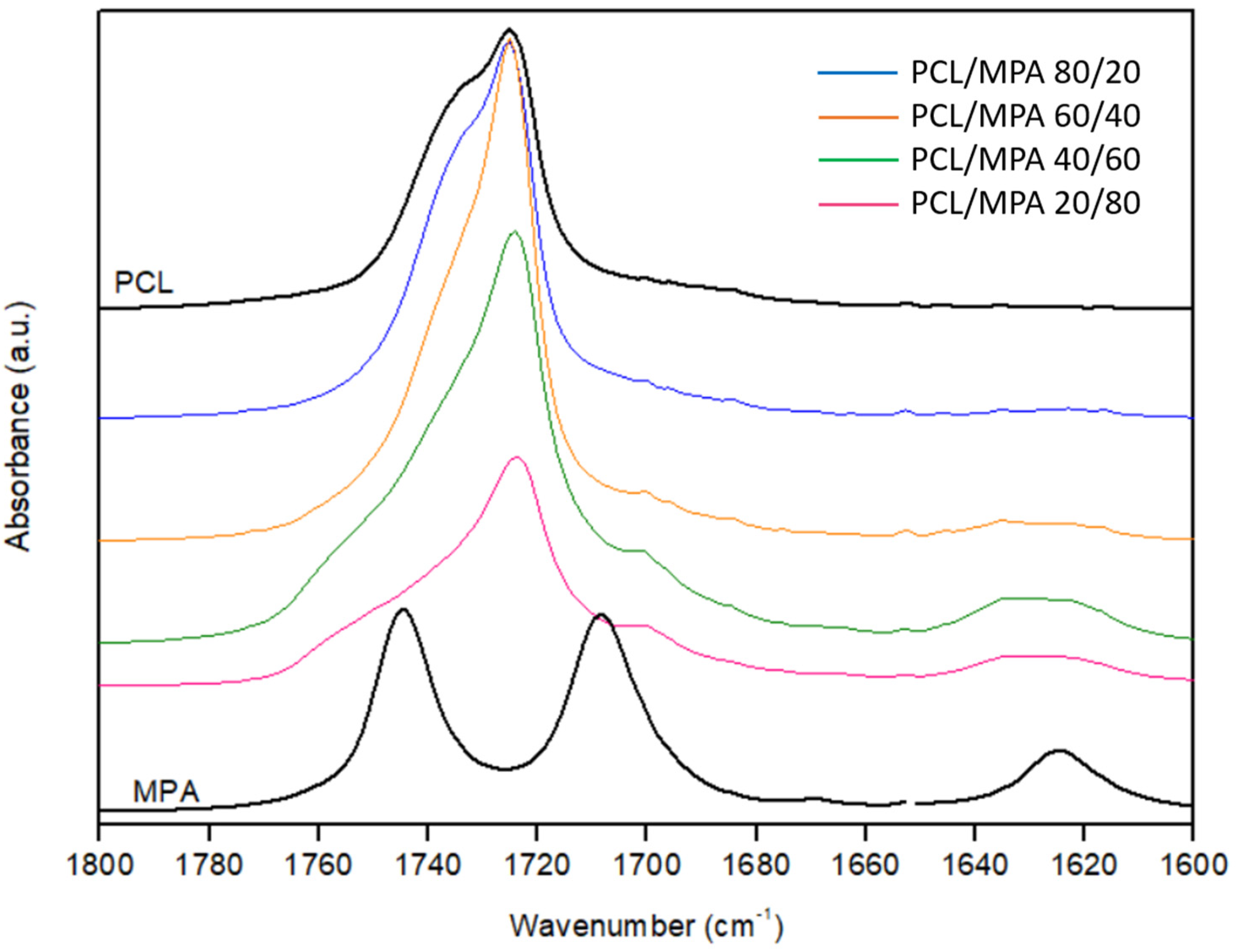
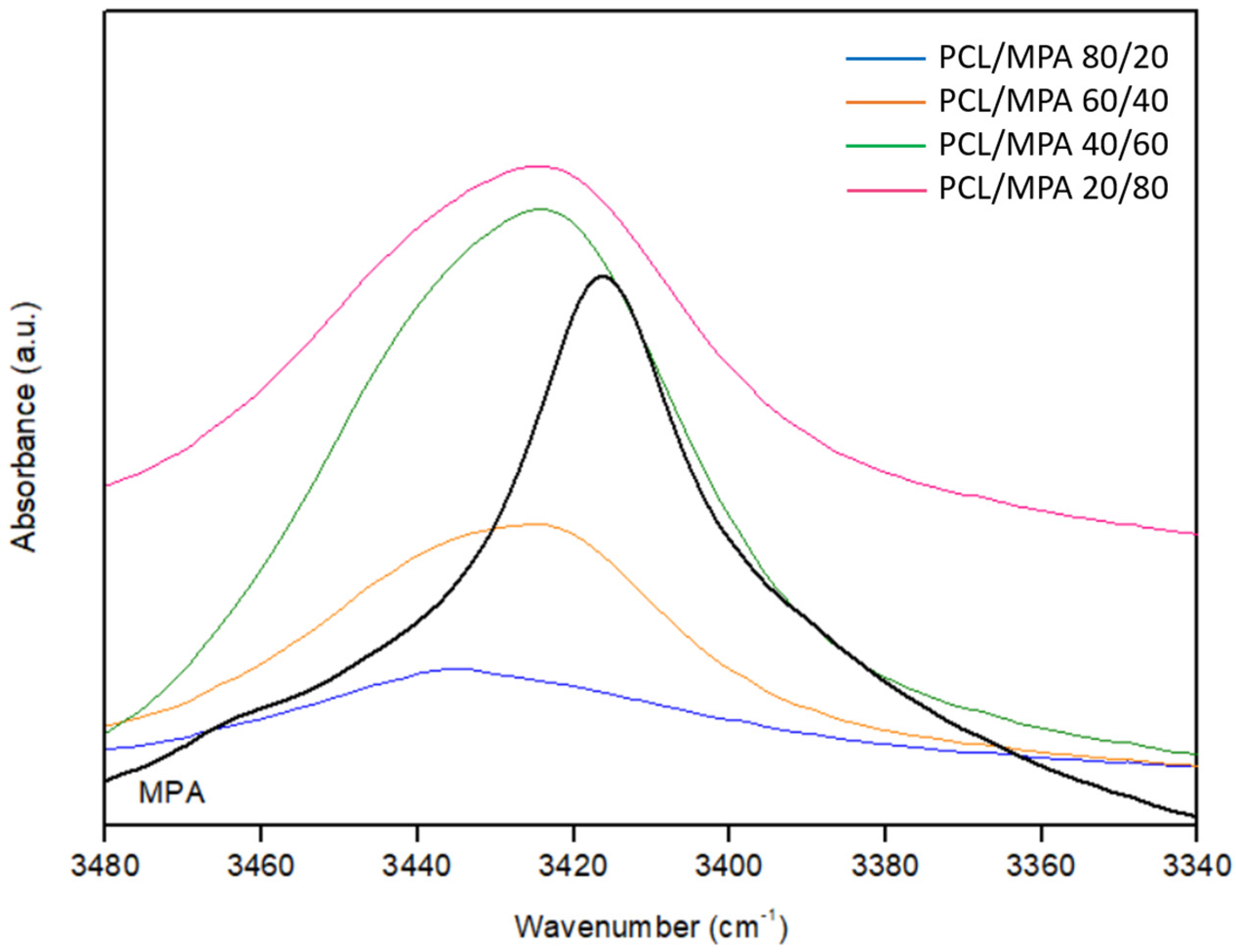
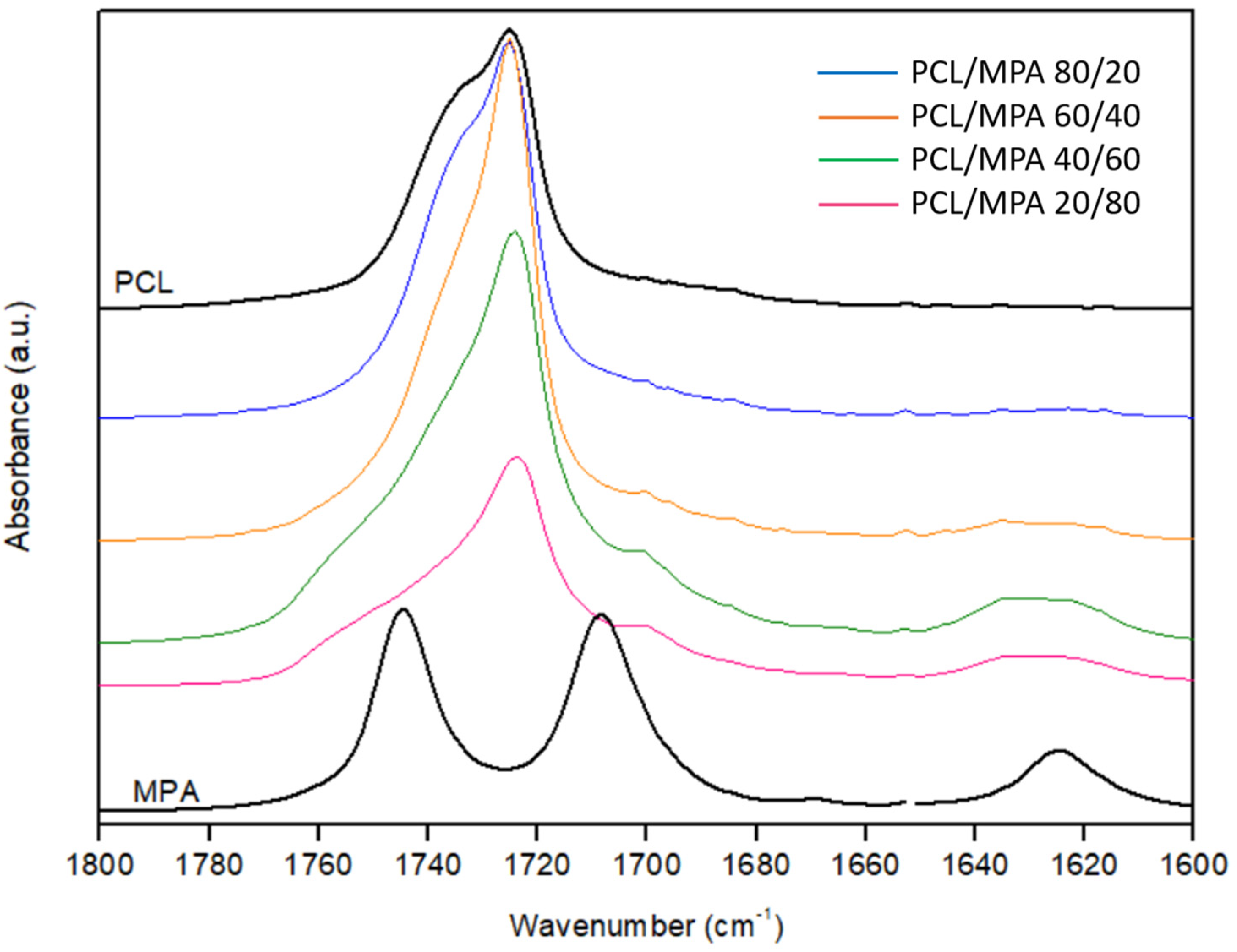
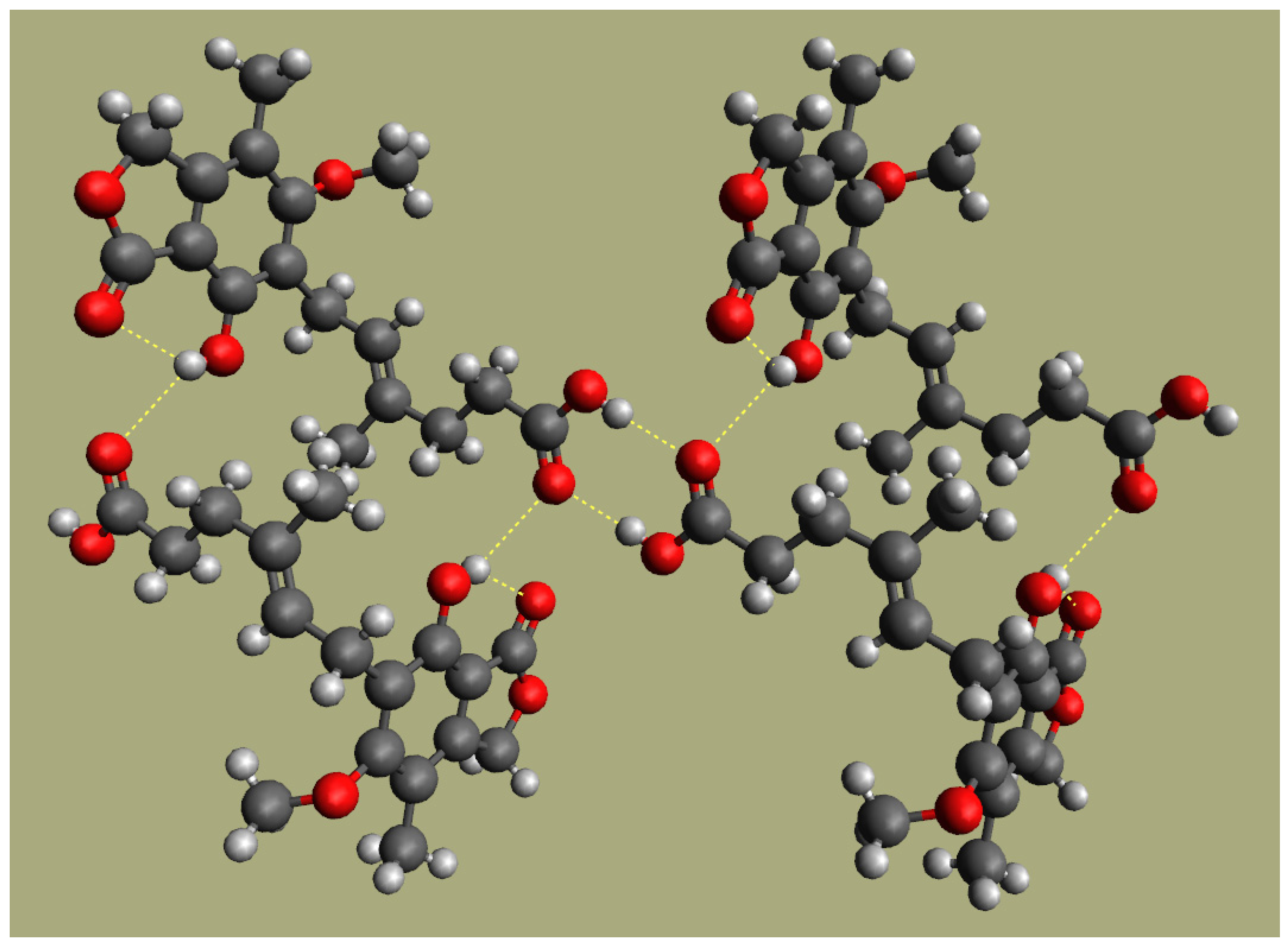
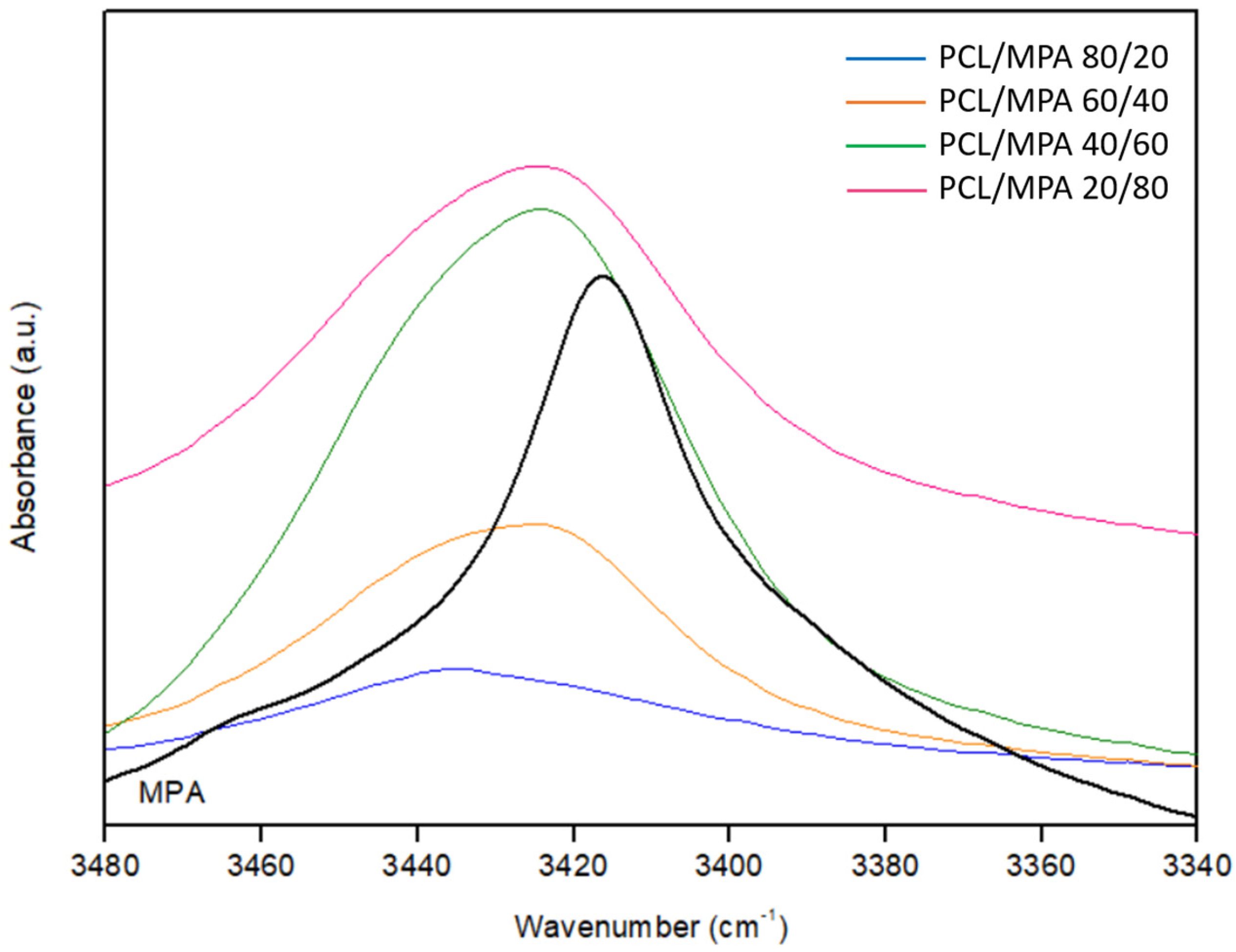
3.3. Fourier Transform Infrared Spectroscopy (FTIR)
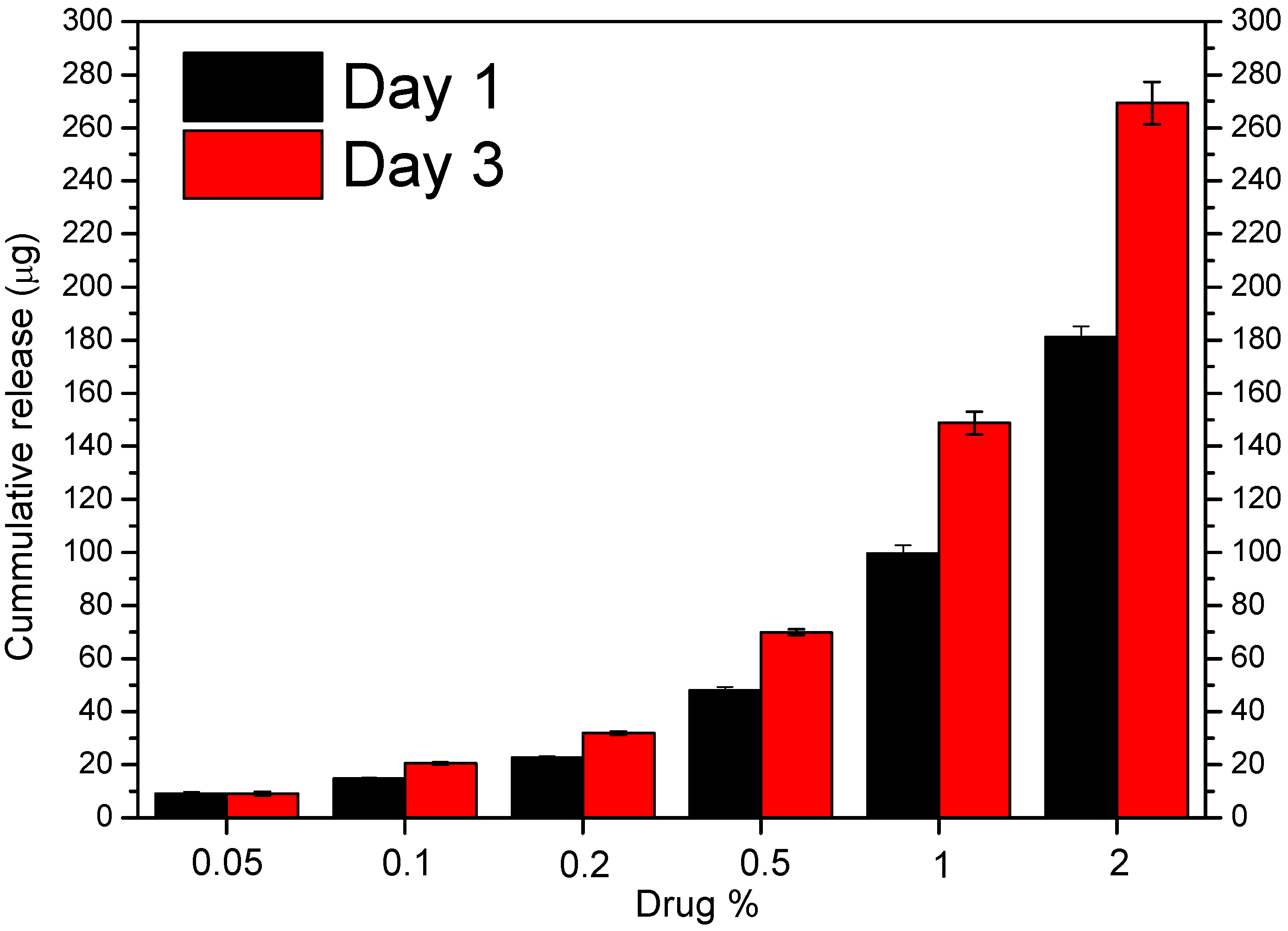
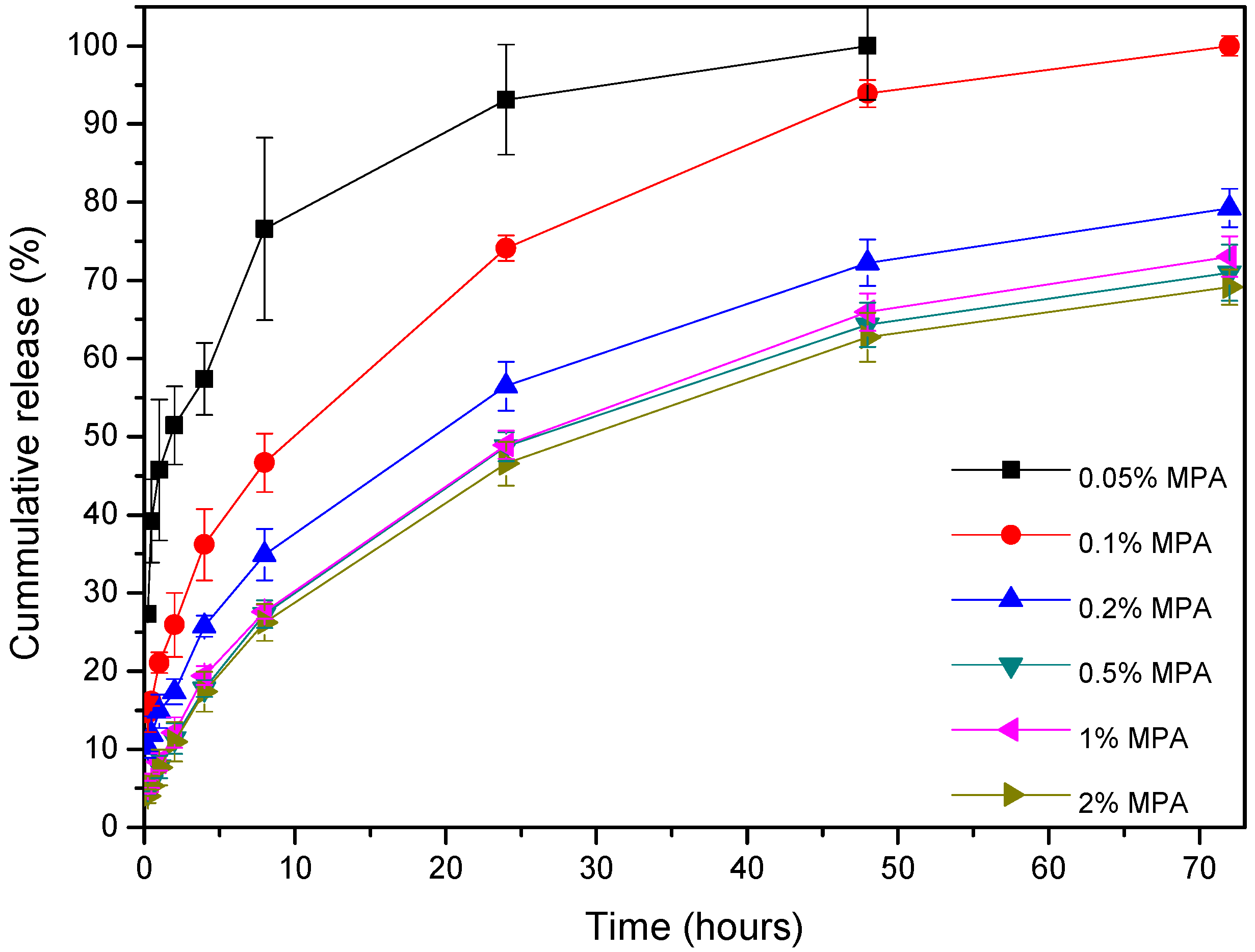
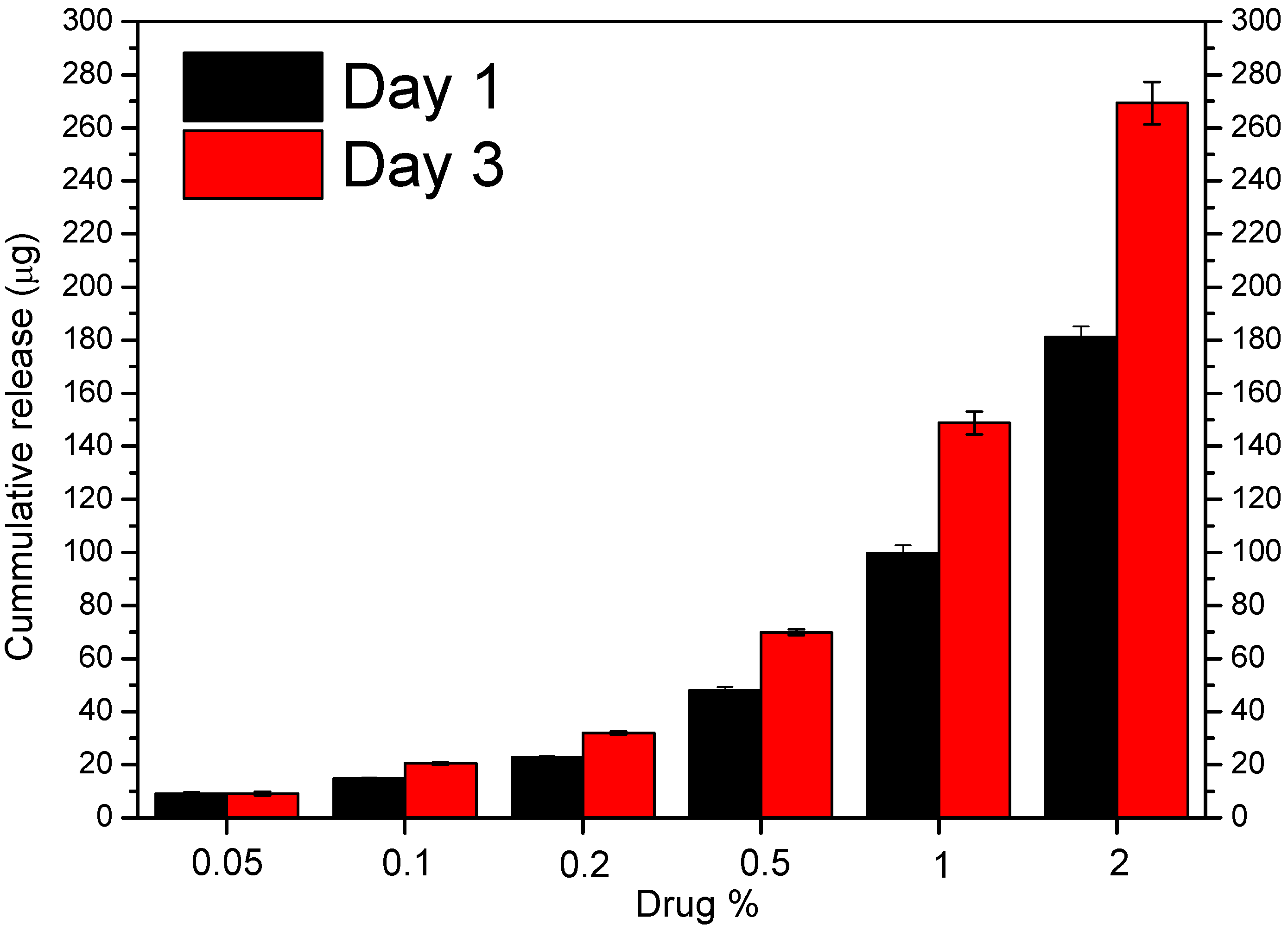
3.4. In Vitro Drug Release
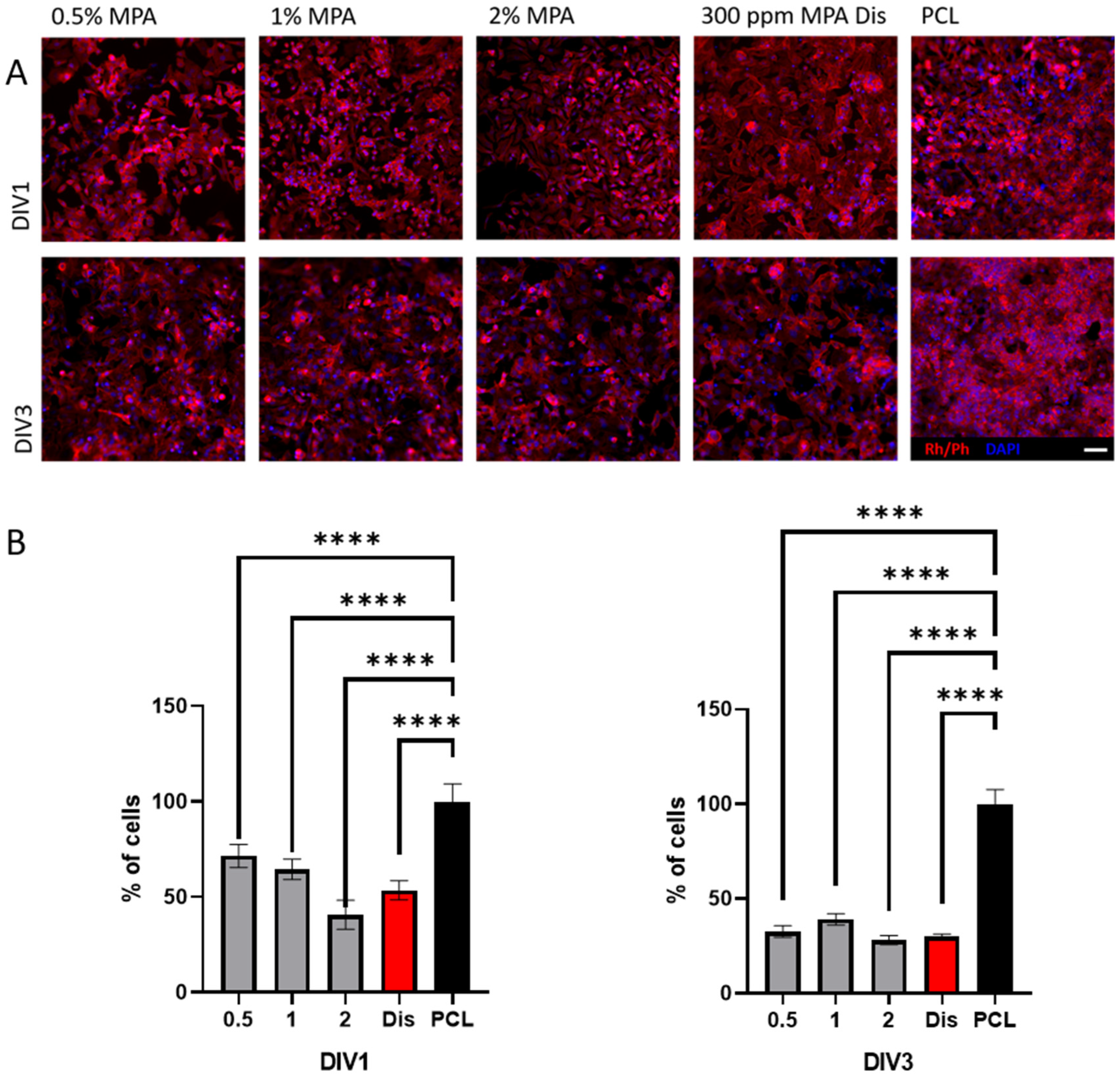
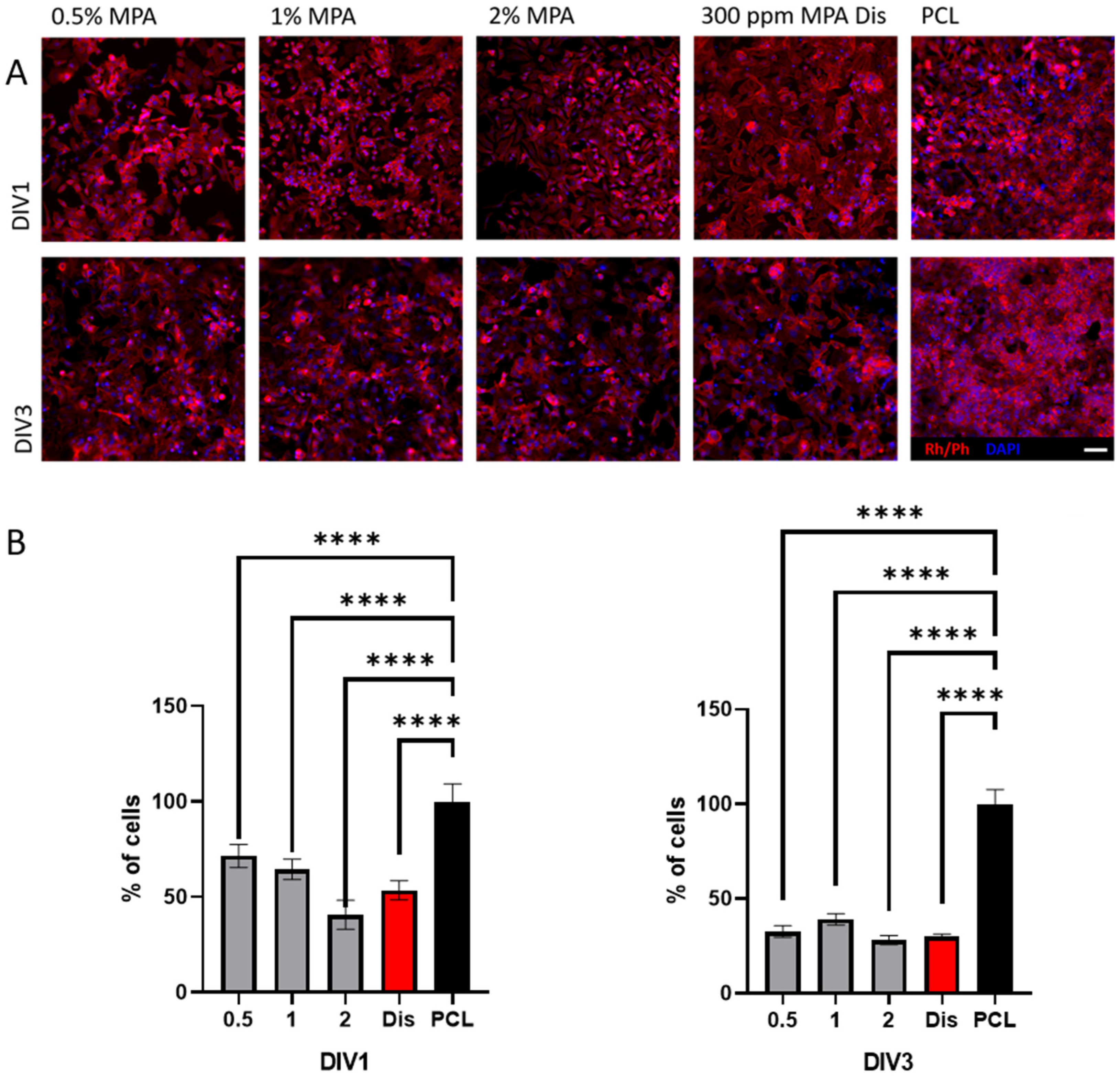
3.5. Cell Viability with HeLa Immortalized Cancer Cells
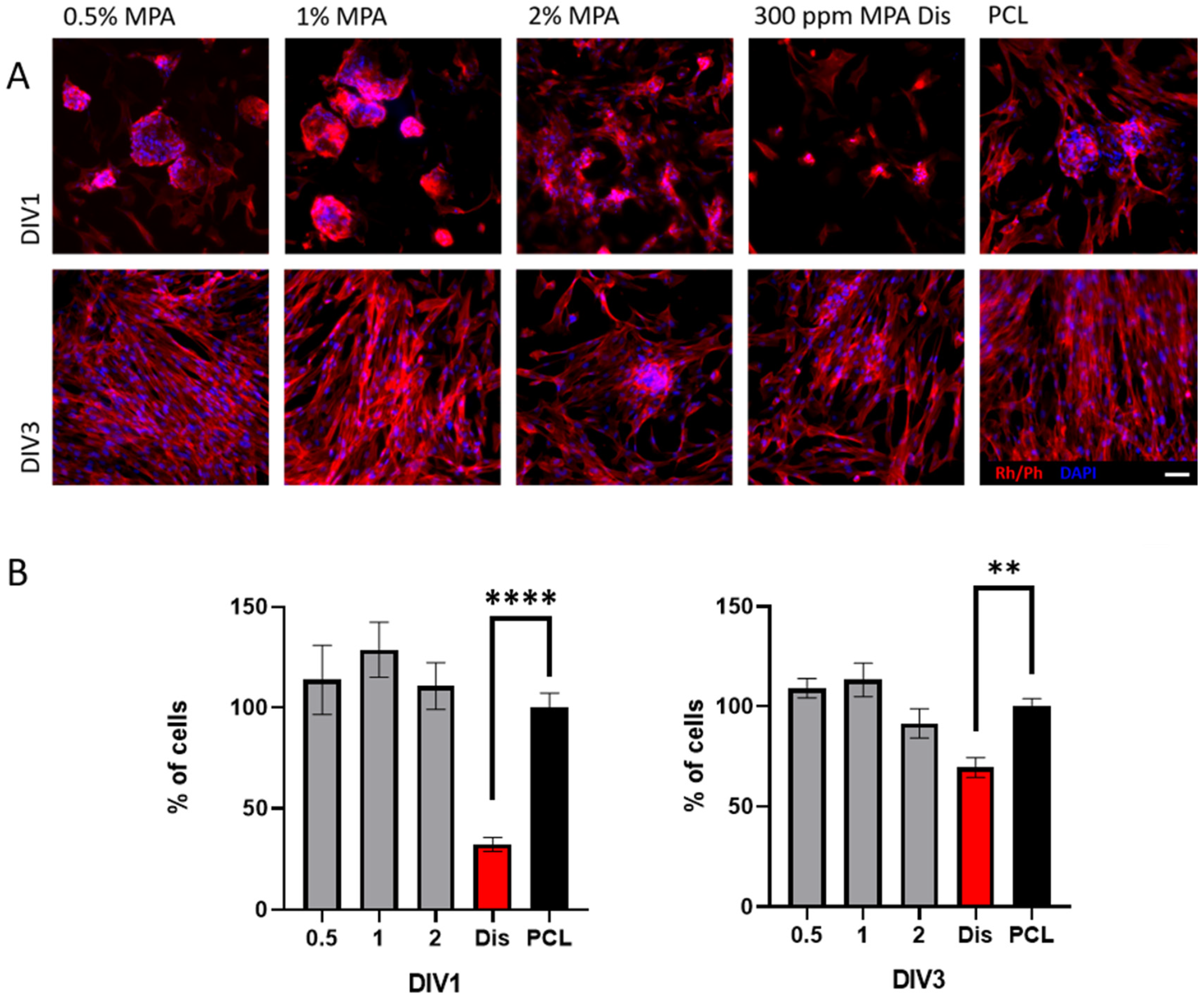
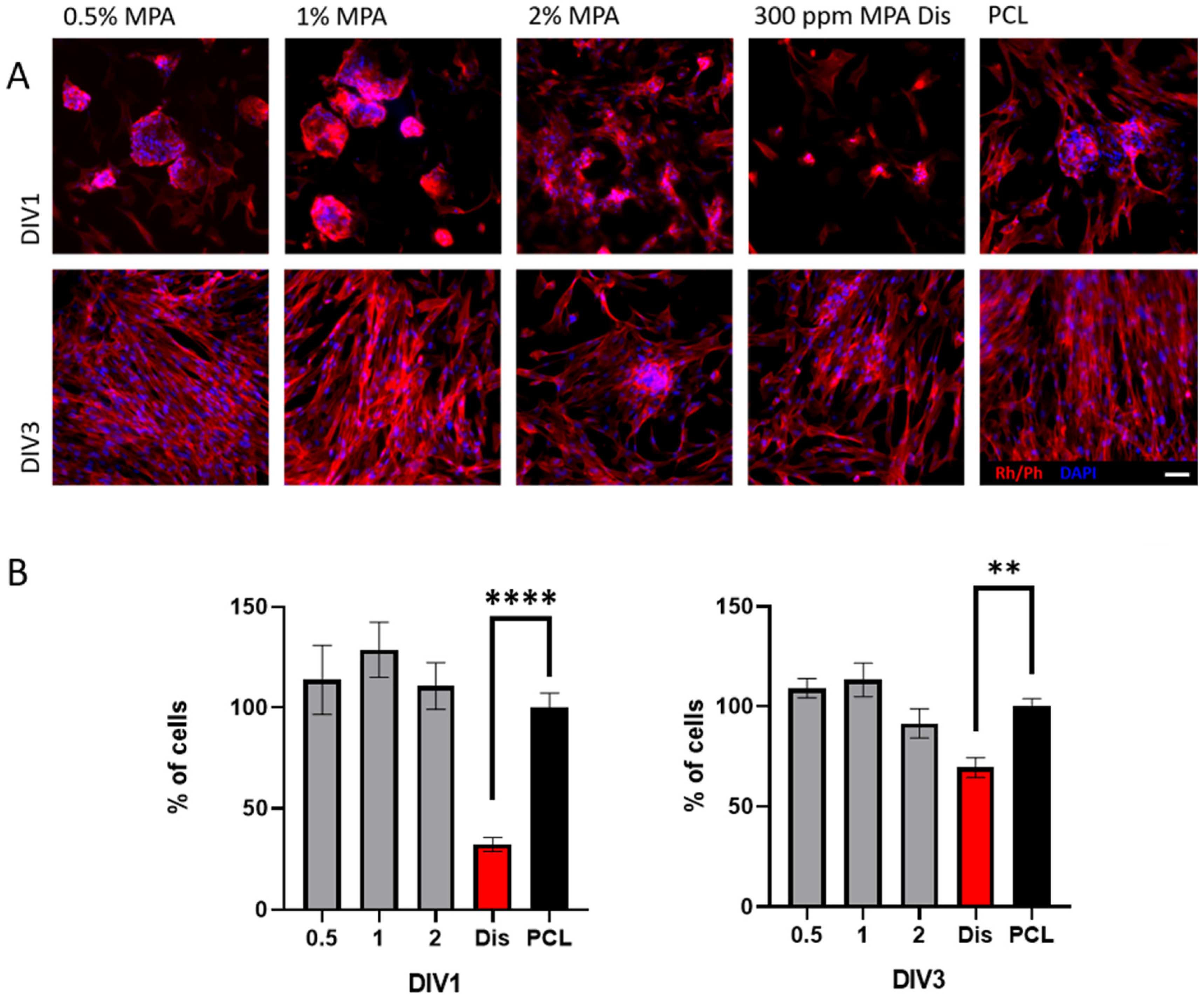
3.6. Cell Viability with Fibroblasts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef]
- Hodgson, J. ADMET—Turning chemicals into drugs. Nat. Biotechnol. 2001, 19, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Amidon, G.L.; Amidon, G.E. Physiological Parameters for Oral Delivery and in vitro Testing. Mol. Pharm. 2010, 7, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Li, F.; Yeh, S.; Wang, Y.; Xin, J. Physical stability of amorphous pharmaceutical solids: Nucleation, crystal growth, phase separation and effects of the polymers. Int. J. Pharm. 2020, 590, 119925. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Wang, Y.; Moinuddin, S.M.; Feng, X.; Ahsan, F. Co-amorphous Drug Delivery Systems: A Review of Physical Stability, in vitro and in vivo Performance. AAPS PharmSciTech 2022, 23, 259. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Riegelman, S. Pharmaceutical Applications of Solid Dispersion Systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Parulski, C.; Gresse, E.; Jennotte, O.; Felten, A.; Ziemons, E.; Lechanteur, A.; Evrard, B. Fused deposition modeling 3D printing of solid oral dosage forms containing amorphous solid dispersions: How to elucidate drug dissolution mechanisms through surface spectral analysis techniques? Int. J. Pharm. 2022, 626, 122157. [Google Scholar] [CrossRef] [PubMed]
- Bhanushali, J.S.; Dhiman, S.; Nandi, U.; Bharate, S.S. Molecular interactions of niclosamide with hydroxyethyl cellulose in binary and ternary amorphous solid dispersions for synergistic enhancement of water solubility and oral pharmacokinetics in rats. Int. J. Pharm. 2022, 626, 122144. [Google Scholar] [CrossRef] [PubMed]
- Becelaere, J.; Van Den Broeck, E.; Schoolaert, E.; Vanhoorne, V.; Van Guyse, J.F.; Vergaelen, M.; Borgmans, S.; Creemers, K.; Van Speybroeck, V.; Vervaet, C.; et al. Stable amorphous solid dispersion of flubendazole with high loading via electrospinning. J. Control. Release 2022, 351, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, A.G.; Singh, M.; Mali, A.R.; Serrano, D.R.; Kumar, R.; Healy, A.M.; Agrawal, A.K.; Kumar, D. Continuous Manufacturing and Molecular Modeling of Pharmaceutical Amorphous Solid Dispersions. AAPS PharmSciTech 2022, 23, 249. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.P.; Knopp, M.M.; Berthelsen, R.; Löbmann, K. Supersaturated amorphous solid dispersions of celecoxib prepared in situ by microwave irradiation. Int. J. Pharm. 2022, 626, 122115. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.F.; Dhareshwar, S.S.; Kesisoglou, F. Amorphous Solid Dispersions or Prodrugs: Complementary Strategies to Increase Drug Absorption. J. Pharm. Sci. 2016, 105, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Van den Mooter, G. The influence of hydrogen bonding between different crystallization tendency drugs and PVPVA on the stability of amorphous solid dispersions. Int. J. Pharm. 2023, 646, 123440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, X.; Tao, W. Curcumin amorphous solid dispersions benefit from hydroxypropyl methylcellulose E50 to perform enhanced anti-inflammatory effects. Int. J. Biol. Macromol. 2023, 252, 126507. [Google Scholar] [CrossRef] [PubMed]
- Van Den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.-Y.; Xu, Y.; Fusetani, N. Natural products as antifouling compounds: Recent progress and future perspectives. Biofouling 2009, 26, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Olesen, N.E.; Huang, Y.; Holm, R.; Rades, T. Statistical Analysis of a Method to Predict Drug-Polymer Miscibility. J. Pharm. Sci. 2016, 105, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Klueppelberg, J.; Handge, U.A.; Thommes, M.; Winck, J. Composition Dependency of the Flory–Huggins Interaction Parameter in Drug–Polymer Phase Behavior. Pharmaceutics 2023, 15, 2650. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Knipp, G.; Zografi, G. Assessing the performance of amorphous solid dispersions. J. Pharm. Sci. 2012, 101, 1355–1377. [Google Scholar] [CrossRef] [PubMed]
- DeBoyace, K.; Wildfong, P.L.D. The Application of Modeling and Prediction to the Formation and Stability of Amorphous Solid Dispersions. J. Pharm. Sci. 2018, 107, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Sasaki, M.; Yoshino, H.; Kofuji, S.; Sasaki, A.T.; Steckl, A.J. In-vitro evaluation of MPA-loaded electrospun coaxial fiber membranes for local treatment of glioblastoma tumor cells. J. Drug Deliv. Sci. Technol. 2017, 40, 45–50. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Yang, Y.; Lu, X.H.; Zhang, H.; Shui, X.X.; Liu, C.; He, X.B.; Jiang, Q.; Zhao, B.H.; Si, S.Y. Mycophenolic acid induces adipocyte-like differentiation and reversal of malignancy of breast cancer cells partly through PPARγ. Eur. J. Pharmacol. 2011, 658, 68. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R. Mycophenolic Acid: A One Hundred Year Odyssey from Antibiotic to Immunosuppressant. Chem. Rev. 2000, 100, 3801–3826. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ha, H.; Seo, J.; Kim, M.S.; Kim, H.J.; Huh, K.H.; Park, K.; Kim, Y.S. Mycophenolic Acid Inhibits Platelet-Derived Growth Factor-Induced Reactive Oxygen Species and Mitogen-Activated Protein Kinase Activation in Rat Vascular Smooth Muscle Cells. Am. J. Transplant. 2004, 4, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Floryk, D.; Huberman, E. Mycophenolic acid-induced replication arrest, differentiation markers and cell death of androgen-independent prostate cancer cells DU145. Cancer Lett. 2006, 231, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Hackl, A.; Ehren, R.; Weber, L.T. Effect of mycophenolic acid in experimental, nontransplant glomerular diseases: New mechanisms beyond immune cells. Pediatr. Nephrol. 2017, 32, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rexach, E.; Meaurio, E.; Iturri, J.; Toca-Herrera, J.L.; Nir, S.; Reches, M.; Sarasua, J.R. Miscibility, interactions and antimicrobial activity of poly(ε-caprolactone)/chloramphenicol blends. Eur. Polym. J. 2018, 102, 30–37. [Google Scholar] [CrossRef]
- Sanchez-Rexach, E.; de Arenaza, I.M.; Sarasua, J.R.; Meaurio, E. Antimicrobial poly(ε-caprolactone)/thymol blends: Phase behavior, interactions and drug release kinetics. Eur. Polym. J. 2016, 83, 288–299. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices—Part 5: Tests for in vitro Cytotoxicity. ISO Standards: Geneva, Switzerland, 2009.
- Rashid, F.; Saeed, A.; Iqbal, J. In Vitro Anticancer Effects of Stilbene Derivatives: Mechanistic Studies on HeLa and MCF-7 Cells. Anti-Cancer Agents Med. Chem. 2021, 21, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aguinagalde, O.; Lejardi, A.; Meaurio, E.; Hernández, R.; Mijangos, C.; Sarasua, J.-R. Novel Hydrogels of Chitosan and Poly(vinyl alcohol) Reinforced with Inorganic Particles of Bioactive Glass. Polymers 2021, 13, 691. [Google Scholar] [CrossRef] [PubMed]
- Pezzoli, R.; Lyons, J.G.; Gately, N.; Higginbotham, C.L. Investigation of miscibility estimation methods between indomethacin and poly(vinylpyrrolidone-co-vinyl acetate). Int. J. Pharm. 2018, 549, 50–57. [Google Scholar] [CrossRef]
- Hernandez-Montero, N.; Ugartemendia, J.M.; Amestoy, H.; Sarasua, J.R. Complex phase behavior and state of miscibility in Poly(ethylene glycol)/Poly(l-lactide-co-ε-caprolactone) Blends. J. Polym. Sci. B Polym. Phys. 2014, 52, 111–121. [Google Scholar] [CrossRef]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Sánchez-Aguinagalde, O.; Meaurio, E.; Lejardi, A.; Sarasua, J.-R. Amorphous solid dispersions in poly(ε-caprolactone)/xanthohumol bioactive blends: Physicochemical and mechanical characterization. J. Mater. Chem. B 2021, 9, 4219–4229. [Google Scholar] [CrossRef] [PubMed]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Chapter 9—Carbonyl Compounds, 3rd ed.; Wiberley, E., Ed.; Academic Press: San Diego, CA, USA, 1990; pp. 289–325. [Google Scholar] [CrossRef]
- Harrison, W.; Shearer, H.M.M.; Trotter, J. Crystal structure of mycophenolic acid. Journal of the Chemical Society. Perkin Trans. 1972, 2, 1542–1544. [Google Scholar] [CrossRef]
- Covarrubias, A.; Zúñiga-Villarreal, N.; González-Lucas, A.; Díaz-Domínguez, J.; Espinosa-Pérez, G. Crystal Structure of Mycophenolic Acid: 6-(4-Hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)-4-methyl-hex-4-enoic Acid. Anal. Sci. 2000, 16, 783–784. [Google Scholar] [CrossRef]
- Zeng, Q.Z.; Ouyang, J.; Zhang, S.; Zhang, L. Structural characterization and dissolution profile of mycophenolic acid cocrystals. Eur. J. Pharm. Sci. 2017, 102, 140–146. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Baishya, H. Application of Mathematical Models in Drug Release Kinetics of Carbidopa and Levodopa ER Tablets. J. Dev. Drugs 2017, 6, 1000171. [Google Scholar] [CrossRef]
- Klangjorhor, J.; Chaiyawat, P.; Teeyakasem, P.; Sirikaew, N.; Phanphaisarn, A.; Settakorn, J.; Lirdprapamongkol, K.; Yama, S.; Svasti, J.; Pruksakorn, D. Mycophenolic acid is a drug with the potential to be repurposed for suppressing tumor growth and metastasis in osteosarcoma treatment. Int. J. Cancer 2020, 146, 3397–3409. [Google Scholar] [CrossRef] [PubMed]
- Dun, B.; Sharma, A.; Teng, Y.; Liu, H.; Purohit, S.; Xu, H.; Zeng, L.; She, J.X. Mycophenolic acid inhibits migration and invasion of gastric cancer cells via multiple molecular pathways. PLoS ONE 2013, 8, e81702. [Google Scholar] [CrossRef] [PubMed]
- Dun, B.; Sharma, A.; Xu, H.; Liu, H.; Bai, S.; Zeng, L.; She, J.X. Transcriptomic changes induced by mycophenolic acid in gastric cancer cells. Am. J. Transl. Res. 2014, 6, 28–42. [Google Scholar]
- Dun, B.; Xu, H.; Sharma, A.; Liu, H.; Yu, H.; Yi, B.; Liu, X.; He, M.; Zeng, L.; She, J.X. Delineation of biological and molecular mechanisms underlying the diverse anticancer activities of mycophenolic acid. Int. J. Clin. Exp. Pathol. 2013, 6, 2880–2886. [Google Scholar] [PubMed]
- Howgate, E.M.; Yeo, K.R.; Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of in vivo drug clearance from in vitro data. I: Impact of inter-individual variability. Xenobiotica 2006, 36, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Rambhia, K.J.; Ma, P.X. Controlled drug release for tissue engineering. J. Control. Release 2015, 219, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Morath, C.; Reuter, H.; Simon, V.; Krautkramer, E.; Muranyi, W.; Schwenger, V.; Goulimari, P.; Grosse, R.; Hahn, M.; Lichter, P.; et al. Effects of mycophenolic acid on human fibroblast proliferation, migration and adhesion in vitro and in vivo. Am. J. Transplant. 2008, 8, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Cao, W.; Li, J.; Sprengers, D.; Hernanda, P.Y.; Kong, X.; van der Laan, L.J.; Man, K.; Kwekkeboom, J.; Metselaar, H.J.; et al. Differential sensitivities of fast-and slow-cycling cancer cells to inosine monophosphate dehydrogenase 2 inhibition by mycophenolic acid. Mol. Med. 2015, 21, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.J.; Jacobs, V.; Bruneau, P.; Ple, P. Glucuronidation by human colorectal adenocarcinoma cells as a mechanism of resistance to mycophenolic acid. Adv. Enzym. Regul. 1995, 35, 91–100. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCL/MPA | Tg Experimental (°C) | Tg Theoretical (Fox) (°C) | Tm PCL (°C) | ΔHf PCL (J/g) |
|---|---|---|---|---|
| PCL | −60.0 | - | 57.2 | 66.4 |
| 80/20 | −44.1 | −48.8 | 51.7 | 49.8 |
| 60/40 | −36.3 | −36.3 | 46.4 | 25.9 |
| 40/60 | −25.7 | −22.4 | - | - |
| 20/80 | −10.2 | −6.8 | - | - |
| MPA | 11.1 | - | - | - |
| MPA wt% | Tm (°C) | ||
|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | |
| 100 | 139.2 | 140.5 | 140.9 |
| 95 | 139.6 | 139.9 | 138.3 |
| 90 | 138.4 | 138.1 | 137.4 |
| 85 | 137.4 | 138.9 | 136.8 |
| 80 | 135.8 | 135.7 | 135.9 |
| MPA % | Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | |
|---|---|---|---|---|---|
| 0.05 | R2 = 0.64 | R2 = 0.94 | R2 = 0.87 | R2 = 0.81 | n = 0.58 |
| 0.1 | R2 = 0.86 | R2 = 0.99 | R2 = 0.98 | R2 = 0.92 | n = 0.5 |
| 0.2 | R2 = 0.74 | R2 = 0.88 | R2 = 0.94 | R2 = 0.95 | n = 0.46 |
| 0.5 | R2 = 0.75 | R2 = 0.85 | R2 = 0.94 | R2 = 0.99 | n = 0.5 |
| 1 | R2 = 0.77 | R2 = 0.89 | R2 = 0.95 | R2 = 0.99 | n = 0.5 |
| 2 | R2 = 0.77 | R2 = 0.87 | R2 = 0.95 | R2 = 0.99 | n = 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Aguinagalde, O.; Sanchez-Rexach, E.; Polo, Y.; Larrañaga, A.; Lejardi, A.; Meaurio, E.; Sarasua, J.-R. Physicochemical Characterization and In Vitro Activity of Poly(ε-Caprolactone)/Mycophenolic Acid Amorphous Solid Dispersions. Polymers 2024, 16, 1088. https://doi.org/10.3390/polym16081088
Sánchez-Aguinagalde O, Sanchez-Rexach E, Polo Y, Larrañaga A, Lejardi A, Meaurio E, Sarasua J-R. Physicochemical Characterization and In Vitro Activity of Poly(ε-Caprolactone)/Mycophenolic Acid Amorphous Solid Dispersions. Polymers. 2024; 16(8):1088. https://doi.org/10.3390/polym16081088
Chicago/Turabian StyleSánchez-Aguinagalde, Oroitz, Eva Sanchez-Rexach, Yurena Polo, Aitor Larrañaga, Ainhoa Lejardi, Emilio Meaurio, and Jose-Ramon Sarasua. 2024. "Physicochemical Characterization and In Vitro Activity of Poly(ε-Caprolactone)/Mycophenolic Acid Amorphous Solid Dispersions" Polymers 16, no. 8: 1088. https://doi.org/10.3390/polym16081088
APA StyleSánchez-Aguinagalde, O., Sanchez-Rexach, E., Polo, Y., Larrañaga, A., Lejardi, A., Meaurio, E., & Sarasua, J.-R. (2024). Physicochemical Characterization and In Vitro Activity of Poly(ε-Caprolactone)/Mycophenolic Acid Amorphous Solid Dispersions. Polymers, 16(8), 1088. https://doi.org/10.3390/polym16081088