1. Introduction
Typically, apple cultivation involves the intensive cultivation of dwarf rootstocks with high-density planting. Dwarf rootstocks with high-density planting due to a reduction in tree size, the facilitation of crop management practices, an increase in land use efficiency, and early flowering and fruiting could lead to an increase in profits [
1]. In apple cultivation, rootstocks are classified into the following four categories according to tree height and the scion meridian: dwarf, semi-dwarf, intermediate, and vigorous [
2,
3]. The M9 is a dwarfing rootstock that belongs to the British M series, and the canopy of the scion varieties grafted on them are ca. 50% smaller compared to other standard rootstocks [
4]. The SH series rootstocks were selected and bred after 16 years of research (1978–1993) by crossing Guoguang × Henan Begonia by the Fruit Tree Research Institute of the Shanxi Academy of Agricultural Sciences in China. The SH40 is a semi-dwarfing rootstock with a good cold tolerance and is widely used in northern China, and the canopy of the scion varieties grafted on them are ca. 70% smaller compared to other standard rootstock. [
5,
6]. The application of apple dwarfing rootstock mainly includes rootstock and interstock. In China, the SH series is basically used as an interstock due to its environmental adaptability and difficult rooting and reproduction [
7,
8].
Transcription factors (TFs) can specifically bind to W-box cis-acting elements on target gene promoters, thereby regulating the target gene expression. Recent studies have shown that WRKY, AP2/EREBP, bHLH, C2H2, NAC, and ARF TFs regulate plant growth and cause dwarfing. WRKY TFs represent one of the most abundant TF families unique to plants discovered in the past 20 years [
9]. WRKY TFs in apple [
10], tomato [
11], and rice [
12] have been identified in genome-wide analyses and the results suggest that they could play multiple roles. WRKY TFs participate in plant growth and development through self-regulation or by regulating downstream hormones and the related pathways [
13].
WRKY TFs play important regulatory roles in the growth and development of many plants. Imran et al. [
14] reported that AtWRKY62 negatively affects
Arabidopsis growth. Abeysinghe et al. (2019) obtained a transgenic dwarf phenotype of
Arabidopsis tetraploids by transforming the AtWRKY18 overexpression vector [
15]. Chen et al. [
16] reported that CsWRKY7 might participate in plant growth through flowering in tea trees. Zhang et al. [
17] reported that OsWRKY78 regulates rice stem elongation and seed development. Zhao et al. [
18] ectopically expressed MdWRKY31 in
Arabidopsis and tobacco and found that MdWRKY31 overexpression increased the sensitivity of apple roots and apple callus to abscisic acid (ABA). In the apple dwarf rootstock M26, MdWRKY9 can inhibit the expression of the brassinolide synthesis gene, eventually causing plant dwarfing [
19]. These findings provide strong evidence for the regulation of TFs to cause dwarfing in apple dwarfing rootstocks.
This study focused on the utilization mode (interstock) of apple production to evaluate the effect of SH40 and M9 on the scion. This study aimed to analyze the regulatory mechanism underlying dwarfing under this utilization mode, providing a reference for the early screening and evaluation of interstock selection and cultivation measures for apple growth regulation. It is anticipated that apple dwarfing is governed by a complex multi-gene regulatory mechanism closely related to gene expression regulation by the TFs. Nonetheless, the detailed metabolic regulatory network remains unclear and warrants further in-depth study.
2. Materials and Methods
2.1. Plant Materials
The test materials were 2-year-old Fuji/M9/Mr, Fuji/SH40/Mr, and Fuji/Mr/Mr. All test materials were potted in plastic pots with a diameter of 35 cm and a height of 40 cm, planted using washed river sand as the matrix, and cultivated in a greenhouse. The temperature was 18 ± 2 °C. The daily light–dark irradiation period was set to 16 h/8 h, with a photon flux density of 200 μmol/m2·s. The test materials of different stock and scion combinations were cultured using Hoagland’s complete nutrient drip irrigation.
The shoot tips and new shoot phloems of different stock combinations were collected in the autumn shoot growth period in late August. The single plant plot was repeated three times. After collection, the samples were wrapped in tin foil, quickly frozen in liquid nitrogen, and stored in a −80 °C refrigerator for later use.
2.2. Extraction of Plant Total RNA and cDNA Synthesis
The total RNA was extracted using the Plant RNA Kit (Tiangen, Beijing, China). The concentration of each RNA sample was determined using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA) and 1 μg RNA was used to obtain the first-strand cDNA by reverse transcription with the PrimeScriptTM RT Reagent Kit with gDNA Erase (TransGen Biotech, Beijing, China).
2.3. RNA-seq and Enrichment Analysis
The RNA integrity was evaluated on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and samples with an RNA integrity number (RIN) ≥ 7 were used in subsequent analyses. Equal amounts of RNA from shoot tips and new shoot phloems were used to produce cDNA libraries using the TruSeq Stranded mRNA LTSample Prep Kit (Illumina Inc., San Diego, CA, USA). These libraries were then sequenced using the Illumina HiSeq 2500 sequencing platform, and 125–150-bp paired-end reads were generated.
For the transcript-level quantification, the FPKM and read count values of each transcript (protein coding) were calculated using bowtie2 [
20] and eXpress [
21]. The DEGs were identified using the DESeq functions estimateSizeFactors and nbinomTest [
22]. Significantly differential expression was defined as
p-value < 0.05 and a fold-change > 2 or < 0.5. The hierarchical cluster analysis of the DEGs was performed to explore the transcript expression patterns. Gene Ontology (GO) enrichment analyses of the DEGs were performed using GOseq software [
23].
2.4. Quantitative Real-Time PCR
The primer sequences used to amplify the genes related to heat stress by quantitative real-time PCR (qRT-PCR) analysis are shown in
Table S1. The qRT-PCR analyses were performed on a Roche LC480 device (Roche Diagnostics GmbH, Mannheim, Germany), and fluorescence quantification was carried out using a TransStart Top Green qPCR SuperMix (TransGen Biotech, Beijing, China). We initially used a two-step PCR amplification procedure, with pre-denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 34 s. The amplification, dissolution, and standard curves were automatically generated by the Roche LC480 software. To calculate the gene expression level, we used the 2
−△△CT method. The relative expression levels of candidate genes were calculated using the β-actin gene product as the template of the internal reference gene standard, and the average ΔCT value of the group with Mr as the interstock was calculated. Each procedure was conducted using three biological replicates.
2.5. Gene Cloning
The full-length ORFs of
MdWRKY24 were retrieved and cloned by transcript BLAST with the Genome Database Rosaceae (GDR) data (
https://www.rosaceae.org/; 10 October 2020). The primers used were MdWRKY24-MF/MR (
Table S1). The PCR detection validation was performed using universal primers M13-47/48. The sequences were compared using DNAMAN (Lynnon Biosoft, San Ramon, CA, USA), aligned using ClustalW, and analyzed using MEGA version 7.0 [
24].
2.6. Subcellular Localization
The subcellular localization expression vector was pCAMBIA2300:eGFP, and the primers used were MdWRKY24-F1/R1. The subcellular localization expression vector pCAMBIA2300:MdWRKY24:eGFP was constructed using the Kpn1/Xba1 enzymes. A heat shock protocol was used to transform E. coli DH5α cells (TransGen Biotech, Beijing, China). Recombinant plasmids were identified by colony PCR and confirmed by sequencing. The constructed vector plasmid was then transferred into Agrobacterium GV3101. Finally, Agrobacterium suspension was injected into the tobacco seedlings (Nicotiana rustica var. pavonii). The plants were cultured under low-light conditions for 2 d, after which the injected tobacco leaves were observed and imaged using a confocal microscope (C2-ER; Nikon, Minato City, Japan). The excitation and emission wavelengths of the chloroplast fluorescence signal were 640 and 675 nm, respectively, whereas those of the GFP fluorescent proteins were 488 and 510 nm, respectively.
2.7. Protein and Gene Analysis
2.8. Arabidopsis Genetic Transformation
The primers used were MdWRKY24-F1/R1. The expression vector for genetic transformation, pCAMBIA2300:MdWRKY24, was constructed using KpnI/XbaI enzymes. The inflorescence dip method was used to transform Arabidopsis; positive seedlings were screened using 30 μg/mL of hygromycin 1/2 MS plates and verified by PCR. The sterilized T3 seeds of Arabidopsis were evenly planted on MS plates (pH 5.8). The plants were grown in a controlled environment with a relative humidity of 60%, a constant temperature of 20–22 °C, and a 14 h/10 h light/dark cycle; the light intensity was 200 μmol/m2/s.
2.9. Yeast Two-Hybrid Analysis
The nuclear system BD vector was pGBKT7 and the nuclear system AD vector was PGADT7. The gene with the vector linker was cloned using seamless cloning and the vector was recombined in one step. pGBKT7:MdWRKY24 did not have a self-activation phenomenon, so the subsequent screening experiments could be performed.
In this experiment, we found homologous genes in Arabidopsis thaliana according to the evolutionary tree relationships. RGL and FT genes may interact with MdWRKY24, as verified by the experiments.
2.10. Statistical Analysis
The data were processed using Microsoft Excel 2007 (Microsoft Corp., Redmond, WA, USA) and the differences were evaluated using the two-tailed t-tests or Duncan’s multiple-range test. A p-value < 0.05 was considered statistically significant. * Mean p-value < 0.05; ** mean p-value < 0.01.
3. Results
3.1. Transcriptomic Analysis
Differentially expressed genes (DEGs; namely, MYB, NAC, and WRKY TFs) in the transcriptome data were screened under the conditions of |log
2 (fold-change)| > 1 and
p-value < 0.05. The pairwise comparisons were performed on the three test materials (M9, M26, and SH40), and the DEGs were obtained by sequencing.
Table S2 shows the results of comparing M9 with Baleng Crab (Mr). Thirteen TFs were differentially expressed, eight were upregulated and five were downregulated. All the TFs belonged to ten gene families.
Table S3 shows the results of comparing SH40 with Mr. Forty-five TFs were differentially expressed, with nine upregulated and thirty-six downregulated TFs. All the TFs belonged to 20 gene families.
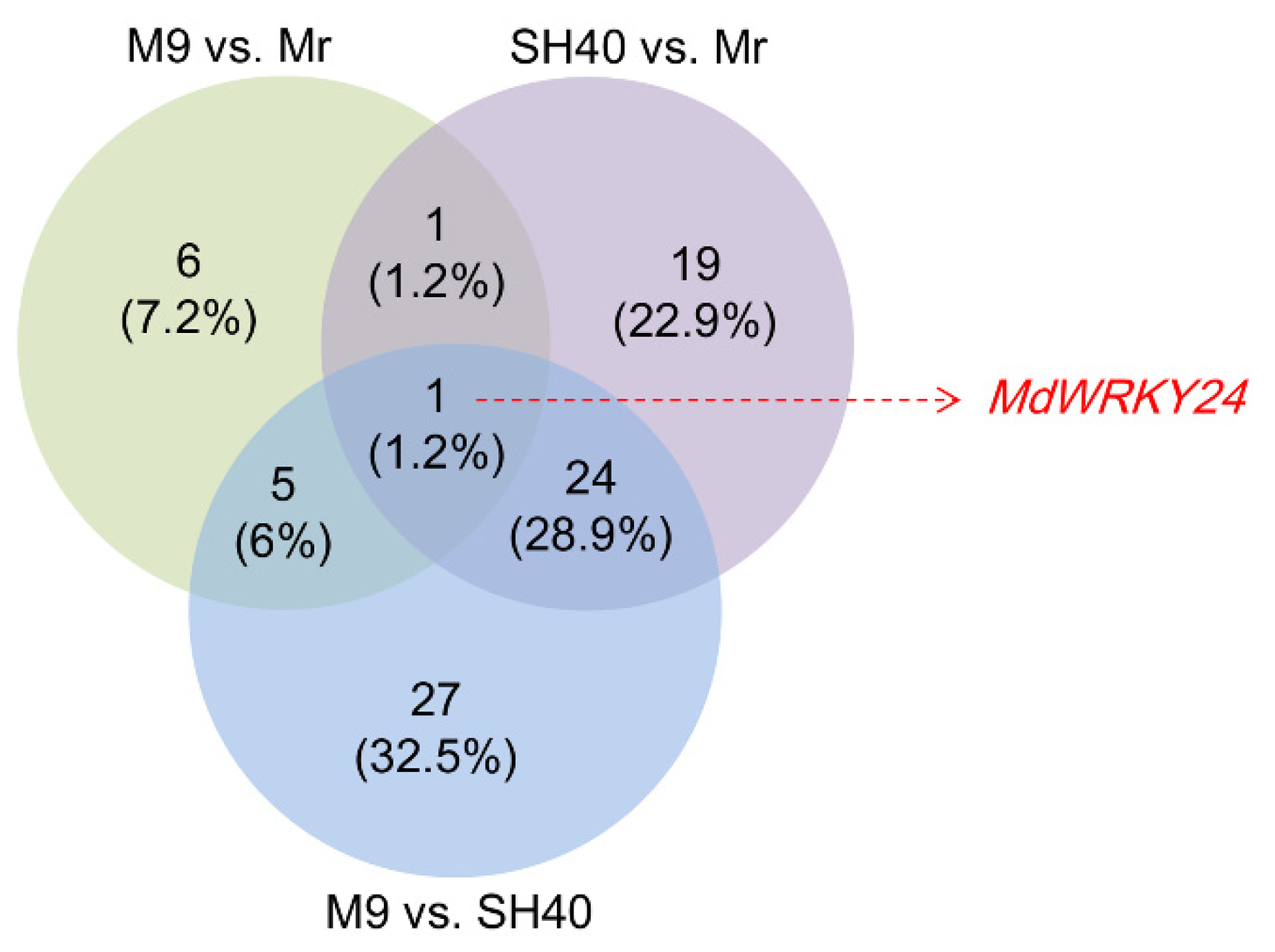
Table S4 shows the results of comparing SH40 with M9. Fifty-seven TFs were differentially expressed; among them, twenty were upregulated and thirty-seven were downregulated. All the TFs belonged to 20 gene families. The MdWRKY24 (gene ID: 103417614) was the only common differentially expressed gene in the pooled comparison of the three groups (
Figure 1), so the following experiments focused on the WRKY family and the functionality of MdWRKY24.
3.2. Expression Analysis and GO Enrichment of MdWRKY Family Members
By comparing the transcriptome sequencing results in the GDR data, 151 TFs containing conserved WRKY domains were obtained (
Table S5). Overall, 125 TFs of the MdWRKY family were differentially expressed in different rootstocks (
Table S5). Among them, 112, 115, and 120 WRKY TFs were expressed in the groups with SH40, M9, and 120 Mr as the interstock. The selectively expressed genes of the
MdWRKY family in different stock and scion combinations are shown in
Table S6. Gene 103402322 was only expressed in Fuji/SH40/Mr; genes 103402297, 103407769, and 103413747 were not expressed in Fuji/SH40/Mr. Genes 103404419, 103409645, 10341136, 103417041, and 103448476 were only expressed in Fuji/M9/Mr; genes 103411844, 103423637, 103425538, and 103426879 were not expressed only in Fuji/M9/Mr. Genes 103417878, 103425943, 103439393, 103443683, 103450758, and 103451854 were only expressed in Fuji/Mr.
GO enrichment of the 125 differentially expressed WRKY TFs between the three groups with SH40, M9, and Mr as interstocks identified 34 functional categories of biological processes, cellular components, and molecular functions (
Figure 2). Among them, more than 100 MdWRKY gene family members were enriched in six functional categories, including the cell biological process, biological regulation, metabolic process, biological process regulation, binding function, and nucleic acid-binding TF activity. The first four functional categories (the cellular process, biological regulation, metabolic process, and regulation of biological process) belong to biological processes, and the latter two functional categories (binding and nucleic acid-binding transcription factor activity) belong to molecular functions.
3.3. Analysis of MdWRKY24
The
MdWRKY gene family was screened according to |log
2 (fold-change)| > 1 and a
p-value < 0.05. Ten MdWRKY TFs were obtained with different interstocks and distributed on different chromosomes (
Table 1).
Table 1 shows that
MdWRKY24 expression was significantly upregulated in the scion with M9 as the interstock, compared with the other two groups. The expression of
MdWRKY40,
MdWRKY70,
MdWRKY49,
MdWRKY33,
MdWRKY50, and
MdWRKY35 was significantly upregulated in the scion with SH40 as the interstock compared with the other two groups.
MdWRKY7,
MdWRKY14, and
MdWRKY13 were significantly downregulated in the scion with SH40 and M9 as the interstock compared to that with Mr as the interstock.
Most MdWRKY TFs have a typical WRKYGQK domain, and some core domains are mutated to WRKYGKK. As shown in
Figure S1, the amino acid sequences of ten WRKY TFs with significant differences were aligned. Of these, nine WRKY TFs had conserved domains, contained C2H2-type zinc finger structures, and belonged to the WRKY class II subfamily. One protein (gene ID: 103433764) had a conserved WRKY domain, contained a C2HC-type zinc finger structure, and belonged to the WRKY class III subfamily. In the phylogenetic tree of ten significantly differentially expressed TFs,
MdWRKY24 (gene ID: 103417614) and
MdWRKY9 (gene ID: 103433719) were the closest (
Figure 3).
3.4. Verification of Differential Expression of MdWRKY Transcription Factors
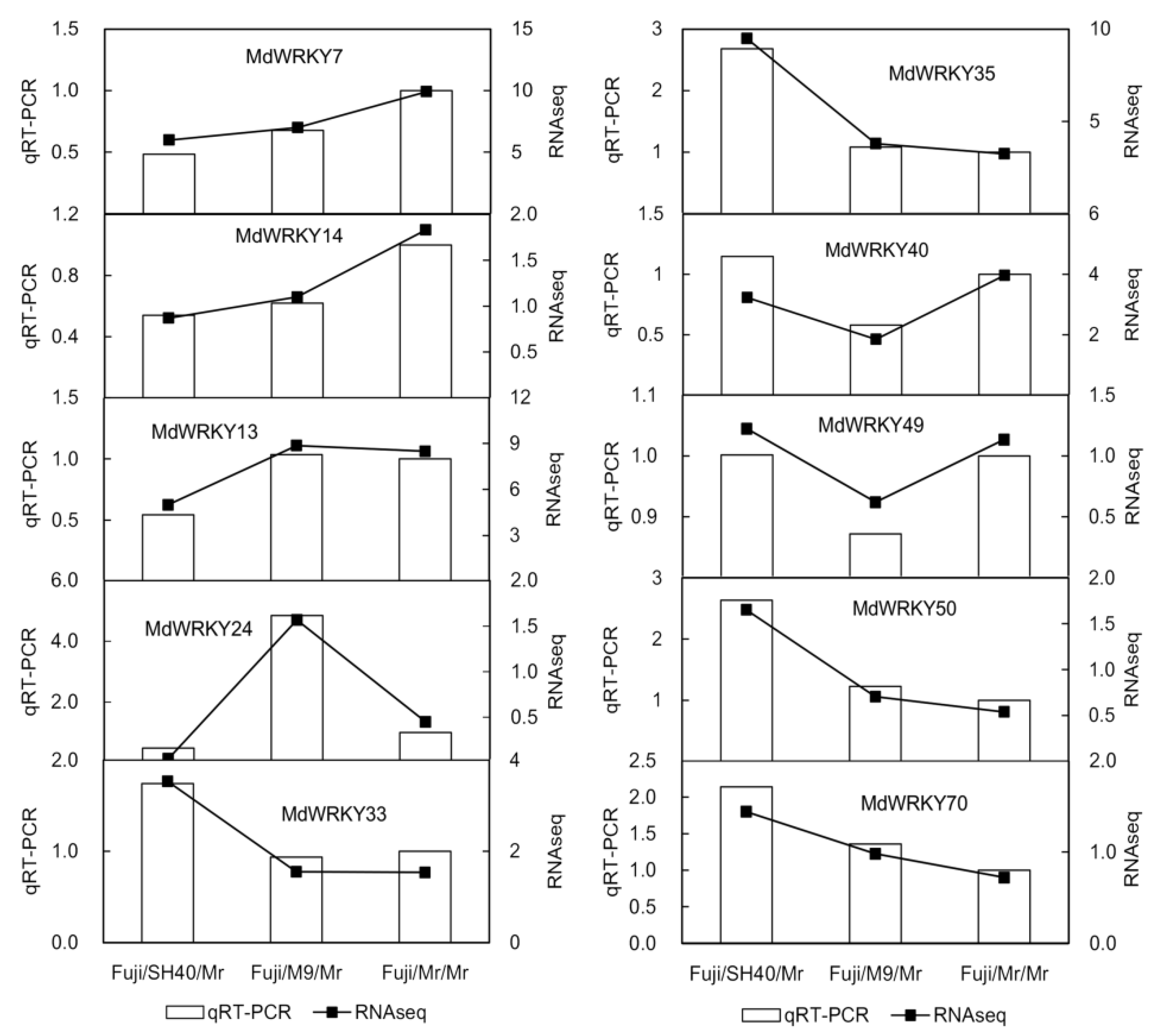
A real-time fluorescence quantification of ten significantly different TFs was performed to verify the reliability of the transcriptome sequencing results (
Figure 4). The two methods (qRT-PCR and RNA-seq) had the same overall trend, indicating that the transcriptome sequencing results were reliable and that the follow-up gene screening and experiments could be carried out based on the results.
3.5. Bioinformatics Analysis of MdWRKY24
The full-length open reading frame (ORF) region of
MdWRKY24 is 657 bp, encoding 218 amino acids (
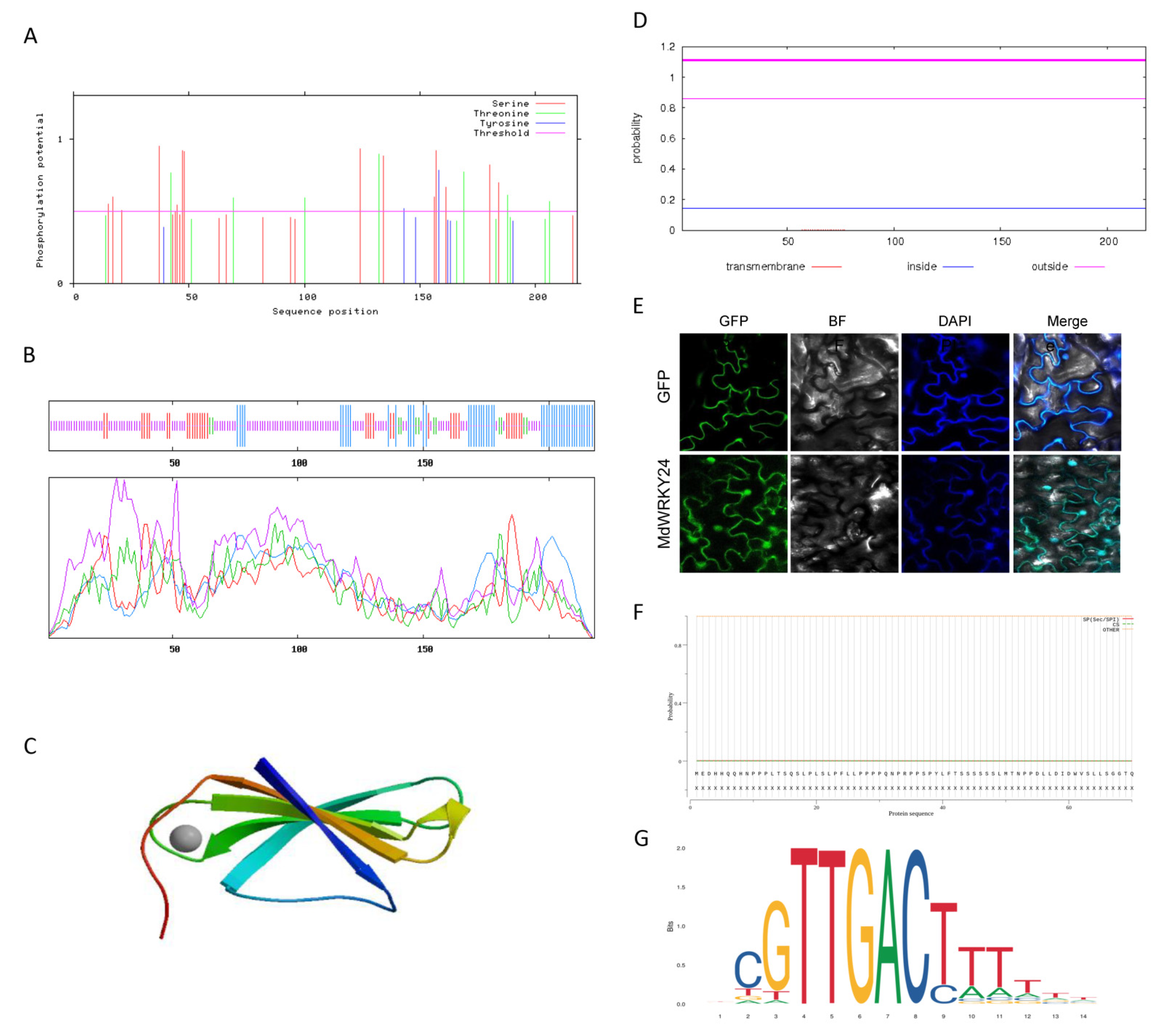
Figure S2). The MdWRKY24 phosphorylation sites were analyzed by NetPhos3.1 (
Figure 5A). The MdWRKY24 protein has a total of 43 phosphorylation sites, of which 23 are serine phosphorylation sites, 13 are amino acid phosphorylation sites, and 1 is a tyrosine phosphorylation site, indicating that the phosphorylation modifications of the protein are mainly serine, followed by threonine. The secondary structure of the MdWRKY24 protein was predicted using SOPMA (
Figure 5B). The protein composition consisted of 22.02% α-helix (Hh), 16.51% extended link (Ee), 5.50% β-turn (Tt), and 55.96% random coil (Cc).
Figure 5C shows the prediction model of the MdWRKY24 protein tertiary structure using SWISS-MODEL. The GMQE value of the protein molecular structure encoded by the MdWRKY24 model was 0.12, the QMN4 value (a comprehensive scoring function for model quality assessment) was −1.22, the modeling method was X-ray, the resolution was 1.60 A, and the sequence similarity was 48% (127–200), with a sequence coverage of 34%. The MdWRKY24 protein is mainly composed of four β-sheets and random coil groups. As shown in
Figure 5D, there is no transmembrane helix outside the protein membrane and no transmembrane region, indicating that the MdWRKY24 protein is an intramembrane protein. Furthermore, MdWRKY24 was localized in the nucleus (
Figure 5E).
Figure 5F shows the prediction of the signal peptide of the amino acid sequence of
MdWRKY24 using SignalP5. As no signal peptide was found at the N-terminus of the protein, the
MdWRKY24 gene was presumed to encode a non-secreted protein.
Figure 5G shows the prediction of the MdWRKY24 protein binding motif (T)TGAC (C/T), known as W-box, using PlantTFDB.
3.6. Homologous Sequence Alignment and Phylogenetic Tree Analysis of MdWRKY24 Protein
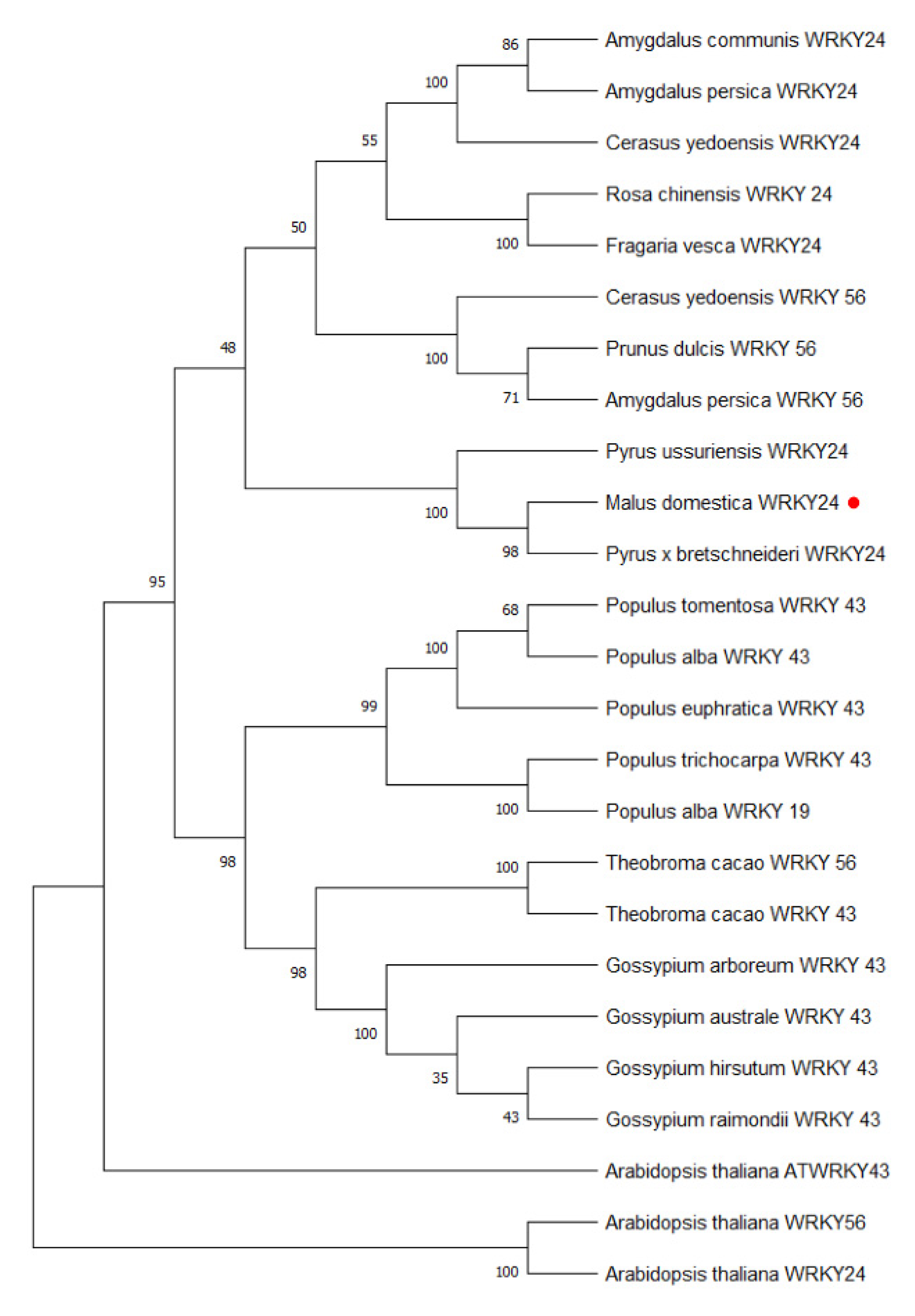
The amino acid sequences of the MdWRKY24 gene were aligned using Blastp. The protein sequences of 14 similar species were obtained, namely white pear (Pyrus × bretschneideri), European sweet cherry (Cerasus avium), plum (Armeniaca mume), peach (Amygdalus persica), Tokyo cherry blossom (Cerasus × yedoensis), wild strawberry (Fragaria vesca ssp.), rose (Rosa chinensis), cocoa (Theobroma cacao), upland cotton (Gossypium hirsutum), longan (Dimocarpus longan), Myrica rubra, mulberry (Morus notabilis), white oak (Quercus lobata), and almond (Amygdalus communis).
The 15 matched proteins belonged to the WRKY family and contained the WRKY domain, which is a 60-amino acid region defined by the conserved amino acid sequence WRKYGQK (in the red box) at the N-terminus; they had a novel C2H2-type zinc finger motif (positioned by the black arrow) and belonged to the WRKY class II subfamily (
Figure S3). MdWRKY24 binds specifically to the DNA sequence motif (T)(T)TGAC(C/T), known as the W-box. The MdWRKY24 protein highly overlaps with the multiple functional domains of the WRKY proteins of other species (the blue part in the figure). The MdWRKY24 and PbrWRKY24 of white pear were the closest phylogenetically, as they were in the same branch and had the highest protein sequence similarity (
Figure 6).
3.7. MdWRKY24 Promoter Region Analysis and Upstream Promoter Binding Gene Prediction
In the upstream 1600 bp region of the
MdWRKY24 gene, a number of homeopathic elements or motifs related to plant hormone response were searched, including ABA-related homeopathic elements, ABRE and G-box, methyl jasmonate regulatory motifs, TGACG-motif, and CGTCA-motif (
Table 2).
According to the promoter sequence of the MdWRKY24 gene and the plant transcriptional regulation network data platform, 21 TFs were predicted to bind upstream, including one AP2 gene family protein, one GATA gene family protein, two BBR_BPC gene family proteins, one C2H2 gene family protein, one GRAS gene family protein, two TALE gene family proteins, two NAC gene family proteins, two MYB related genes family proteins, four MYB gene family proteins, and five MIKC_MADS gene family proteins. This indicates that the MdWRKY24 gene may combine with other TFs to regulate downstream target genes.
3.8. Expression of MdWRKY24 in Different Tissues
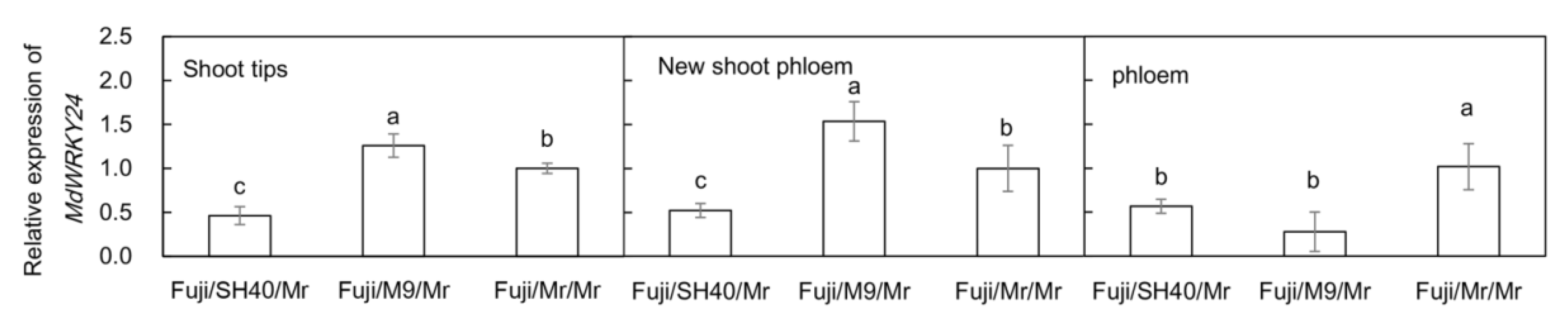
As shown in
Figure 7, the expression of the
MdWRKY24 gene in the shoot tip and new shoot phloem of the group with M9 as the interstock was significantly higher than that of Fuji/SH40/Mr (about 3 folds) and Fuji/Mr/Mr (1.2–1.5 folds). The expression level of the
MdWRKY24 gene in the phloem of Fuji/Mr/Mr was significantly higher than that of Fuji/SH40/Mr (about 2 folds) and Fuji/M9/Mr (about 3.5 folds).
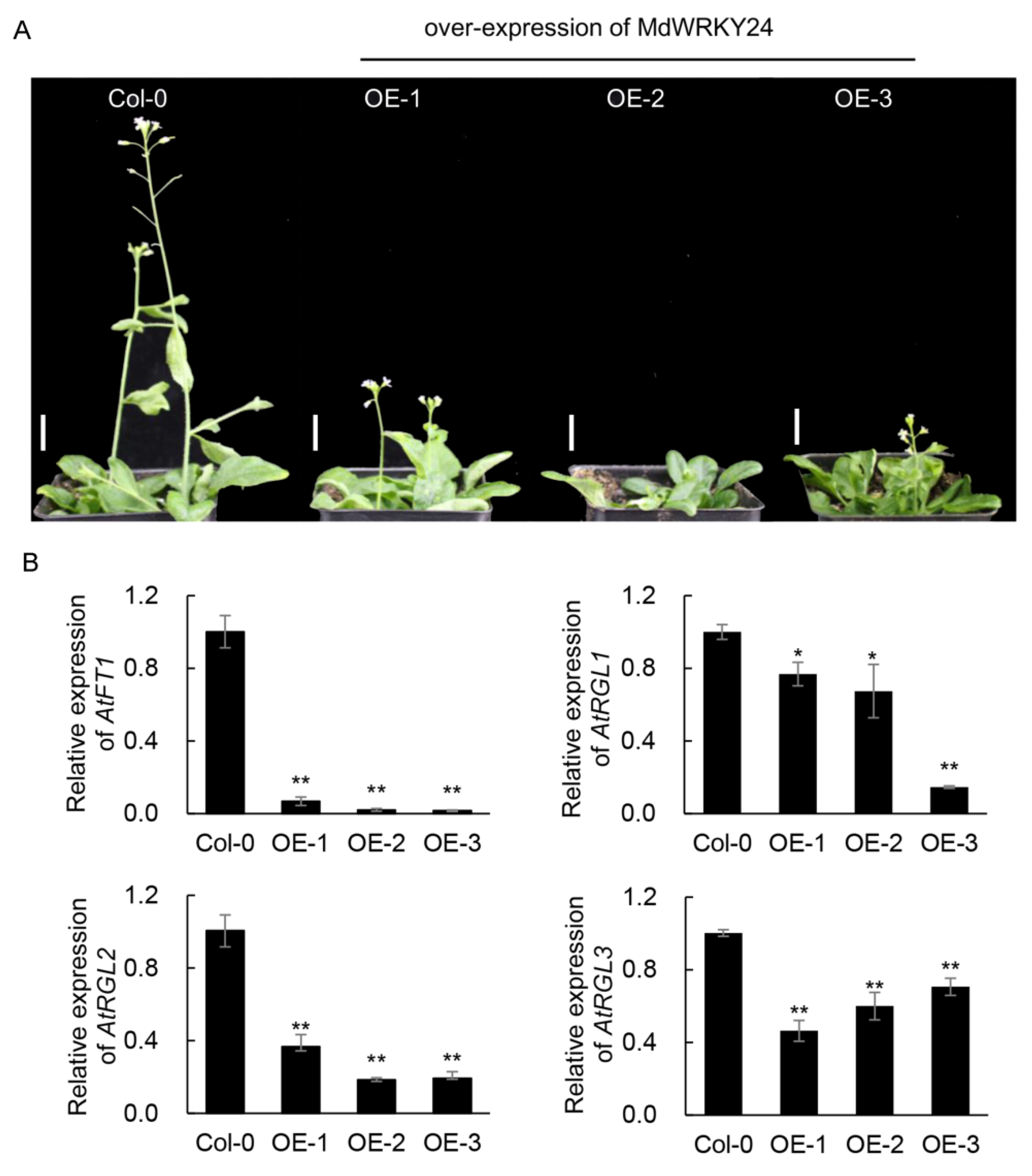
3.9. Functional Analysis of MdWRKY24 after Heterologous Expression in Arabidopsis
MdWRKY24 overexpression resulted in dwarfing traits and delayed flowering in
Arabidopsis. The expression of AtFT1, AtRGL1, AtRGL2, and AtRGL3 in MdWRKY24-overexpressing
Arabidopsis was significantly lower than that in the wild-type (
Figure 8).
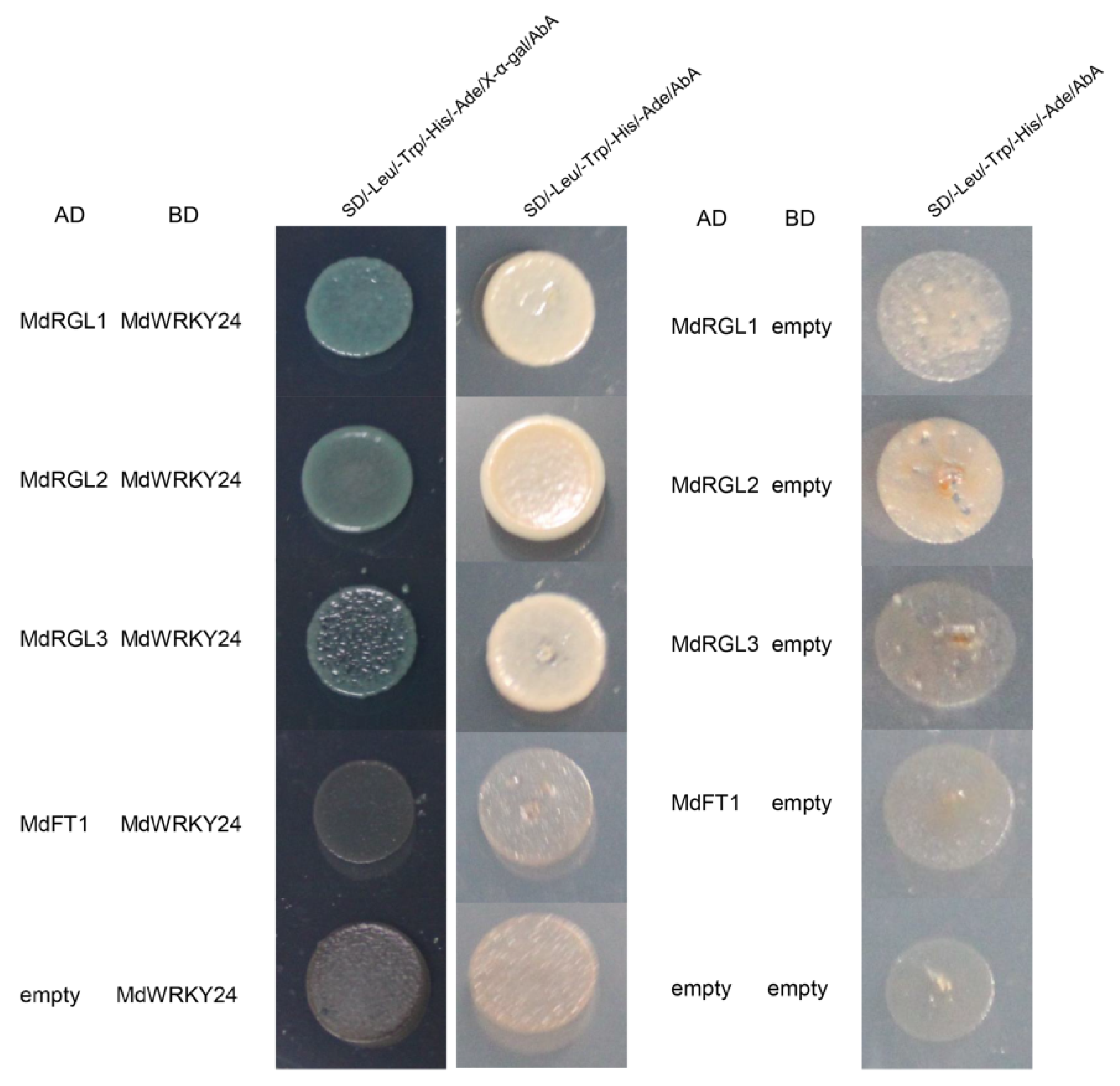
3.10. Analysis of MdWRKY24-Interacting Proteins
MdRGL1-AD+MdWRKY24-BD, MdRGL2-AD+MdWRKY24-BD, and MdRGL3-AD+MdWRKY24-BD were grown on SD/-Leu/-Trp/-His/-Ade plates (containing X-α-gal), which turned blue; while MdRGL1-AD+empty-BD, MdRGL2-AD+empty-BD, MdRGL3-AD+empty-BD, null-AD+MdWRKY24-BD, and MdFT1-AD+MdWRKY24-BD could not grow on the SD/-Leu/-Trp/-His/-Ade plates (
Figure 9). There was no obvious color change on the SD/-Leu/-Trp/-His/-Ade plates (containing X-α-gal). The above results indicate that MdRGL1, MdRGL2, and MdRGL3 interact with MdWRKY24, but MdFT1 does not interact with MdWRKY24.
4. Discussion
In the present study, 150 possible WRKY TFs were obtained by analyzing the RNA-seq data. The MdWRKY TFs were selectively expressed in different stock and scion combinations. The expression analysis of MdWRKY24 showed different expression patterns in different stock and scion combinations. Differences between groups were significant, consistent with the trend of differential expression in the shoot tip and new shoot phloem, indicating that the differential regulation of TF expression inherent in rootstocks may yield expression differences in the scion, regulating the growth of the tree.
The pairwise comparison of the WRKY TFs of different stock-scion combinations revealed that the MdWRKY24 gene was differentially expressed. The CDs sequence of the cloned MdWRKY24 gene was 657 bp, containing the conserved WRKYGQK domain and the C2H2-type zinc finger structure, thus belonging to the WRKY class II subfamily. The downstream protein binding motif was W-box. MdWRKY24 had the highest similarity with the PbrWRKY24 protein in white pear; however, the function of PbrWRKY24 is still unclear. Phylogenetic tree analysis of apple family members revealed that MdWRKY24 and MdWRKY9 were most closely related.
WRKY TFs have a wide range of functions and play important roles in plant growth and stress responses. Previous studies have shown that MdWRKY9 overexpression in the M26 rootstock can inhibit the expression of the brassinolide synthesis gene
MdDWF4, resulting in a decrease in the content of brassinolide in M26 and a dwarf phenotype [
19] in
Arabidopsis thaliana. Analysis of the AtWRKY62 loss-of-function mutants confirmed that AtWRKY62 adversely affected the
Arabidopsis growth [
14]. After the transfection of the AtWRKY18 overexpression vector into the
Arabidopsis tetraploids, the growth of the transgenic
Arabidopsis was reduced, and a dwarf phenotype appeared [
15]. Here, we noted that dwarfing traits emerged after the MdWRKY24 overexpression, supporting that MdWRKY TFs are related to plant dwarfing. These findings provide strong evidence that the dwarfing-related TFs induced by dwarfing rootstocks regulated growth in apple trees.
MdWRKY45 interacts with the DELLA protein RGA-like 1 (RGL1), a repressor of the gibberellin signaling pathway, and WRKY45, which actively regulates age-induced leaf senescence. RGL1 is a key component of the gibberellic acid (GA)-mediated signaling pathway [
27]. Both in vivo and in vitro biochemical analyses demonstrated that WRKY75 interacts with DELLA proteins [
28]. We found that both RGL1 and GA insensitive (GAI) inhibited the activation ability of AtWRKY75, thereby attenuating the expression of its regulator [
28]. Therefore, we also analyzed the relationship between MdWRKY24 and
RGL genes and found that MdWRKY24 and MdRGL1/2/3 can interact, indicating that the MdWRKY24-MdRGL pathway mediates the GA signaling pathway to regulate plant growth and the dwarfing phenotype.
In the present paper, the RGL gene was identified as a DELLA-like protein, metabolized by gibberellin, and related to apple dwarfing. Zhang et al. [
29] found that the IAA export protein PIN played a key role in the dwarfing of M9. MdNAC1 plays a role in plant dwarfing traits, potentially by regulating ABA and BR production [
30]. The results of the studies above indicate that hormones (gibberellin, IAA, ABA, and BR) are related to the mechanism of dwarfing rootstocks. Therefore, whether the dwarfing traits of plants depend on a single hormone or the synergistic effects of multiple hormones warrants further research
Chen et al. [
16] found that CsWRKY7 may be involved in plant growth in tea plants. They studied the flowering time integrator gene and floral meristem characteristic genes of WRKY7-overexpressing lines. Furthermore, mutations in the
AtWRKY75 gene lead to delayed flowering in
Arabidopsis, and chromatin immunoprecipitation analysis showed that AtWRKY75 directly binds to the promoter of
MdFT1 [
28]. Genetic analysis showed that AtWRKY75 positively regulates flowering in a FLOWERING LOCUS T (FT)-dependent manner [
28]. Here, we found that MdWRKY24 overexpression delayed flowering, and yeast two-hybrid mining revealed no interaction between MdWRKY24 and the flowering gene
MdFT1, indicating that MdWRKY24 may affect plant flowering in other ways. TFs regulate the specific spatiotemporal transcription of downstream target genes by binding to specific cis-elements (hormone-related and photosynthetic-related homeopathic elements), thereby precisely regulating the timing of plant development and rapidly responding to external stresses. The present study found that the
MdWRKY24 gene promoter has 21 binding protein motifs for AP2, GATA, BBR_BPC, C2H2, GRAS, TALE, NAC, MYB, and MIKC_MADS TFs. These results indicate that the
MdWRKY24 gene may be mobilized by hormones, photosynthesis, or TFs. Follow-up research should explore more in-depth molecular regulation pathways related to MdWRKY24-mediated regulation of dwarfing mechanisms. Nevertheless, the current study provides a new path for the molecular-assisted breeding of apple dwarf rootstocks, offering technical support for further developing apple cultivation techniques.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}