
Metabolomic and Transcriptomic Analyses Provide New Insights into Health-Promoting Metabolites from Cannabis Seeds Growing in the Bama Region of China

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
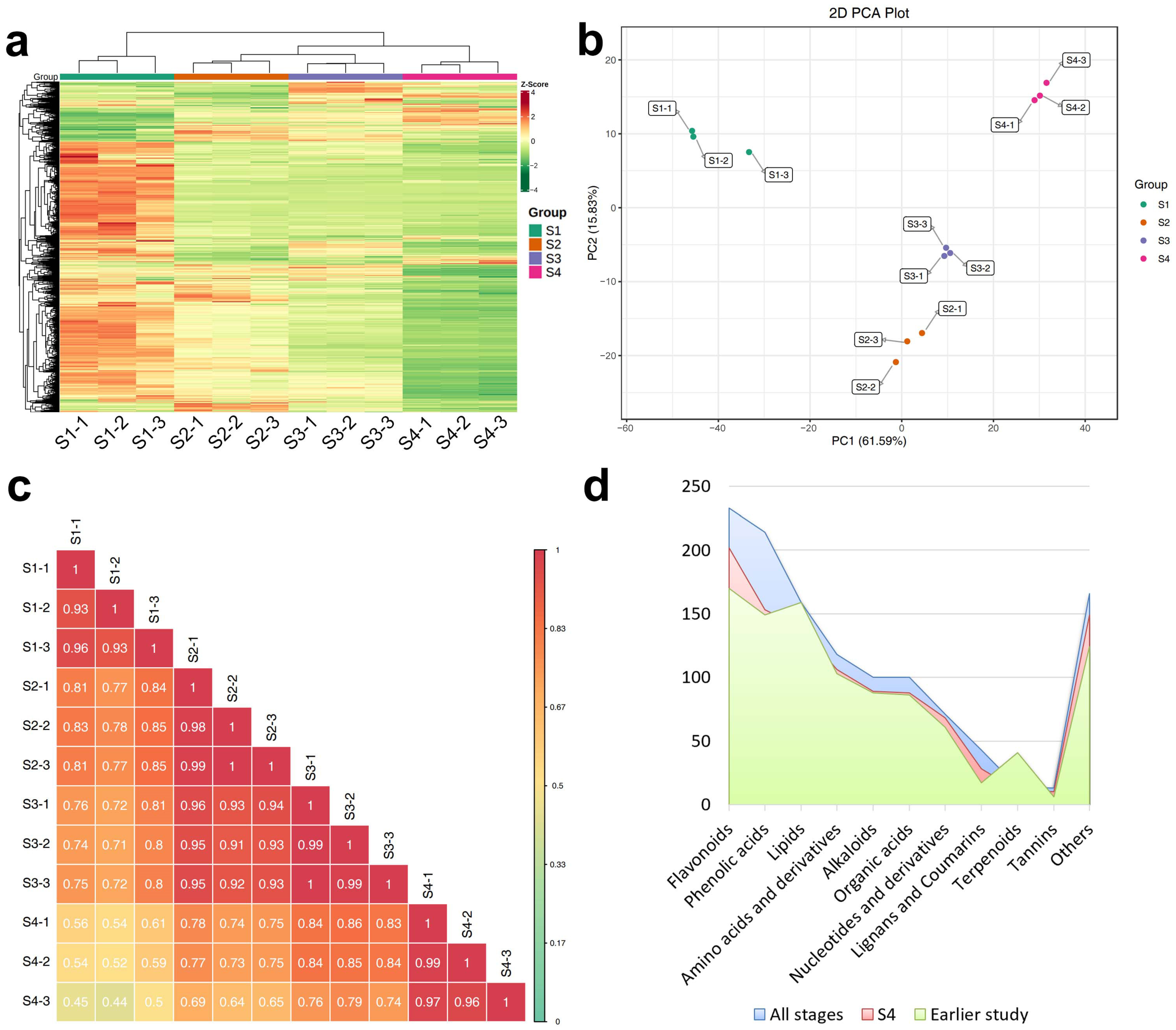
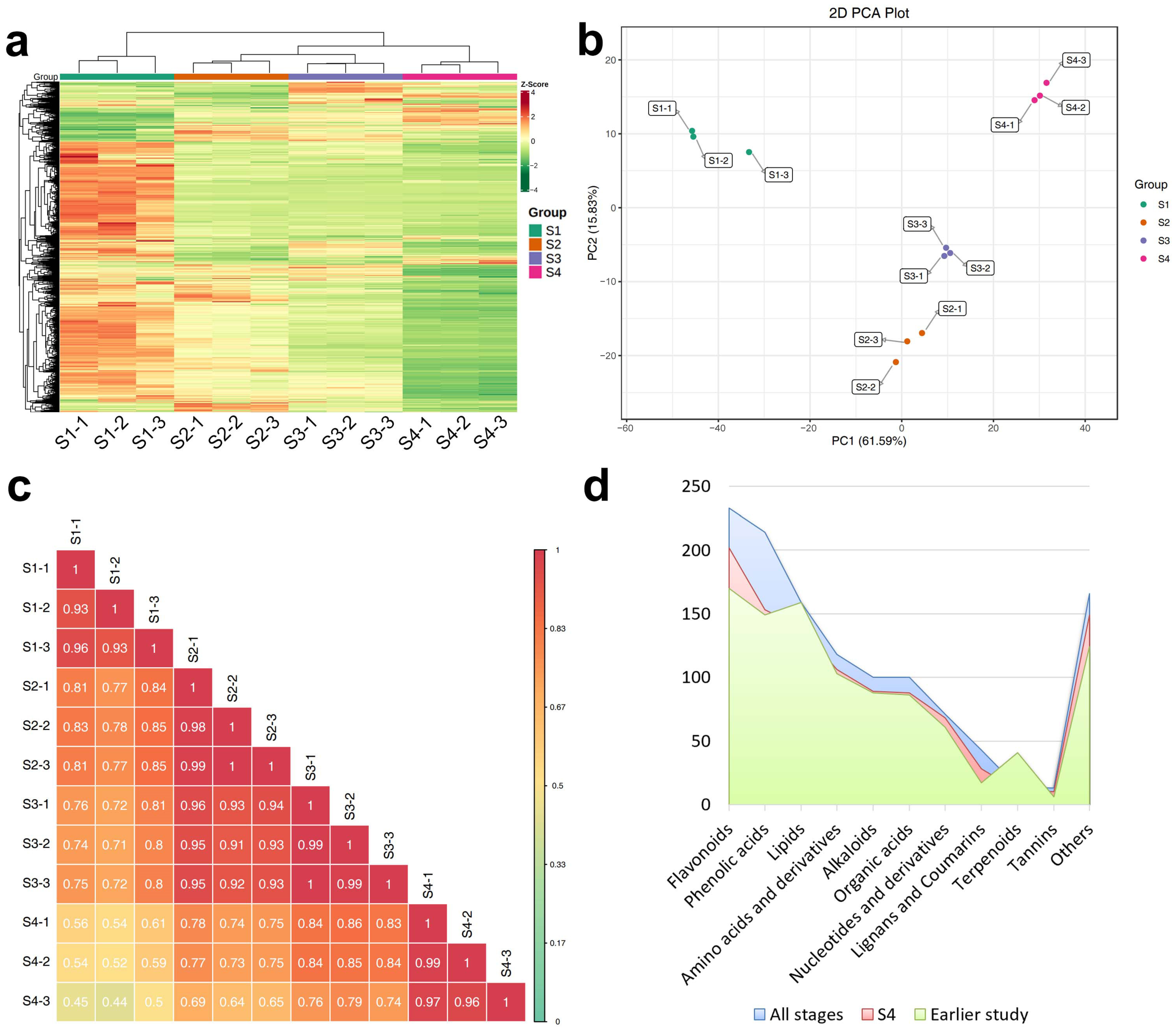
2.1. Metabolome of Hemp Seeds at Different Maturity Stages
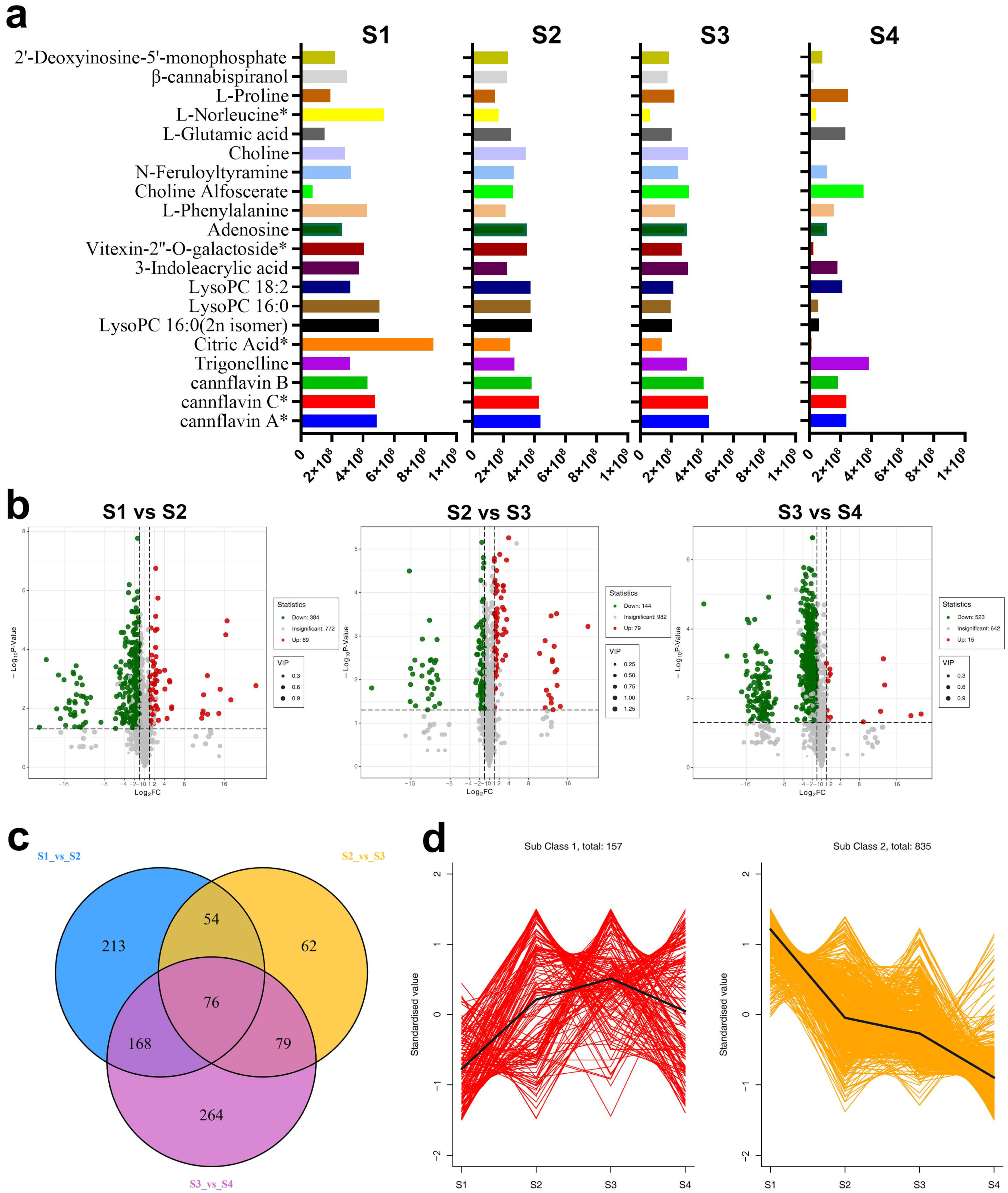
2.1.1. Composition of Metabolites
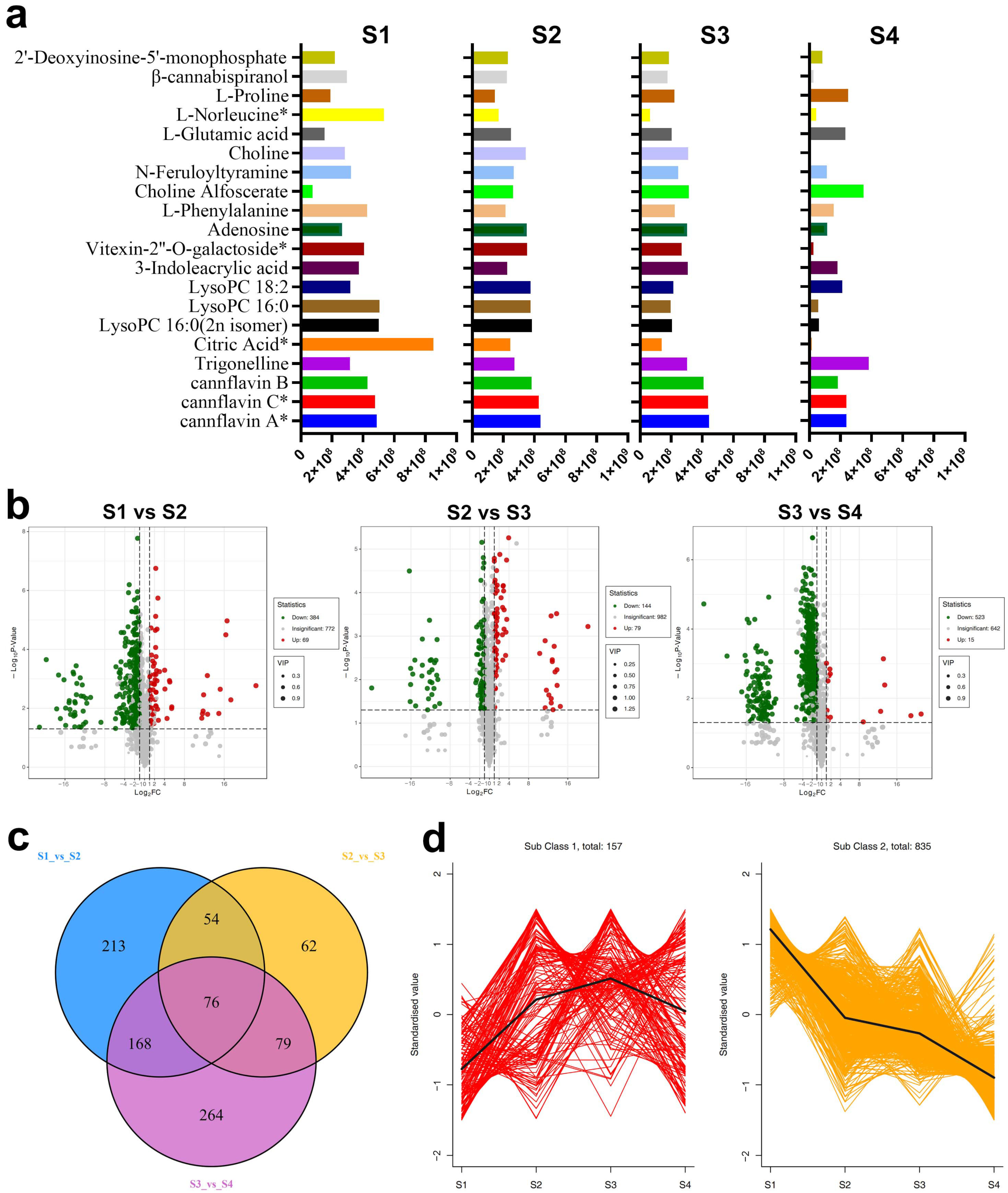
2.1.2. Differentially Accumulated Metabolites
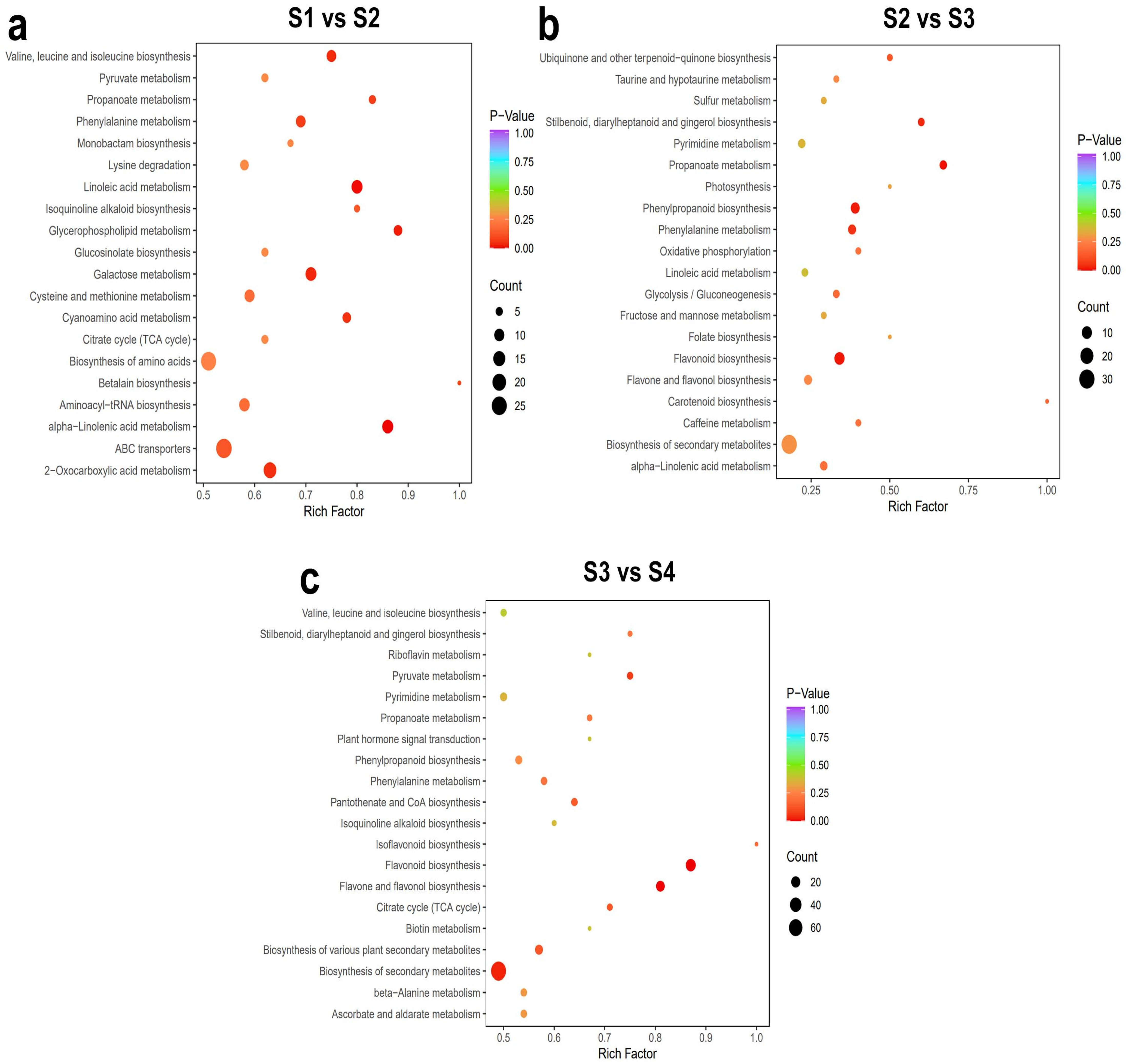
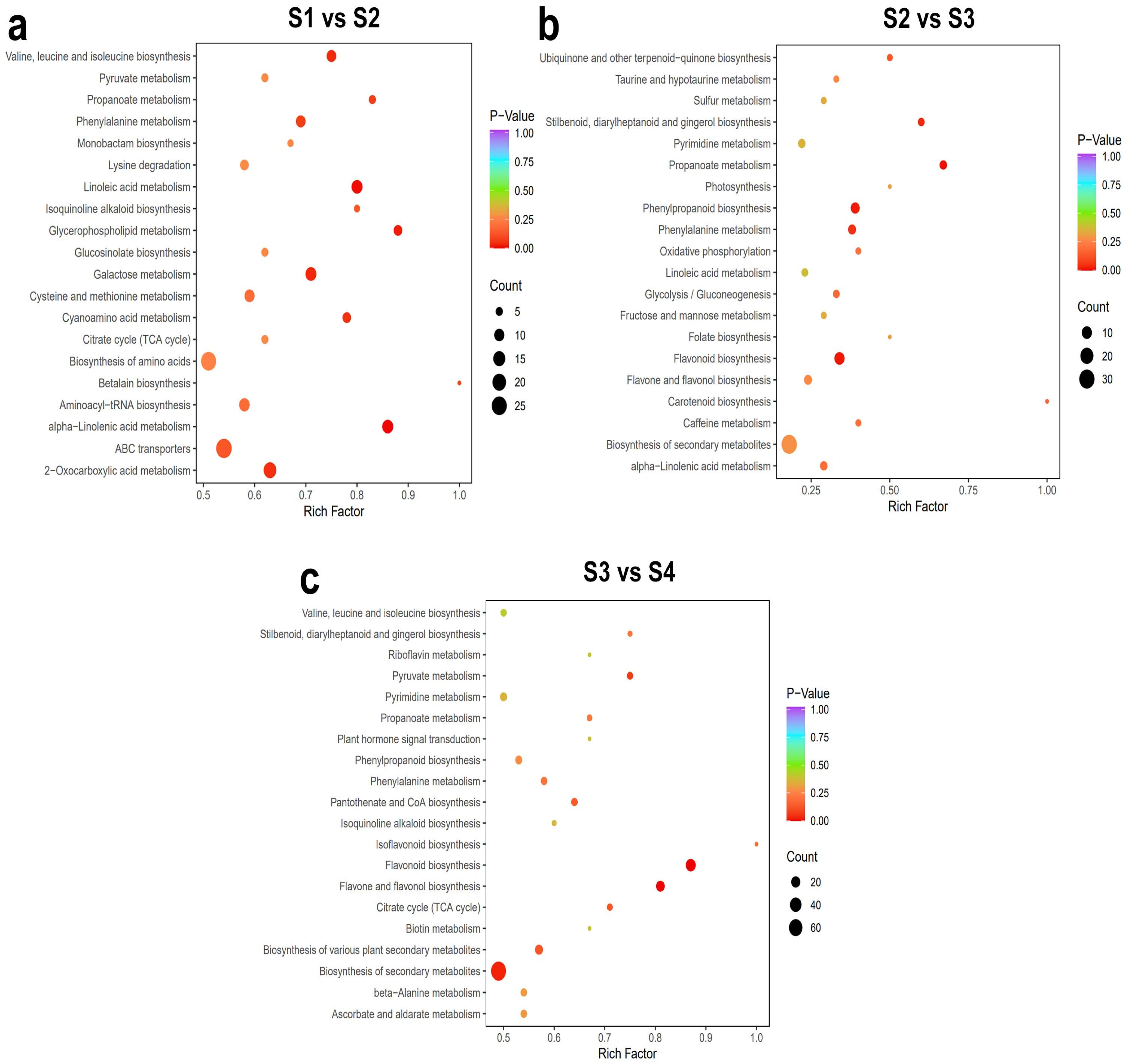
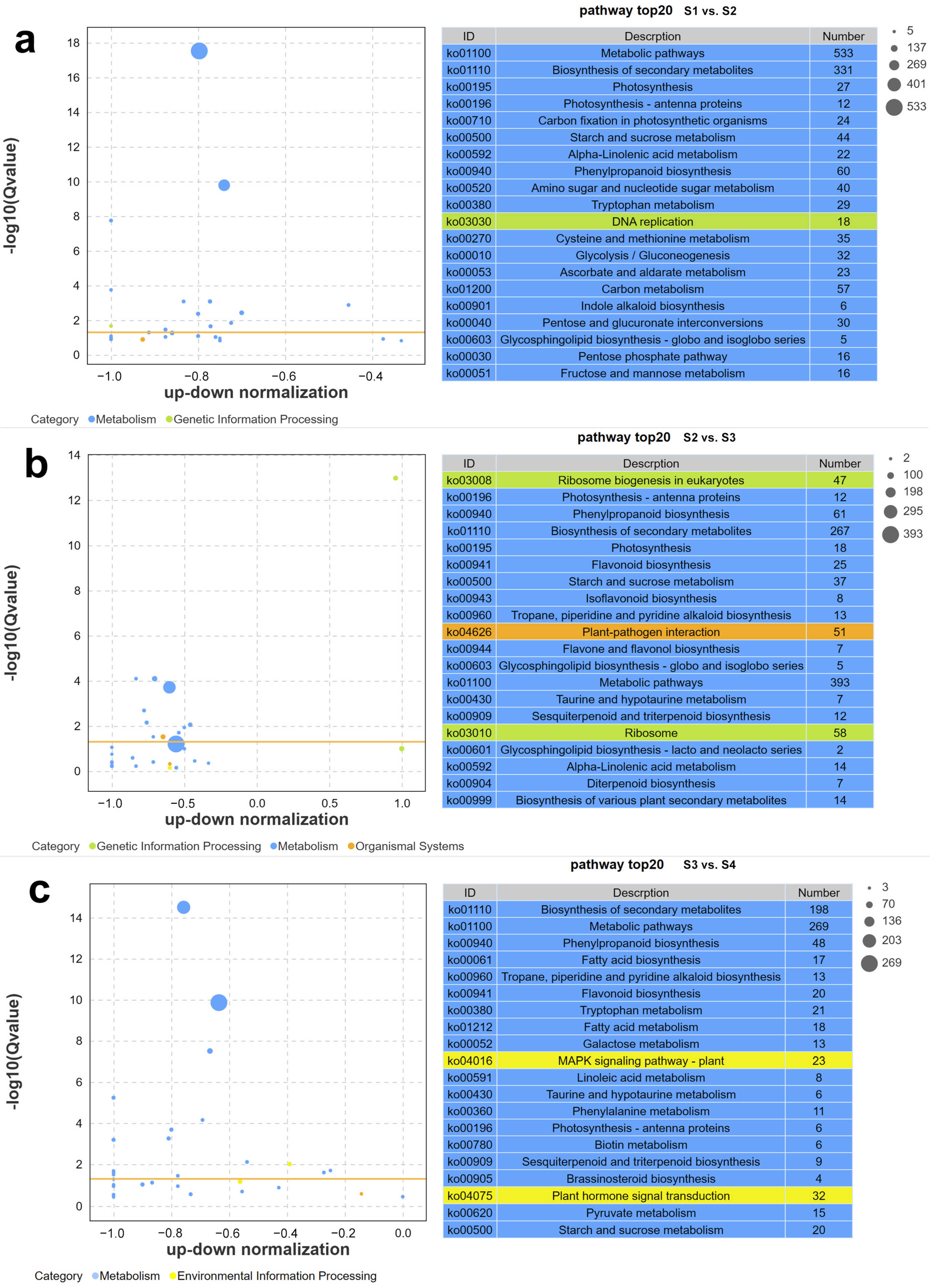
2.1.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
2.2. Transcriptome Analysis of Hemp Seeds at Different Stages of Maturity
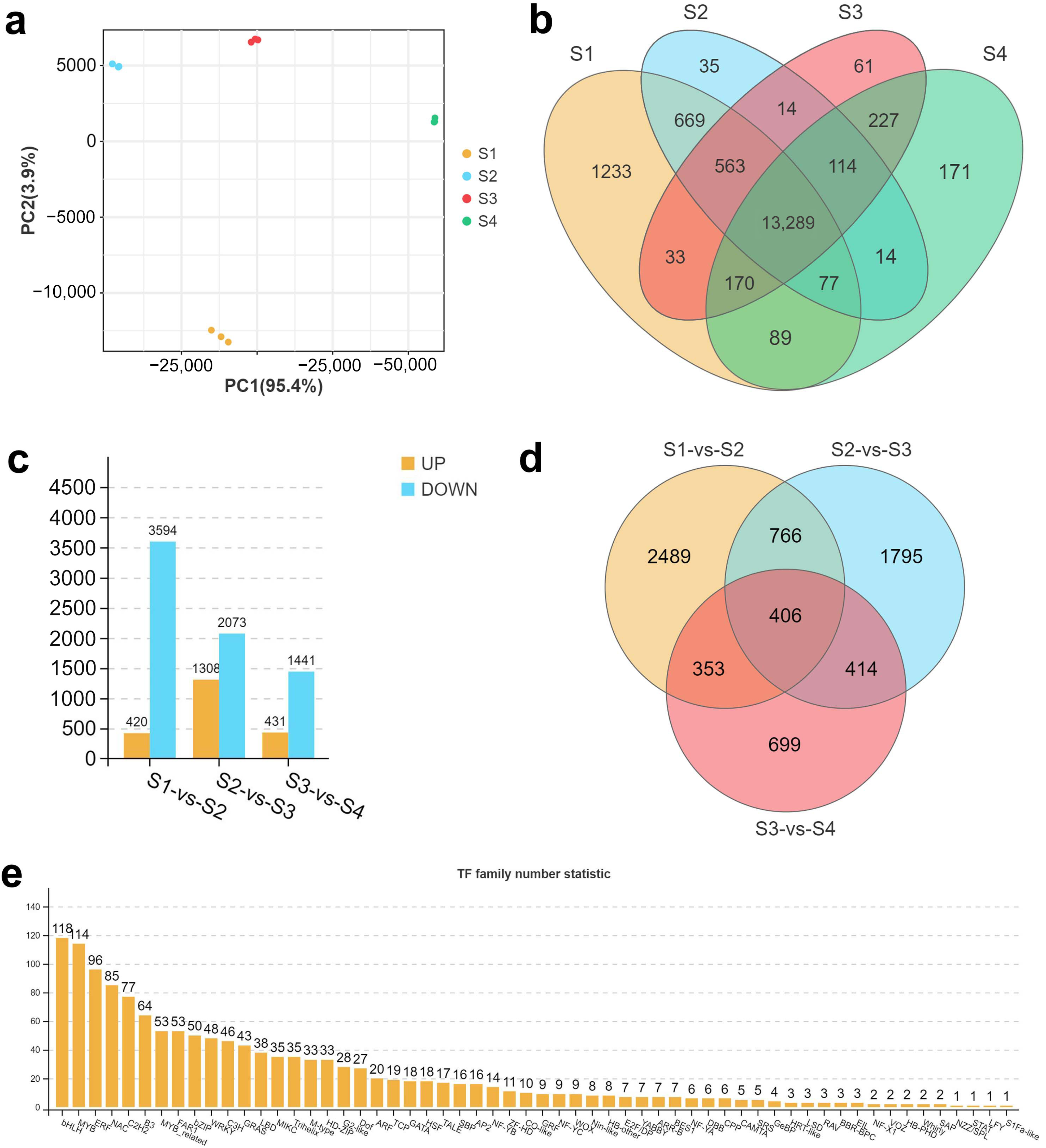
2.2.1. Sequencing Statistics
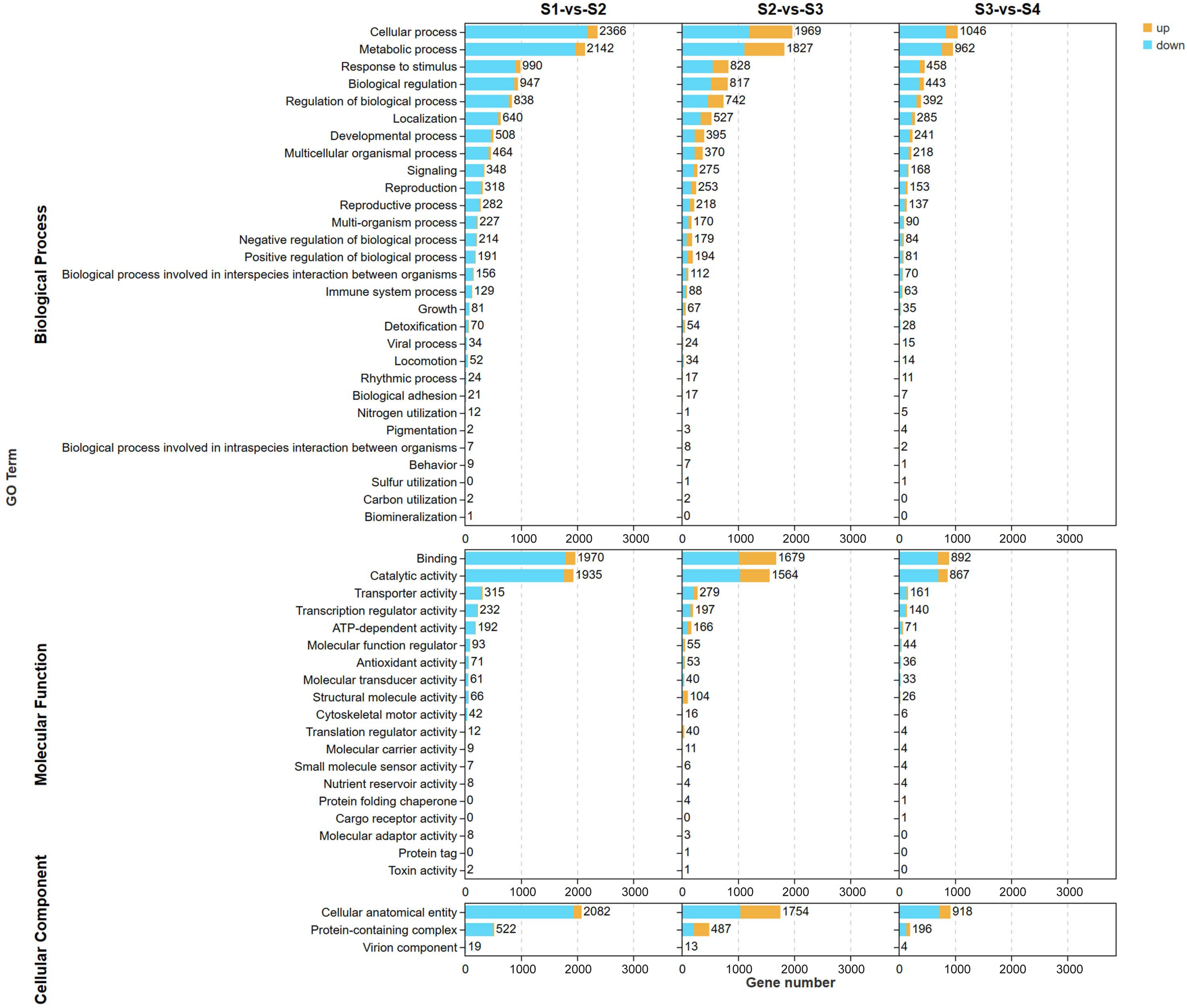
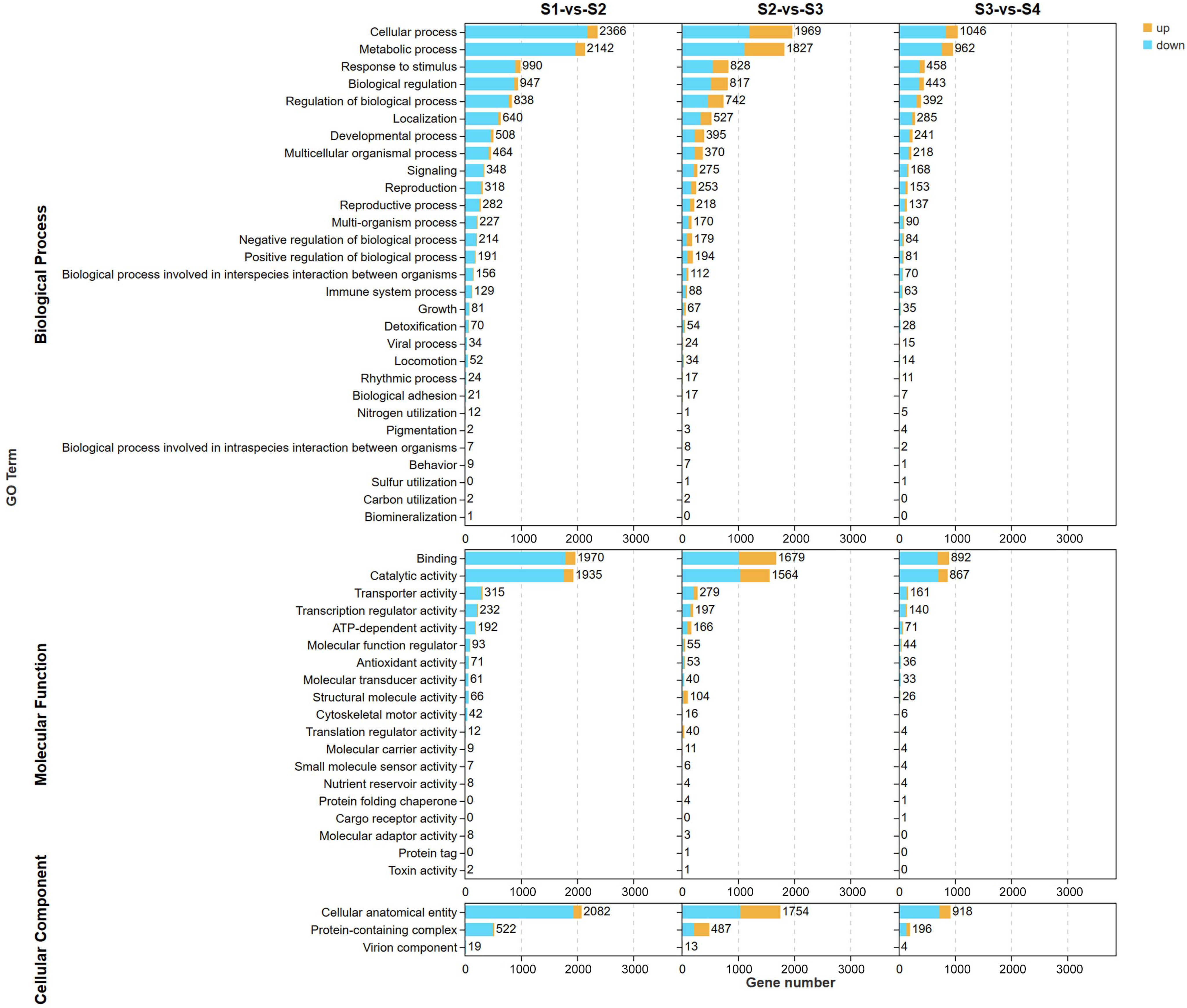
2.2.2. Gene Ontology (GO) and KEGG Analysis
2.3. Correlation Analysis
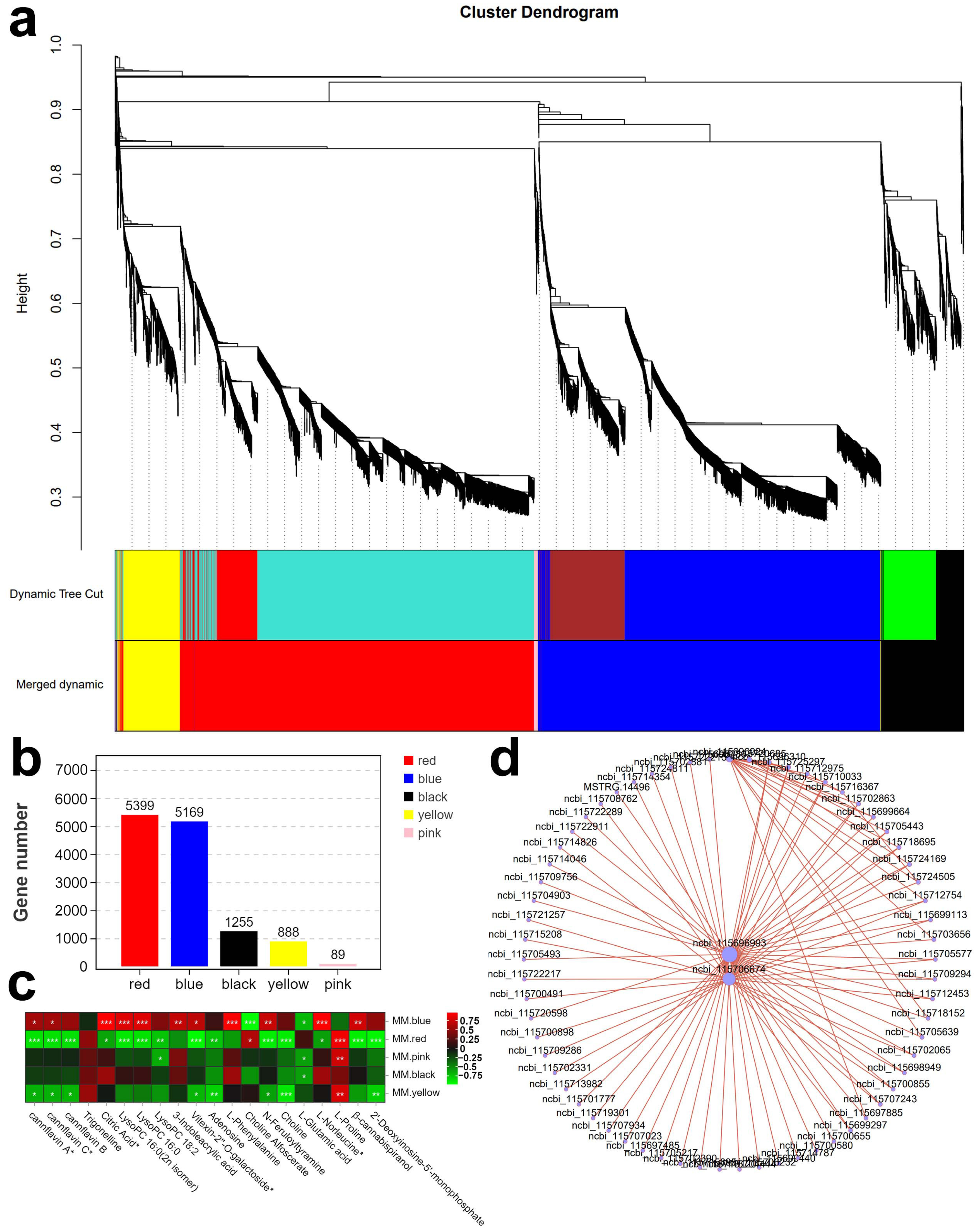
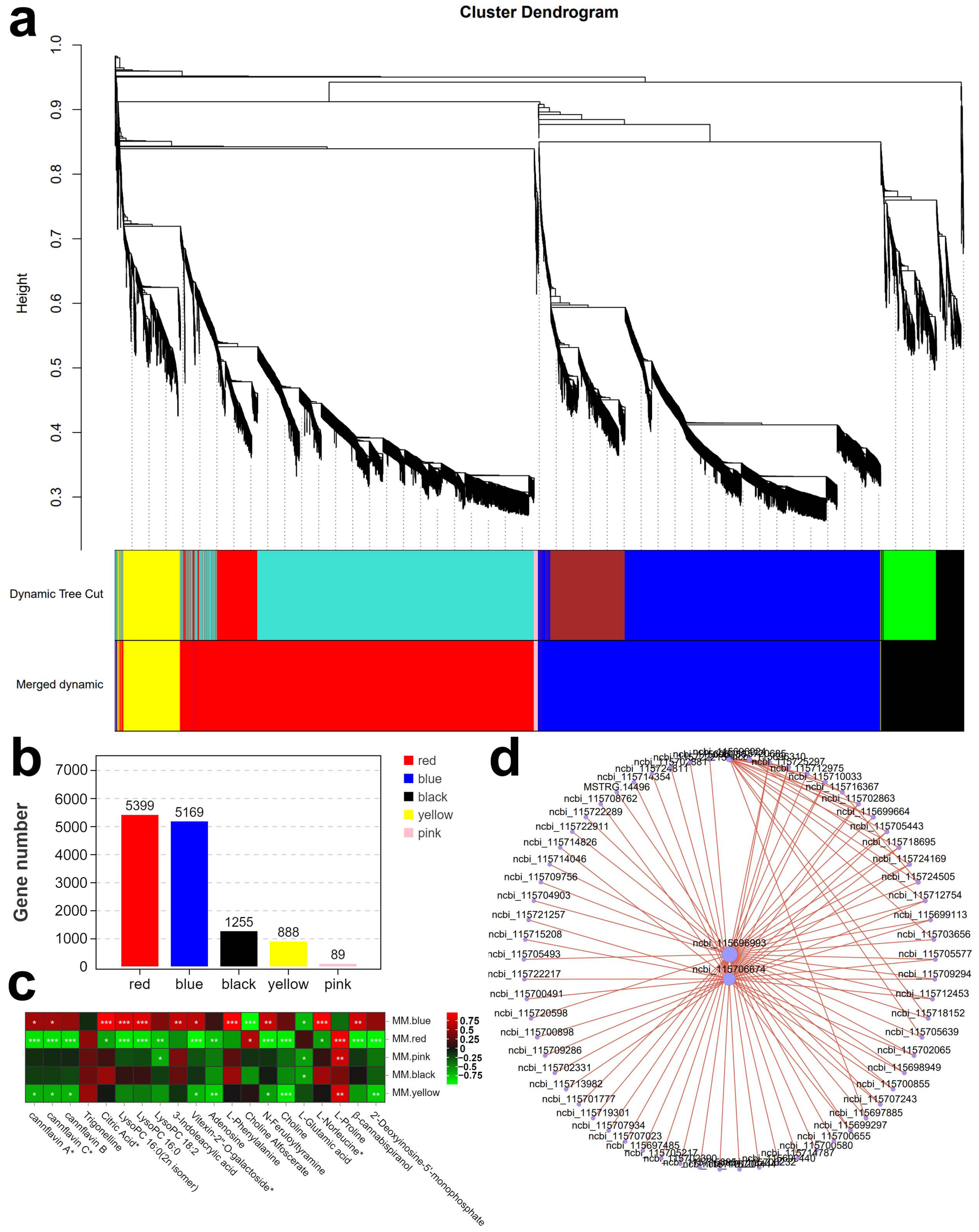
2.3.1. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.3.2. TF Genes Related to Metabolite Classes in Hemp Seeds Revealed by K-Means Clustering Analysis
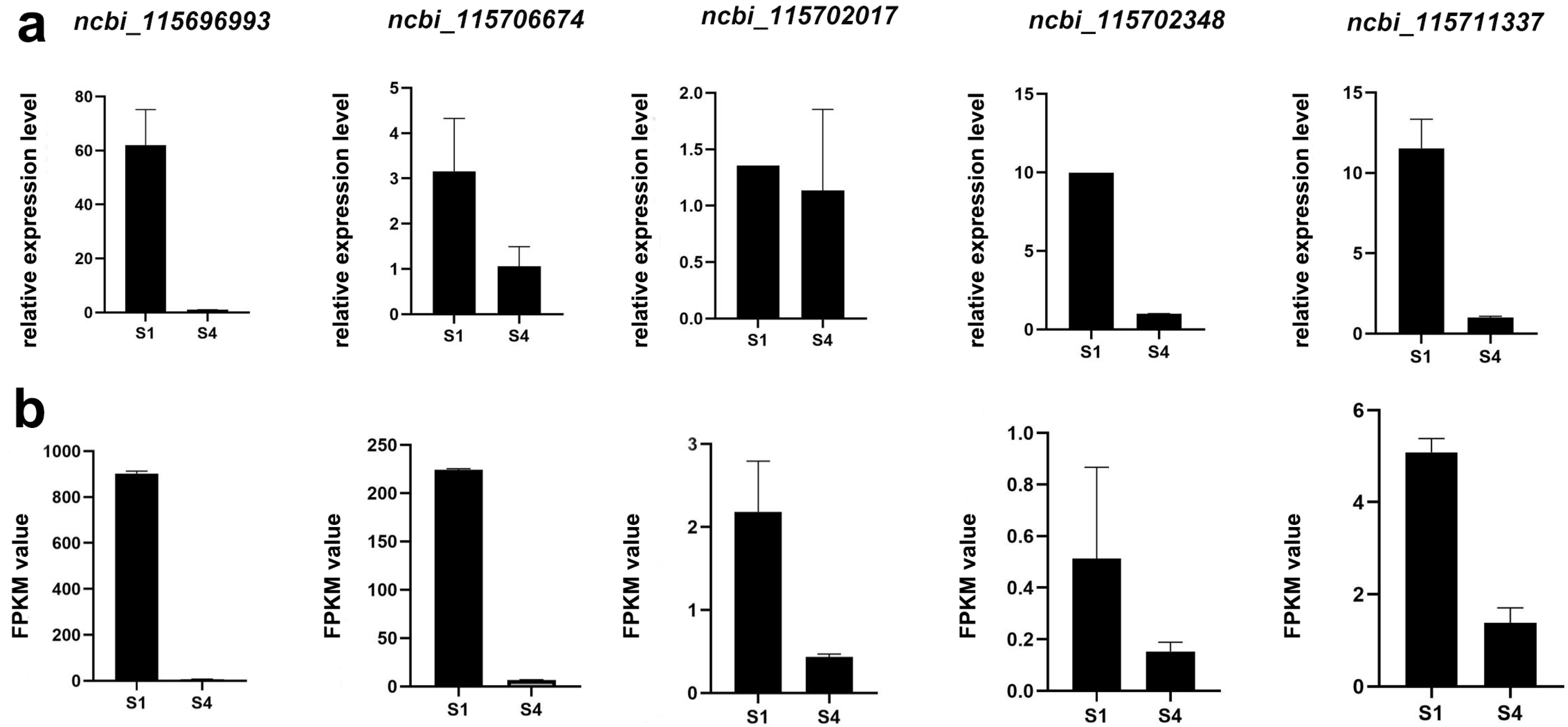
2.4. qPCR Valid
3. Discussion
4. Materials and Methods
4.1. Sampling and Experimental Procedures
4.2. Targeted Metabolome Analysis
4.3. Transcriptome Sequencing
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.5. Transcription Factor (TF) and K-Means Clustering Analysis of Metabolites
4.6. Quantitative Polymerase Chain Reaction (qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Small, E. Evolution and Classification of Cannabis sativa (Marijuana, Hemp) in Relation to Human Utilization. Bot. Rev. 2015, 81, 189–294. [Google Scholar] [CrossRef]
- Eržen, M.; Košir, I.J.; Ocvirk, M.; Kreft, S.; Čerenak, A. Metabolomic Analysis of Cannabinoid and Essential Oil Profiles in Different Hemp (Cannabis sativa L.) Phenotypes. Plants 2021, 10, 966. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, J.; Zhao, J.; Wang, W.; Griffin, J.; Li, Y.; Bean, S.; Tilley, M.; Wang, D. Hempseed as a nutritious and healthy human food or animal feed source: A review. Int. J. Food Sci. Technol. 2021, 56, 530–543. [Google Scholar] [CrossRef]
- Brand, E.J.; Zhao, Z. Cannabis in Chinese Medicine: Are Some Traditional Indications Referenced in Ancient Literature Related to Cannabinoids? Front. Pharmacol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Hou, C.; Wei, X.; Zhou, Y.; Zhang, S.; Chen, Y.; Yu, H.; Dong, L.; Chen, S. Metabolomics Analysis Revealed the Characteristic Metabolites of Hemp Seeds Varieties and Metabolites Responsible for Antioxidant Properties. Front. Plant Sci. 2022, 13, 904163. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Yuan, L. Challenges towards Revitalizing Hemp: A Multifaceted Crop. Trends Plant Sci. 2017, 22, 917–929. [Google Scholar] [CrossRef] [PubMed]
- House, J.D.; Neufeld, J.; Leson, G. Evaluating the Quality of Protein from Hemp Seed (Cannabis sativa L.) Products through the use of the Protein Digestibility-Corrected Amino Acid Score Method. J. Agric. Food Chem. 2010, 58, 11801–11807. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Ali, V.; Khajuria, M.; Faiz, S.; Gairola, S.; Vyas, D. GC-MS based metabolomic approach to understand nutraceutical potential of Cannabis seeds from two different environments. Food Chem. 2021, 339, 128076. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.J.; Feng, B.; Ahmad, M.H.; Tahir, M.; Aslam, M.Z.; Khalid, M.F.; Hussain, S.; Zhong, R.; Ali, Q.; Xu, Q.; et al. LC-MS/MS-based metabolomics approach identified novel antioxidant flavonoids associated with drought tolerance in citrus species. Front. Plant Sci. 2023, 14, 1150854. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vizcaino, F.; Fraga, C.G. Research trends in flavonoids and health. Arch. Biochem. Biophys. 2018, 646, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.J.; Duan, M.; Yang, M.; Fan, H.; Shen, S.; Hu, L.; Wang, L. Novel insights into anthocyanin metabolism and molecular characterization of associated genes in sugarcane rinds using the metabolome and transcriptome. Int. J. Mol. Sci. 2022, 23, 338. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.F.; Xiao, Z.T.; Zhang, H.Y. Where do health benefits of flavonoids come from? Insights from flavonoid targets and their evolutionary history. Biochem. Biophys. Res. Commun. 2013, 434, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Chen, Y.C. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013, 138, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, A.C.; Aucott, L.; Reid, D.M.; Macdonald, H.M. Associations between dietary flavonoid intakes and bone health in a scottish population. J. Bone Miner. Res. 2011, 26, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.H.; Jiang, Y.M.; Shi, J.; Tomás-Barberán, F.A.; Datta, N.; Singanusong, R.; Chen, S.S. Flavonoids in Food and Their Health Benefits. Plant Foods Hum. Nutr. 2004, 59, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Gómez Candela, C.; Bermejo López, L.M.; Loria Kohen, V. Importance of a balanced omega 6/omega 3 ratio for the maintenance of health: Nutritional recommendations. Nutr. Hosp. 2011, 26, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The Importance of the Omega-6/Omega-3 Fatty Acid Ratio in Cardiovascular Disease and Other Chronic Diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.; Jurgoński, A. The Effect of Hemp (Cannabis sativa L.) Seeds and Hemp Seed Oil on Vascular Dysfunction in Obese Male Zucker Rats. Nutrients 2021, 13, 2575. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; He, S. The co-evolution of therapeutic landscape and health tourism in bama longevity villages, China: An actor-network perspective. Health Place 2020, 66, 102448. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, H. Therapeutic landscapes and longevity: Wellness tourism in Bama. Soc. Sci. Med. 2018, 197, 24–32. [Google Scholar] [CrossRef]
- Ren, M.; Li, H.; Fu, Z.; Li, Q. Centenarian-Sourced Lactobacillus casei Combined with Dietary Fiber Complex Ameliorates Brain and Gut Function in Aged Mice. Nutrients 2022, 14, 324. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Ma, L.; Zhang, R.; Liu, F.; Jiang, P.; Xu, J.; Cao, H.; Du, X.; Lin, F.; Cheng, L.; et al. Plasma proteomic and autoantibody profiles reveal the proteomic characteristics involved in longevity families in Bama, China. Clin. Proteom. 2019, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, F.; Sun, D.W.; Li, Q. Intestinal Lactobacillus community structure and its correlation with diet of Southern Chinese elderly subjects. J. Microbiol. 2016, 54, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yu, T.; Huang, G.; Cai, D.; Liang, X.; Su, H.; Zhu, Z.; Li, D.; Yang, Y.; Shen, P.; et al. Gut Microbiota Community and Its Assembly Associated with Age and Diet in Chinese Centenarians. J. Microbiol. Biotechnol. 2015, 25, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, S.; Yu, G.; Zou, Y.; Lu, H.; Wei, Y.; Tang, J.; Long, B.; Tang, X.; Yu, D.; et al. Comparations of major and trace elements in soil, water and residents’ hair between longevity and non-longevity areas in Bama, China. Int. J. Environ. Health Res. 2021, 31, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Erridge, S.; Mangal, N.; Salazar, O.; Pacchetti, B.; Sodergren, M.H. Cannflavins—From plant to patient: A scoping review. Fitoterapia 2020, 146, 104712. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.A.; Casaretto, J.A.; Al-Abdul-Wahid, M.S.; Sukumaran, A.; Geddes-McAlister, J.; Rothstein, S.J.; Akhtar, T.A. Biosynthesis of cannflavins A and B from Cannabis sativa L. Phytochemistry 2019, 164, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.J.; Duan, M.; Wei, X.; Zuo, H.; Ma, L.; Tahir ul Qamar, M.; Li, M.; Han, S.; Hu, L.; Wang, L. LC-MS/MS-based metabolomics approach revealed novel phytocompounds from sugarcane rind having promising pharmacological value. J. Sci. Food Agric. 2022, 102, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Ashihara, H.; Ludwig, I.A.; Katahira, R.; Yokota, T.; Fujimura, T.; Crozier, A. Trigonelline and related nicotinic acid metabolites: Occurrence, biosynthesis, taxonomic considerations, and their roles in planta and in human health. Phytochem. Rev. 2015, 14, 765–798. [Google Scholar] [CrossRef]
- Zhou, J.; Chan, L.; Zhou, S. Trigonelline: A Plant Alkaloid with Therapeutic Potential for Diabetes and Central Nervous System Disease. Curr. Med. Chem. 2012, 19, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Meneguzzo, F.; Delisi, R.; Pagliaro, M. Citric acid: Emerging applications of key biotechnology industrial product. Chem. Cent. J. 2017, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Babaei, F.; Moafizad, A.; Darvishvand, Z.; Mirzababaei, M.; Hosseinzadeh, H.; Nassiri-Asl, M. Review of the effects of vitexin in oxidative stress-related diseases. Food Sci. Nutr. 2020, 8, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Min, J.-W.; Kong, W.-L.; He, X.-H.; Li, J.-X.; Peng, B.-W. A review on the pharmacological effects of vitexin and isovitexin. Fitoterapia 2016, 115, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Catanesi, M.; d’Angelo, M.; Antonosante, A.; Castelli, V.; Alfonsetti, M.; Benedetti, E.; Desideri, G.; Ferri, C.; Cimini, A. Neuroprotective potential of choline alfoscerate against β-amyloid injury: Involvement of neurotrophic signals. Cell Biol. Int. 2020, 44, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, C. A review of redox signaling and the control of MAP kinase pathway in plants. Redox Biol. 2017, 11, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Girolomoni, L.; Ferrante, P.; Berteotti, S.; Giuliano, G.; Bassi, R.; Ballottari, M. The function of LHCBM4/6/8 antenna proteins in Chlamydomonas reinhardtii. J. Exp. Bot. 2017, 68, 627–641. [Google Scholar] [CrossRef]
- Rao, M.J.; Tahir ul Qamar, M.; Wang, D.; Ali, Q.; Ma, L.; Han, S.; Duan, M.; Hu, L.; Wang, L. A high-throughput lipidomics and transcriptomic approach reveals novel compounds from sugarcane linked with promising therapeutic potential against COVID-19. Front. Nutr. 2022, 9, 988249. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.J.; Duan, M.; Wang, J.; Han, S.; Ma, L.; Mo, X.; Li, M.; Hu, L.; Wang, L. Transcriptomic and Widely Targeted Metabolomic Approach Identified Diverse Group of Bioactive Compounds, Antiradical Activities, and Their Associated Genes in Six Sugarcane Varieties. Antioxidants 2022, 11, 1319. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wen, L.; Chen, Z.; Zhang, Z.; Pang, X.; Deng, Z.; Liu, T.; Guo, Y. Study on metabolic variation in whole grains of four proso millet varieties reveals metabolites important for antioxidant properties and quality traits. Food Chem. 2021, 357, 129791. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–d334. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Miller, J.R.; Andre, R. Quantitative polymerase chain reaction. Br. J. Hosp. Med. 2014, 75, 188–192. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Class | p-Value | TF Family | NR Description |
|---|---|---|---|---|
| ncbi_115718555 | 1 | 0.016 | Dof | XP_030503214.1 dof zinc finger protein DOF2.4 [Cannabis sativa] |
| ncbi_115712978 | 1 | 0.049 | TALE | XP_030497288.1 BEL1-like homeodomain protein 2 isoform X1 [Cannabis sativa] |
| ncbi_115711337 | 2 | <0.001 | GRAS | XP_030495535.1 scarecrow-like protein 3 [Cannabis sativa] |
| ncbi_115702017 | 2 | <0.001 | MYB | XP_030485339.1 transcription factor MYB62 [Cannabis sativa] |
| ncbi_115698497 | 2 | 0.001 | MYB | XP_030481432.1 transcription factor MYB30-like [Cannabis sativa] |
| ncbi_115698755 | 2 | 0.001 | NAC | XP_030481781.1 NAC domain-containing protein 83 [Cannabis sativa] |
| ncbi_115702348 | 2 | 0.001 | NAC | XP_030485672.1 NAC domain-containing protein 30 [Cannabis sativa] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, M.; Rao, M.J.; Li, Q.; Zhao, F.; Fan, H.; Li, B.; He, D.; Han, S.; Zhang, J.; Wang, L. Metabolomic and Transcriptomic Analyses Provide New Insights into Health-Promoting Metabolites from Cannabis Seeds Growing in the Bama Region of China. Agronomy 2024, 14, 787. https://doi.org/10.3390/agronomy14040787
Duan M, Rao MJ, Li Q, Zhao F, Fan H, Li B, He D, Han S, Zhang J, Wang L. Metabolomic and Transcriptomic Analyses Provide New Insights into Health-Promoting Metabolites from Cannabis Seeds Growing in the Bama Region of China. Agronomy. 2024; 14(4):787. https://doi.org/10.3390/agronomy14040787
Chicago/Turabian StyleDuan, Mingzheng, Muhammad Junaid Rao, Qing Li, Falin Zhao, Hongzeng Fan, Bo Li, Dandan He, Shijian Han, Jiangjiang Zhang, and Lingqiang Wang. 2024. "Metabolomic and Transcriptomic Analyses Provide New Insights into Health-Promoting Metabolites from Cannabis Seeds Growing in the Bama Region of China" Agronomy 14, no. 4: 787. https://doi.org/10.3390/agronomy14040787